用于生产目标化合物的方法和设备
文献发布时间:2024-04-18 19:58:30
技术领域
本发明涉及根据相应独立专利权利要求的前序部分的用于生产目标化合物的方法和设备。
背景技术
具有二至四个碳原子的煤油的氧化脱氢(ODH)是众所周知的。在ODH过程中,所述煤油尤其用氧转化以得到相应的烯烃和水。本发明特别涉及乙烷氧化脱氢为乙烯,下文也称为ODHE。然而,本发明原则上不限于乙烷的氧化脱氢,而是还可以扩展到其他煤油例如丙烷或丁烷的氧化脱氢(ODH)。以下解释相应地适用于这种情况。
ODH(E)可能优于确立已久的用于生产烯烃的方法,例如蒸汽裂解或催化脱氢。例如,由于所涉及反应的放热性质以及几乎不可逆的形成水,因此不存在热力学平衡限制。ODH(E)可以在相对较低的反应温度下进行。原则上,不需要对所使用的催化剂进行再生,因为氧的存在能够或引起原位再生。最后,与蒸汽裂解相比,形成更少量的无价值的副产物,例如焦炭。
对于关于ODH(E)的进一步细节,可以参考相关文献,例如Ivars,F.and LópezNieto,J.M.,Light Alkanes Oxidation:Targets Reached and Current Challenges,inDuprez,D.and Cavani,F.(eds.),Handbook of Advanced Methods and Processes inOxidation Catalysis:From Laboratory to Industry,London 2014:Imperial CollegePress,第767-834页,或
特别地,基于MoVNb的催化剂体系已显示出用于ODH(E)的前景,如例如在F.Cavaniet al,"Oxidative dehydrogenation of ethane and propane:How far fromcommercial implementation?",Catal.Today,2007,127,113-131中提到的。还可以使用另外包含Te的催化剂体系。当本文提及“基于MoVNb的催化剂体系”或“基于MoVTeNb的催化剂体系”时,这应理解为具有作为混合氧化物的所提及元素的催化剂体系,也分别表示为MoVNbO
在ODH过程中,特别是当在工业相关反应条件下使用基于MoVNb(Te)O
在ODH(E)中特别是在低稀释条件下需要使用氧作为氧化剂,其原则上会导致设备内产生爆炸性混合物。此处可用于避免某些设备部分或反应器区域中的爆炸性混合物的混合概念是已知的,并且例如在用于商业壳管式反应器的EP 3 476 471 A1中进行了描述;然而,除了结构措施(例如,火焰或爆炸屏障设备、最小化自由体积、耐压设计)之外,这些或其他方法特别要求在实际反应器之前远未达到相应的点火温度。
根据现有技术,ODH(E)优选在固定床反应器中进行,特别是在冷却的壳管式反应器中,例如采用熔盐冷却。对于高放热反应,即特别是还包括ODH(E)的氧化反应,具有多个区域的反应器床的使用通常是已知的。例如,申请人在WO 2019/243480 A1中描述了基本原理。该文献公开了使用不同的催化剂床或相应的反应区的原理,不同催化剂床或相应反应区的每空间单元具有不同的催化剂负载和/或催化剂活性。
根据现有技术,进一步的加热仅在反应器中进行,并且更精确地仅在相应的反应管本身中进行,以便达到活性(单层或多层)催化剂床的入口温度(例如,根据已经引用的WO2019/243480 A1)。为此,在入口区域引入足够长的惰性床以确保气流的有效加热。下面解释这方面的细节。
然而,此外,在相应方法的设计中还必须考虑可能包含在反应进料中的痕量组分的特别影响。与所有非均相催化过程一样,氧化方法并且特别是ODH(E)也是如此,此处使用的催化剂,某些组分充当所谓的催化剂毒物并导致催化剂的活性和/或选择性逐渐降低(“催化剂中毒”),直至并包括完全失活。
这些效应最初并且特别发生在活性催化剂装填开始时。不受理论的过多束缚,在被称为催化剂中毒的催化剂失活机制中,特别地,在用于催化期望的主反应(在这种情况下为ODH(E))的催化剂的活性位点上或处发生特定的反应或反应进料中物质的积累。随后,至少在期望的主反应所需的条件下,这些物质不可逆地与对主反应有活性的催化剂位点结合,结果它们不再可用于主反应。
因此,催化剂中毒的特征特别在于以下事实:失活前沿在流动方向上推过相应的催化剂床,即,当催化剂床的初始区域(即,催化剂床中的失活前沿的上游)部分已经完全或几乎完全失活,失活前沿下游(紧邻)的区域仍然显示其完全或几乎完全的活性。
在运行期间——在氧化方法例如ODH(E)的情况下通常显著超过1年且高达数年,因此通常存在催化剂的逐渐失活或催化剂活性的降低,这通常在操作期间通过借助于相应指定的操作参数逐渐升高反应器中的温度来补偿。由于最初进料到反应器的绝对活性催化剂质量保持恒定,这通常意味着在相同反应速率下对有价值产物的选择性损失。
作为非详尽的列表,典型的干扰性痕量组分特别是硫化合物、磷化合物、氮化合物、金属及其化合物(特别是碱金属、碱土金属和重金属)、铝硅酸盐、卤化物(特别是氯化物)和含卤素化合物以及重烃。在实践中,操作剂如油、润滑剂和油脂也经常进入反应器,从而产生相应的负面后果。所提到的碱土金属和重金属可特别包括Na、K、Cs、Mg、Ca、Al、Si、Fe、Cr、Ni、As、Sb、Hg、Pb和V。此处要考虑的是,含氧和氮的化合物(例如含氧化合物、醇、羰基化合物、胺、氮氧化物等)以及重烃(特别是不饱和化合物和芳香族化合物)的进入也会产生负面影响。
痕量组分的来源主要在于氧、乙烷和水的来源或供应中。一方面,这些组分或其衍生物源自相应的原材料源,即,在ODH(E)的情况下,来自乙烷或相关气体源。另一方面,对于碱金属和碱土金属(特别是Na、K以及Mg和Ca)以及卤化物(特别是氯化物)来说,如果相应的水处理设计不充分,或者由于操作故障导致干扰性微量组分的浓度增加,蒸汽发生系统也可能导致蒸汽污染。
然而,特别地,蒸汽也可包含计量化学物质,其例如用于抑制腐蚀,并且特别地,还可以包含(高挥发性)含氮组分。此外,氧源也可能导致干扰性痕量组分的进入,例如来自环境空气或来自与过程相关的污染。
因此,相应的进料制备或处理通常在实际反应器的上游进行,并且可以包括例如吸附、吸收和/或蒸馏步骤。
特别地,通常被设计为固定床并填充有合适的防护材料的所谓的防护床用于去除痕量组分。这里可以使用吸附作用机制和反应作用机制。根据要去除的痕量组分的类型和数量,防护床可以是可再生的以及不可再生的。因此,这里需要额外的设备和方法步骤来用于进料制备。
该进料制备通常涉及烃进料,而对于蒸汽,在蒸汽生产的上游进行相应的水处理。然而,原则上,根据本发明的ODH(E)方法所需的氧也可能被杂质污染,这使得相应的额外预纯化是必要的。
在大规模应用中通常用于ODH(E)的具有多达数万个平行管的壳管式反应器是复杂且成本密集的构造或装置。因此,出于设计和成本原因,大小和尺寸必须尽可能紧凑。这里的一个重要参数是单个反应管的长度,必须尽可能有效地利用它,这意味着其长度应保持尽可能短。特别是,仅填充惰性材料的体积应保持尽可能小,否则它们没有商业用途。
因此,如下所述,惰性材料的质量应当最小化或至少以有用的方式替换。然而,然而,相应地,特别是也应尽可能避免催化剂填充物的“超尺寸”(例如为了能够补偿老化和/或中毒效应)。这种惰性体积和/或“超尺寸”的催化剂填充物的减少另外导致反应器上压降的减少,这是因为各个反应管的长度也相关减少,这对于“低压”方法(例如ODH(E))尤其重要,该方法通常在低于10巴(绝对)或低于6巴(绝对)的压力下进行。
原则上,如果观察到最低温度,也可以想象扩大催化剂床以实现更长的运行时间。原则上,可以在运行开始(SOR)时选择稍低的温度,以便通过升高温度来补偿失活。然而,由于已经提到的对催化剂床长度的影响以及相关的选择性损失,这在实践中仅是部分有利的。
另一方面,此类反应器的排空和填充过程也相对复杂。因此,在两次催化剂进料之间实现尽可能长的运行时间是有利的。
因此,根据本发明,目的是保持两次催化剂更换之间的时间间隔尽可能长,同时在运行时间期间实现经济生产,即实现尽可能高且最恒定的选择性或有价值产物的产率。为此,有必要最大长度降低对反应条件例如反应温度的调整。因此,有必要实现反应条件的稳定。
如上所述,通常使用用于进料处理的上游方法步骤和惰性预热区二者,然而,该区域已经位于实际反应器内,特别是相应的反应管内。然而,特别是在壳管式反应器的情况下,这意味着该惰性预热区位于结构相对复杂的区域中,并且反应器的体积因反应技术方面未使用的空间而增加。该预热区的必要尺寸由特定的热传递(特别取决于颗粒几何形状、流速、反应气体组成以及反应气体的密度、粘度和热容量)以及反应器入口处的进料混合物与实际反应区开始处所需的反应温度之间的温差决定。
因此,重要的是保持该区域尽可能短,因此其体积尽可能小和/或有利地使用它。然而,与此同时,用于进料处理的上述上游措施也意味着额外的设备支出,其也必须最小化。
发明内容
上述目的通过具有各独立专利权利要求的特征的用于生产目标化合物的方法和设备实现。优选实施方案在每种情况下都是从属权利要求和以下描述的主题。
用于多相催化、特别是用于氧化过程且尤其是ODH(E)的典型催化剂需要一定的最低温度,即所谓的起燃温度,以发生相当大的反应。根据现有技术,该起燃温度特别取决于所使用的催化活性材料。由于常规使用的催化剂床中的起燃温度高于进入所用反应器的进料混合物的进料温度,因此使用上述惰性材料的预热区。
在一个实施方案中,本发明现在利用这样的事实:特定催化剂材料的活性以及与之相关的起燃温度可以受到生产并且特别是受到单个生产步骤的影响。已经发现,特别是对于有利使用的MoVNb(Te)O
在上述实施方案中,本发明通过采用有利地具有相同类型和元素组成的催化剂来利用这一点,所述催化剂在壳管式反应器的反应管的区域(下文也称为“第一管段”)中具有相应较低的起燃温度和相关较高的活性,所述区域先前用于加热至相应区域下游的起燃温度并填充有惰性材料。如下文所解释的,因此可以全部或部分省去用于预热的惰性区,并且至少大部分可以避免布置在下游(在本文中称为“第二管段”的管段中)的主催化剂床的失活。
然而,原则上,在本发明的范围内也可以使用以另一种方式提供的具有更高活性并因此具有降低的起燃温度的催化剂。具体实例在下文提到。
总体而言,本发明提出了一种用于生产目标化合物的方法,其中将处于第一温度范围内的温度的进料混合物分配至壳管式反应器的多个平行反应管中,在反应管的第一管段中进行加热至第二温度范围内的温度,并且在布置在反应管的第一管段下游的第二管段中,使用布置在第二管段中的一种或多种催化剂进行氧化催化转化。根据本发明,所述加热至少部分地使用布置在第一管段中并且具有在第一温度范围内的起燃温度的催化剂来进行。换言之,本发明因此提出至少部分地省去具有惰性材料的预热区,而是提供具有较低起燃温度或较高活性的上游催化剂床。在这一点上已经应当指出的是,在本发明的范围内不需要完全省去惰性材料。例如这可以用于在合适的点处将相应的气流均匀分布在反应管的整个横截面上。
在本发明的范围内,第一温度范围可以特别为170℃至280℃,优选200℃至270℃并且特别优选220℃至260℃,第二温度范围优选280℃至450℃,并且特别优选300℃至400℃。不考虑具体值,第一温度范围内的温度(以及相应的起燃温度)特别地比(主床的)第二温度范围内的温度低30至110K,优选40至80K,并且特别优选40至60K。实际上,在各自的温度范围内存在一个曲线,即加热到各自的热点,然后再次下降。“起燃温度”特别应理解为是指催化剂在技术相关条件下转化超过10%的所考虑的反应物(即在ODH的情况下的煤油,在ODH(E)的情况下的乙烷)的温度。
通过使用本发明可以克服开头提到的缺点。特别地,通过使用本发明,与现有技术相比,可以保持较小的体积或者可能完全避免,否则在其他情况下该体积仅填充有惰性材料并且因此没有任何商业利益。由于各个反应管的长度相应减少,惰性体积的减少导致整个反应器的压降减少,这对于例如ODH(E)的方法特别有利。对于进一步的优点,请参考对本发明的目的的解释,其至少部分地通过所提出的措施来实现。
布置在第一管段中的催化剂和布置在第二管段中的多种催化剂中的一种或至少一种有利地至少包含金属钼、钒、铌和任选的碲,特别是以相应的混合氧化物的形式,因为,如根据本发明所证明的,上述有利效果对于相应的催化剂特别显著。
此外,根据本发明,布置在第一管段中的催化剂和布置在第二管段中的多种催化剂中的一种或至少一种可以至少部分地由相应金属的氧化物生产。因此,由于容易获得起始材料,催化剂的生产成本非常低。
如已经讨论的,布置在第一管段中的催化剂和布置在第二管段中的多种催化剂中的一种或至少一种有利地具有相同的元素组成。这使得能够简单地生产相应的催化剂,它们之间的差异仅仅由于制造工艺的不同而导致。根据此处应用的理解,即使不同催化剂之间各元素或其化合物的含量相差不超过10%、5%或1%,也应该仍然存在“相同的元素组成”。
有利地,由于不同的焙烧强度,布置在第一管段中的催化剂的活性比布置在第二管段中的多种催化剂中的一种或至少一种高超过10%。活性也可以高例如20%、30%或40%。相反,由于不同的焙烧强度,布置在第一管段中的催化剂有利地具有低于超过3K、优选低于超过5K、进一步优选低于超过10K、特别是低于超过15K的起燃温度。焙烧强度特别地由焙烧过程来调节处理,但也可以是例如特别强烈的例如持久的焙烧。
因此,根据本发明,反应器中先前常用的具有惰性材料的预热区可以完全或部分地填充有具有相同催化活性材料的催化剂。然而,在这种情况下,有利地选择具有非常低的起燃温度的材料,该起燃温度理想地对应于反应器入口处的反应物混合物的温度。根据本发明,如果需要,可以在反应器管入口处,即在前述具有低起燃温度的非常活性的催化剂层的上游(因此在第一管段的上游)设置短的惰性层,以便在反应器管中实现形成限定的流动轮廓和因此限定的起始条件(所谓的入口路径)。这种入口路径的长度通常是所用惰性颗粒的当量直径的至少10倍,但最通常小于50cm,特别是小于30cm或小于20cm。
可能的惰性入口路径之后是反应性预热路径(以第一管段的形式)。根据本发明提供的这种反应性预热路径的基本功能是,反应物流中可能包含的任何催化剂毒物已经可以在此与催化活性材料反应和/或被吸附,因为预热区中的催化活性材料有利地对应于随后的主反应区(在第二管段中)的催化活性材料。优选地,这些一个或多个随后的主反应区设计有多个催化剂床,这些床的体积活性从反应管起点到反应管末端(即在流动方向上)增加(例如,通过用合适的惰性材料对具有相同基本活性的催化剂颗粒进行不同的稀释;在这方面参见WO 2019/243480 A1,如已经讨论的)。
对于反应性预热路径(即,在第一管段中),特别地根据本发明使用活性催化剂材料,其化学组成对应于随后主反应区(即第二管段)中的材料,但活性甚至更高,如已经用其他方式解释的那样。这种甚至更高活性的MoVTeNbO
在这种情况下,较高的体积活性通常伴随着较高的孔体积和/或较高的BET表面积,并且特别是缺少惰性稀释。BET表面积是质量比表面积,它是根据已知方法由实验数据计算得到的,通常以平方米每克(m
在相应的实施方案中,第一管段中的孔体积和/或BET表面积比第二管中的最大孔体积和/或最大BET表面积高,特别是高出15%至60%。
这种反应性预热路径(即,第一管段)的长度优选为所用催化剂颗粒当量直径的至少十倍,但优选小于40cm或小于30cm,特别优选在5cm至25cm。另外,其中反应性预热相对于主反应区的长度总计小于0.1,优选小于0.07,并且特别优选小于0.04的实施方案与技术设计特别相关。
换言之,第一管段中布置有催化剂的区域的长度在绝对尺寸上小于40cm和/或该长度相对于第二管段中布置有一种或多种催化剂的区域的总长度小于0.1。
在其他方面相同的化学材料的较高基础活性意味着这种材料的活性位点的数量较大。反应性预热路径(即第一管段)中的大量活性位点增加了对可使催化剂失活的物质的吸收能力。因此,在相同反应物摩尔流量的情况下,主反应区(在第二管段中)的使用寿命延长。这一点特别重要,因为该反应性预热区的功能在于保护随后的(下游)主反应区免受使主反应区中的催化剂失活的物质的影响(特别是通过中毒)。
如已经陈述的,在所谓的催化剂中毒的催化剂失活机制中,进料中的特定反应或物质积聚发生在对期望主反应(在这种情况下,特别是氧化脱氢,特别是乙烷的氧化脱氢ODH(E))的实际催化具有活性的催化剂位点处,在这个过程中,至少在期望的主反应所需的条件下,这些物质不可逆地结合至对主反应有活性的催化剂位点上,这意味着所述位点不再可用于实际的主要反应。为了使主反应区上游的床能够实现其功能,如果该催化剂材料在较低温度下起燃,即精确地催化这些反应,则是特别有利的。否则,也不会与可能的催化剂毒物发生特定反应。
然而,令人惊讶的是,根据本发明,尽管使用活性更高的催化剂,但就商业产物的转化率和选择性而言,反应性预热路径对总体反应器性能,即整体反应床(反应性预热区和一个或多个主反应区)的性能没有明显影响,如从图2中可以看出,下文对此进行了解释。这里参考该图和所附的表3。因此,反应性预热区的功能尤其应被视为有利地延长主反应区的使用寿命。如其中所解释的,在本发明的范围内,随着失活的增加,冷却剂温度可以任选地升高。
反应性预热区的功能可以通过记录作为催化剂活性量度的值的装置来监测。特别适合于此目的的是在该反应性预热区的床中的至少一个点处监测该反应性预热区的床温度,但优选分布在预热区的床中的多个点上。类似地,主反应区的活性也可以非常容易地在线检测(当然,此外还可以在线监测反应器出口处的氧含量并完成工艺气体成分的定期分析,尽管这些通常不在线进行)。
特别地,再次总结,根据本发明的催化剂的实施方案可以是这样的,由于制造方式的不同,第一管段中的体积活性高于第二管段中的最大体积活性。
如上所述,本发明可特别与烷烃的ODH结合使用,使得进料混合物有利地包含氧和煤油,特别具有2至6个碳原子的煤油,并且氧化转化作为煤油的氧化脱氢进行。在具有特别优点的ODH中,使用乙烷作为煤油并进行乙烷氧化脱氢(ODHE)。
氧化转化有利地在240℃至500℃、优选280℃至450℃、特别是300℃至400℃范围的催化剂温度下进行。
进料混合物有利地在1巴至10巴(绝对)、特别是2巴至6巴(绝对)的压力范围内的压力下进料到反应器。因此,这是一种在相对较低的压力下操作的方法,其中以特定的方式获得了上面已经提到的缩短的反应管长度的优点。
特别有利的是,在本发明的范围内,进料混合物中的水含量可以设定为5体积%至95体积%,特别是10体积%至50体积%,进一步特别是14体积%至35体积%。还如例如在申请人的EP 3 558 910 B1中公开的,还可以例如确定指示该催化剂或其中一种催化剂的活性的至少一个特征变量,并且在此基础上,基于至少一个确定的特征变量设定反应进料流中的水量。
特别地,其中进料混合物包含乙烷并且进料混合物中水与乙烷的摩尔比为至少0.23的实施方案可能是有利的。
无论冷却介质如何被引导(即,并流或逆流),都可以应用本发明。当冷却介质、特别是熔盐以逆流方式被引导时,可以实现特别的附加优点,因为在这种情况下,来自主反应的反应热可以在反应性预热区中被部分利用。同样,不同的冷却回路与不同的催化剂层的组合是可以想到的(也如WO 2019/243480A1中更详细地指出的)。
如果反应器以这样的方式设计,即反应器在反应性预热区(即,第一管段)明确地以不同的方式另外冷却,即在所述区域中,存在单独的冷却回路(甚至可能具有不同的冷却剂流动方向)的选项,则具有特别的优点。这样做的优点是反应性预热区中有针对性的温度调节和因此活性调节。因此,该区域例如还可以通过相应的热输入明确地“开启”,或者如果不需要或仅在小程度上需要则“关闭”。
换言之,在一个实施方案中,本发明提出使用在反应管周围流动的一种或多种冷却介质来冷却反应管。在这种情况下,第一管段和第二管段可以以特别有利的方式使用不同的冷却介质、在不同的冷却介质回路中的相同的冷却介质、和/或在不同或相同的流动方向上的相同或不同的冷却介质来冷却。
本发明还涉及一种用于生产目标化合物的设备,其具有壳管式反应器,所述反应器具有多个平行反应管,所述反应管具有第一管段和布置在第一管段下游的第二管段,其中一种或多种催化剂布置在第二管段中,并且所述设备具有被配置用于以下的装置:将温度处于第一温度范围内的进料混合物分配至反应管,将所述进料混合物加热至第二温度范围内的温度,以及在第二管段中使用布置在第二管段中的一种或多种催化剂对所述进料混合物进行氧化催化转化。
根据本发明,针对第一管段中的加热的至少一部分,提供具有在第一温度范围内的起燃温度的催化剂。
对于根据本发明提出的设备的进一步特征和优点,明确参考上述解释。在相应的实施方案中,设备在这方面被特别配置为执行如上面同样在各个实施方案中已经解释的方法。这些解释相应地适用。
实施方案
下面参考对应于本发明的实施方案的实施例和不根据本发明的比较例以及相关的附图和表格来进一步解释本发明。
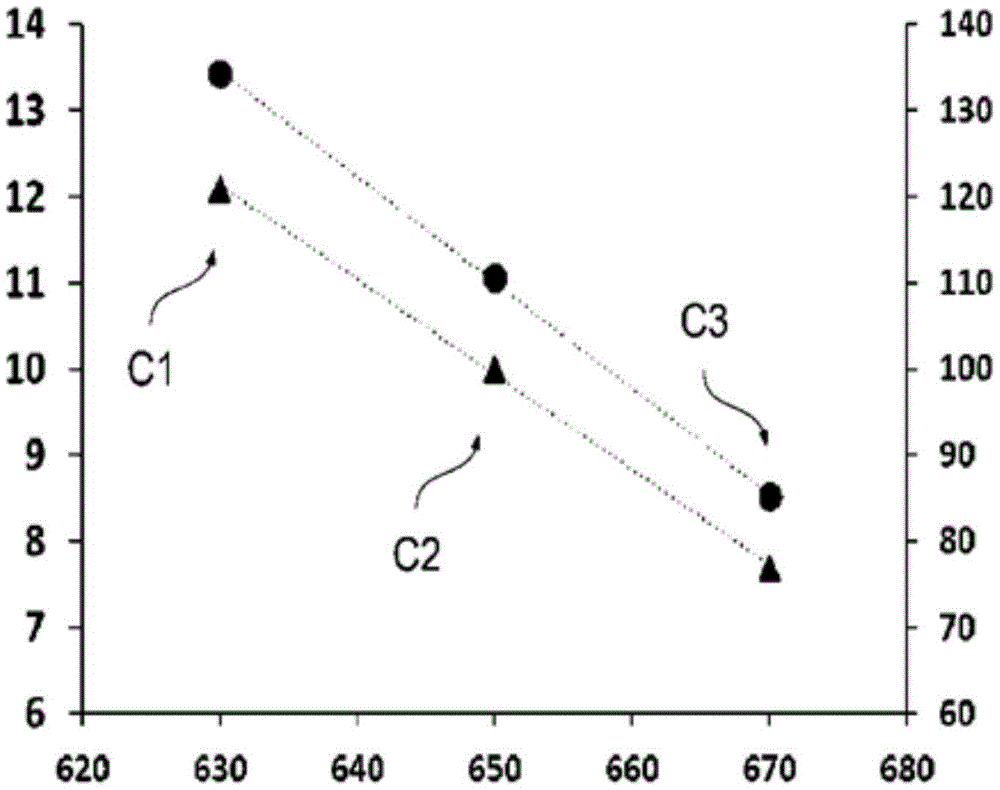
图1说明了在不同焙烧温度下获得的催化剂的不同催化剂活性。
图2说明了根据本发明的一个实施方案的具有反应性预热区的三级催化剂床的温度曲线。
图3以简化示意图示出了根据本发明实施方案的设备。
图4以简化示意图说明了根据本发明实施方案的反应器。
在实施方案中,如上所述,本发明利用了特定催化剂材料的活性以及与之相关的起燃温度可受生产影响的事实。催化活性材料本身在组成方面原则上保持相同并且特别可以从相同的合成方法获得。这种令人惊讶的效果是在MoVNb(Te)O
在本文中,催化剂材料原则上可以如DE 10 2017 000 861 A1中实施例2中所述来生产。在此,在每种情况下可以对合适的金属氧化物进行水热合成。
在DE 10 2017 000 861 A1中使用的方法(其也可在本发明范围内使用)中,将TeO
在80℃下在干燥箱中进行干燥3天,然后将产物在冲击磨机中研磨。获得0.8kg的固体产量。随后在空气中在280℃下进行焙烧4h(加热速率5℃/min空气:1L/min)。在蒸馏器中于600℃下进行2h活化(加热速率5℃/min氮:0.5L/min)。
然而,与上述方法不同,使用表1中列出的分级焙烧温度。此外,表1中列出的催化剂是在回转窑中而不是在蒸馏器中活化的。获得的催化剂表示为1至3。表1中给出的根据BET的比表面积和孔体积是指压片之前的焙烧的催化剂材料。
表1
在完全相同的条件下(填充的催化剂量为46g,系统压力为3.5巴(绝对),反应进料组成为乙烷:氧:水(蒸气)为55.3:20.7:24(在每种情况下为mol%),GHSV为1140(NL
活性等级如图1所示,其中相对于横轴上的焙烧温度,以每升催化剂每小时的摩尔数表示的乙烷转化率形式的活性(即每催化剂体积的活性)描绘在左侧纵轴上(图中的圆圈),并且以百分比表示的相对活性描绘在右侧纵轴(图中的三角形)上。根据表1获得的各催化剂或催化剂样品的值显示为C1、C2和C3。
从图1开始,可以预期这些不同焙烧的催化剂的起燃温度也不同,即,最高活性的材料也具有最低的起燃温度。表1证实了这点。此处起燃温度定义为乙烷转化率为10%时的温度。
对于催化剂样品催化剂1,该起燃温度几乎可以直接从实验数据读取(在250℃反应温度下-这对应于催化剂床起始温度和热油浴温度-乙烷转化率为9.6%),但对于其他两个样品(催化剂2和3)不是这样,因为在那里研究的转化率范围较小。然而,对于表1中列出的所有催化剂样品,转化率是在至少4个不同温度下测定的(参见表1中不同温度水平的数量),并且所有催化剂样品的转化率和温度水平相距足够远。
因此,可以为每个催化剂样品绘制阿伦尼乌斯图,即反应速率常数的自然对数相对于反应温度(开尔文)的倒数的图。阿伦尼乌斯图的创建原则上是本领域技术人员已知的。
阿伦尼乌斯图为每个催化剂样品提供了具有不同参数(斜率和截距)的直线。借助相应的直线方程,可以确定特定乙烷转化率的相关反应速率常数,并由此确定相应的反应温度。针对10%乙烷转化率确定的相应反应温度在表1中的“起燃温度[℃](计算值)=10%乙烷转化率的温度”行中给出。
基于观察到的催化剂1至3的活性和起燃温度随焙烧温度变化的趋势(参见图1和表1),可以认为,至少在一定限度内,具有较低焙烧温度的催化剂的活性可以进一步提高,只要焙烧的温度和持续时间(即焙烧强度)足以形成对于催化目的而言足够稳定的固相或晶相即可。
事实上,对于已经在400℃下焙烧的催化剂(催化剂4),观察到活性的进一步显著增加以及因此起燃温度向较低值的进一步显著移动。该催化剂在测试设备2中用以下测试参数进行测试:测试设备2由可用长度为1m且内径为25mm的管式反应器组成。通过浸入其中的氮流化的盐浴来加热并同时冷却反应器。由于技术原因,使用空气代替纯氧作为氧化剂;此外,该测试设备2只能在大气压下操作。该设置中的其他测试条件如下:填充的催化剂量为337g,乙烷:氮:氧:水(蒸气)的反应进料组成为11.1:46.7:6.8:35.4(各自为mol%),GHSV为418(NL
与催化剂2相比,催化剂4的活性显著更高,这从测试装置2中的直接实验比较中得到证明(参见表2):催化剂2在322℃的盐浴温度下具有约67%的乙烷转化率。另一方面,催化剂4仅需要302℃的盐浴温度即可实现64%的转化率,并且在此温度下的转化率仍明显高于催化剂2在310℃的较高温度下的转化率(催化剂2的乙烷转化率为53%)。
为了估计催化剂4在测试设备1的技术上更相关的条件下的起燃温度,遵循以下程序:使用在表2中给出的盐浴温度和给出的其他测试条件下测定的乙烷转化率,计算对应于该温度的反应速率常数。该过程原则上是本领域技术人员已知的。
该反应速率常数用作确定相应的阿伦尼乌斯直线的起点。由于催化剂4只有一个测量点可用,因此假设表观活化能与测试条件无关,使用与测试设备1的测试条件相同的阿伦尼乌斯直线斜率。
使用为催化剂4确定的该阿伦尼乌斯直线,并考虑到由该程序产生的不精确性,对于测试设备1的技术相关测试条件,估计催化剂4的起燃温度范围为约233-242℃。尽管在技术相关条件下关于催化剂4的起燃温度具有相对较高的不确定性,但可以看出,催化剂4的起燃温度范围明显低于催化剂1的起燃温度;因此,催化剂4也具有最高的活性。
表2
表2中标有星号的值是指纯MoVNbTe氧化物催化剂粉末(压片之前)。对于压片,添加二氧化硅和蜡作为压片赋形剂。二氧化硅的孔隙率共同决定最终催化剂成型体的孔隙率,这意味着所述孔隙率的确切值不同。然而,压片前实际催化剂粉末的氮孔体积与活性相关。
上面解释的根据本发明的发现是令人惊讶的。根据本发明的催化剂样品的不同活性和起燃行为可以令人惊讶地与来自催化剂表征的数据相关(参见表1和2)。通过降低催化剂生产过程中的焙烧温度,可以实现比表面积的增加,并且甚至更显著地,可以实现比孔体积的增加,这是一个新发现。然而,虽然较高的活性通常伴随着选择性的降低,但令人惊讶的是,在本发明的范围内,仍然可以实现整个反应床的高选择性或甚至恒定的高选择性。
图2示出了具有反应性预热区的基于催化2(参见上文)的主反应区的三级催化剂床的温度分布,其中根据特别优选的实施方案,反应性预热区的材料的基本活性为主反应区的催化剂材料的约1.2倍。在每种情况下,在SOR(使用新鲜催化剂的研究期的开始,“运行开始”,实线)和EOR(研究期的结束,“运行结束”,虚线)获得的温度曲线在纵轴上以℃为单位相对于横轴上以米为单位的反应管长度示出。
对于设计,要注意确保反应混合物在反应器管入口处的组成和总质量流量以及因此主反应区的负载(即,主反应区的WHSV)是相同的。在第一种情况(SOR)下,反应性预热区表现出其全部活性,而在第二种情况(EOR)下,反应性预热区完全失活,即惰性(例如由于中毒),而相反地,主反应区的催化剂床持续表现出其全部活性(即,在典型的磨合期后发生的活性),这适用于如上解释的催化剂失活机制“中毒”。两种情况使用相同的冷却剂温度。冷却剂以与反应气体逆流的方式引导。可以看出,在反应性预热路径失活的过程中,可以保持反应器功能不变,而不需要调整操作参数,例如冷却剂温度。通常,随着失活的进行,冷却剂温度必须升高。在本发明的范围内,这可以另外任选地进行,但是在这种情况下,在更晚的时间和/或更小的程度上进行,例如在反应性预热区的广泛失活之后,这延长了相应技术反应器的总体使用寿命。
在下表中,再次以表格形式总结了图2中所示的参数和另外的参数。
表3
图3以高度简化的设备图的形式示出了根据本发明的一个实施方案的用于生产烯烃的设备,该设备整体由1表示。在这种情况下仅示意性地指示设备1。特别地,使用壳管式反应器100中大大放大的反应管11(未按比例绘制)来说明预热区和随后的反应区的基本布置。尽管下面描述了用于ODHE的设备1,但是如上所述,本发明也适用于高级烃的ODH。在这种情况下,以下解释相应地适用。
如所提到的,设备1具有壳管式反应器100,在所示的实例中,将包含乙烷且以任何方式获得的进料混合物A进料到该反应器100。进料混合物A可以包含例如从未示出的精馏单元取出的烃。进料混合物A还可以例如预热并以另一种方式处理。进料混合物A可能已经包含氧和任选的反应调节剂例如水蒸气,但是也可以在壳管式反应器100上游或中添加相应的介质,这未单独示出的。从管式反应器100中取出产物混合物B。
如图4详细所示,壳管式反应器100具有多个平行的反应管10(仅部分指定),其延伸穿过预热区110,然后穿过多个反应区120、130、140,在所示实例中为三个。反应管10被夹套区20包围,在该实例中,所解释类型的冷却剂C被引导通过该夹套区20。因为如上所述,反应管10可以使用在反应管10周围流动的多种冷却介质来冷却,或者不同的管段可以使用不同的冷却介质、在不同的冷却介质回路中的相同的冷却介质、和/或在相同或不同的流动方向上的相同或不同的冷却介质来冷却,所以该图示被极大地简化。
在进料到壳管式反应器中之后,进料混合物A在第一温度范围内的温度下适当地分配到反应管10中。每个反应管均具有位于预热区110中的第一管段11和位于反应区120、130和140中的第二管段12。
在反应管10的第一管段11中加热至第二温度范围内的温度,并且在布置在反应管10的第一管段11下游的第二管段12中,使用布置在第二管段12中的一种或多种催化剂对相应预热的进料混合物A进行氧化催化转化。
加热至少部分地使用布置在第一管段11中的催化剂来进行,该催化剂具有在第一温度范围内的起燃温度并且其使用已经导致部分转化。对于进一步的细节,特别是关于提供附加惰性区的细节,明确地参考上述解释。
未示出后续的方法步骤或设备组件。特别地,可以使工艺气体与洗涤水或合适的水溶液接触,由此可以冷却工艺气体并且可以将乙酸从工艺气体中洗掉。至少大部分不含水和乙酸的工艺气体可被进一步处理并进行乙烯的分离。包含工艺气体中的乙烷可以再循环到反应器100中。
- 用于大麻素化合物的同时生产的设备和方法
- 在从含氧化合物合成烯烃中用于高碳烯烃的回收和利用的方法和设备
- 用于磷酸法活性炭生产的炭化装置、生产设备和生产方法
- 用于共生产乙二醇和三碳化合物的微生物和方法
- 用于生产目标化合物的方法及设施
- 用于生产目标化合物的方法及设施