一种包含SN38的抗体药物偶联物中间体及其制备方法
文献发布时间:2023-06-19 19:32:07
技术领域
本发明涉及抗体药物偶联物领域,具体涉及一种抗体药物偶联物中间体及其制备方法。
背景技术
抗体偶联药物(Antibody-Drug Conjugate,ADC)作为一种新型的生物靶向药物,实现了单抗靶向作用和小分子药物细胞毒性的强强联合,现已成为肿瘤靶向治疗发展最快的领域之一。ADC的三个组成单元(抗体、细胞毒素和连接子)共同组成靶向药物递送系统,其中抗体实现靶向性,连接子保证ADC在血液转运过程中的稳定性,而到达作用靶点后,毒素发挥对癌细胞的杀伤作用。目前有60多种ADC药物正在推进抗肿瘤治疗临床试验,其中绝大多数毒素为微管蛋白抑制剂,少部分为DNA抑制剂。DNA抑制剂相比较微管蛋白抑制剂有两个优势:1)DNA抑制剂(picomolar IC 50values)比微管蛋白抑制剂(sub-nanomolar IC50values)有更高活性,在治疗较少抗原的肿瘤中起到更好的作用;2)能够杀死不在分裂期的癌症细胞,在进行实体瘤治疗方面具有优势。
喜树碱类化合物是临床中应用的拓扑异构酶1(TOP1)抑制剂,对临床上生长缓慢的实体瘤有良好的效果。但是喜树碱的特殊结构导致其水溶性及脂溶性都很差,必须进行水溶性改造。以喜树碱为弹头的ADC药物为解决上述缺陷提供了新的方案。
目前已有2款以喜树碱衍生物为弹头的ADC药物获批上市,为Enhertu(trastuzumab deruxtecan)和Trodelvy(Sacituzumab govitecan),它们在治疗肿瘤尤其是恶性肿瘤方面满足了极大的临床需求。阿斯利康/第一三共研发的Enhertu以组织蛋白酶B活化的GGFG四肽作为linker,并引入一小段自裂解结构,释放出来的是Exatecan的衍生物Dxd。在HER2阳性转移性乳腺癌中,99例患者使用Enhertu的客观缓解率为54.5%,疾病控制率为93.9%。Sacituzumab govitecan的连接子为Mcc-triazole spacer-PEG7-x-lysine-PABC,在细胞中溶酶体(pH在5左右)中分解释放喜树碱(SN38)。Sacituzumab govitecan的三阴乳腺癌临床二期结果显示,其有效率高达30%,69.5%的患者肿瘤都出现了缩小,而三阴乳腺癌临床几乎是“无药可治”。此外,针对多线治疗失败的小细胞肺癌,Sacituzumabgovitecan可以使60%的患者肿瘤缩小;针对化疗、靶向和PD-1治疗都失败的非小细胞肺癌,Sacituzumab govitecan的控制率高达43%。但临床试验中,尽管治疗效果显著,但同时也存在较为严重的ADC毒性引发的安全性问题,如血液系统毒性、神经毒性、肺毒性、皮肤毒性、肝毒性、眼毒性、代谢异常、心脏毒性等,因此改善ADC药物的安全性是目前药物开发中需要高度关注和解决的问题。
此外,部分ADC的连接子及载荷的合成中也存在金属离子反应产物不便于去除问题,如Sacituzumab govitecan所用到的CL2A-SN38在合成过程中涉及到点击化学反应(Click反应)(参见中国专利申请公开号CN102448494A,说明书第50页第[0273]段),因此会导致该反应产物中含有Cu
连接子(Linker)在ADC结构中起着至关重要的作用,其会影响ADC的药代动力学参数、治疗指数和药效。Linker可维持ADC复合物在血流中的稳定性,连接子的设计首先要考虑到稳定性,因为ADC药物的半衰期比较长,所以需要连接子的性质稳定,这样才能防止连接子在血液中被分解提前释放毒素。如果连接子在血液中不够稳定,就会导致ADC在进入肿瘤细胞之前就被分解,药物对肿瘤的打击作用就会降低,甚至会误杀其他细胞。而且,在肿瘤细胞内化ADC的过程中,Linker应能够快速释放细胞毒性药物。由此,连接子对于开发的ADC药品的安全性、有效性具有显著的决定作用。并且根据连接子结构的不同,也极大的影响ADC的制备过程的难度系数、生产成本以及环境友好性,并且极大的影响ADC药品的质量,如ADC的稳定性等,进而会影响ADC药物的安全性。
发明内容
本发明提供了一种包含SN38的抗体药物偶联物中间体及其制备方法,该制备方法步骤简单,节约成本,在反应中不会引入重金属离子,具有更高的环境友好性,并且采用该制备方法合成的抗体药物偶联物中间体具有良好的稳定性,从而显著提升ADC药物的安全性。
本发明涉及一种如式(Ⅰ)所示的抗体药物偶联物中间体:
其中:
X
X
R
R
优选的,所述的X
优选的,所述X
优选的,所述的R
优选的,所述的X
优选的,所述的抗体药物偶联物中间体的结构如式(1)-(16)所示:
/>
/>
/>
/>
本发明还提供了一种抗体药物偶联物中间体的制备方法。
进一步的,所述的抗体药物偶联物中间体的结构为:
/>
所述的R
所述的R
优选的,R
进一步的,所述的式(4)化合物的制备方法为:
反应A:将化合物a和化合物b溶于溶剂中,室温搅拌适当时间后置于低温条件下加入还原剂,再搅拌适当时间后,再置于室温搅拌过夜,反应结束后旋干溶剂并进行萃取、干燥和纯化;
反应B:将SN38和DNPC溶于溶剂中,加入有机碱,室温搅拌适当时间,反应结束后旋干溶剂并进行打浆和过滤;
反应C:将反应B的产物溶于溶剂中,加入反应A的产物及有机碱,室温搅拌适当时间,反应结束后旋干溶剂并进行纯化;
反应D:将反应C的产物溶于溶剂中,加入酸,低温条件下搅拌适当时间,反应结束后旋干溶剂后与Mc-VC-PAB-PNP一起溶于溶剂中,低温条件下再搅拌适当时间,加入有机碱,反应结束后旋干溶剂并进行纯化。
反应进程1:
进一步的,所述的式(12)化合物的制备方法为:
反应A:将化合物a和化合物b溶于溶剂中,室温搅拌适当时间后置于低温条件下加入还原剂后搅拌适当时间,再置于室温搅拌过夜,反应结束后旋干溶剂并进行萃取、干燥和纯化;
反应B:将SN38和DNPC溶于溶剂中,加入有机碱,室温搅拌适当时间,反应结束后旋干溶剂并进行打浆和过滤;
反应C:将反应B的产物溶于溶剂中,加入反应A的产物及有机碱,室温搅拌适当时间,反应结束后旋干溶剂并进行纯化;
反应D:将反应C的产物溶于溶剂中,加入酸,低温条件下搅拌适当时间,反应结束后旋干溶剂后与MP2-VC-PAB-PNP一起溶于溶剂中,低温条件下搅拌适当时间,加入有机碱,反应结束后旋干溶剂并进行纯化。
反应进程2:
进一步的,所述的化合物(I-1)的制备方法为:
反应A:将化合物a和化合物b溶于溶剂中,室温搅拌适当时间后置于低温条件下中加入还原剂后搅拌适当时间,再置于室温搅拌过夜,反应结束后旋干溶剂并进行萃取、干燥和纯化;
反应B:将SN38和DNPC溶于溶剂中,加入有机碱,室温搅拌适当时间,反应结束后旋干溶剂并进行打浆和过滤;
反应C:将反应B的产物溶于溶剂中,加入反应A的产物及有机碱,室温搅拌适当时间,反应结束后旋干溶剂并进行纯化;
反应D:将反应C的产物溶于溶剂中,加入二(对硝基苯)碳酸酯和有机碱,恒温搅拌适当时间,反应结束后旋干溶剂并进行纯化;
反应E:将反应D的产物和胺类化合物溶于溶剂中,加入有机碱,低温条件下搅拌适当时间,反应结束后旋干溶剂并进行纯化;
反应F:将反应E的产物溶于溶剂中,加入酸,低温条件下搅拌适当时间,反应结束后旋干溶剂后与Mc-VC-PAB-PNP一起溶于溶剂中,低温条件下搅拌适当时间,加入有机碱,反应结束后旋干溶剂并进行纯化。
反应进程3:
进一步的,所述的化合物(I-2)的制备方法为:
反应A:将化合物a和化合物b溶于溶剂中,室温搅拌适当时间后置于低温条件下中加入还原剂后搅拌适当时间,再置于室温搅拌过夜,反应结束后旋干溶剂并进行萃取、干燥和纯化;
反应B:将SN38和DNPC溶于溶剂中,加入有机碱,室温搅拌适当时间,反应结束后旋干溶剂并进行打浆和过滤;
反应C:将反应B的产物溶于溶剂中,加入反应A的产物及有机碱,室温搅拌适当时间,反应结束后旋干溶剂并进行纯化;
反应D:将反应C的产物溶于溶剂中,加入二(对硝基苯)碳酸酯和有机碱,恒温搅拌适当时间,反应结束后旋干溶剂并进行纯化;
反应E:将反应D的产物和胺类化合物溶于溶剂中,加入有机碱,低温条件下搅拌适当时间,反应结束后旋干溶剂并进行纯化;
反应F:将反应E的产物溶于溶剂中,加入酸,低温条件下搅拌适当时间,反应结束后旋干溶剂后与MP2-VC-PAB-PNP一起溶于溶剂中,低温条件下搅拌适当时间,加入有机碱,反应结束后旋干溶剂并进行纯化。
反应进程4:
进一步的,所述的化合物(I-1)选自:
/>
/>
所述的化合物(I-2)选自:
/>
进一步的,上述任一反应进程中所述的“低温条件下”为冰水浴下。
进一步的,上述任一反应进程中所述的溶剂可独立地为极性溶剂和/或非极性溶剂,所述的极性溶剂为THF、DMF、DMA、NMP的一种或几种;所述的非极性溶剂为二氯甲烷、四氯化碳的一种或几种。
进一步的,上述任一反应进程中所述的有机碱可独立地为N,N-二异丙基乙胺、三乙胺、吡啶的一种或几种,优选为N,N-二异丙基乙胺以及吡啶中的一种或两种。
进一步的,所述的酸为盐酸、三氟乙酸、柠檬酸中的一种或两种。
进一步的,所述的胺类化合物为伯胺或仲胺。
进一步的,所述的反应A中使用乙酸乙酯进行萃取,所述的纯化采用柱层析法,洗脱剂为二氯甲烷和甲醇。
进一步的,所述的反应B中使用乙酸乙酯、正己烷、二氯甲烷中的一种或多种组合进行打浆。
进一步的,所述的反应C中采用柱层析法进行纯化,洗脱剂为二氯甲烷和甲醇。
进一步的,所述的反应D中采用柱层析法进行纯化,洗脱剂为二氯甲烷和甲醇。
进一步的,所述的反应E中采用柱层析法进行纯化,洗脱剂为二氯甲烷和甲醇。
进一步的,所述的反应F中采用制备液相法进行纯化,流动相A为MeCN,0.1%HCOOH,流动相B为H
进一步的,所述反应均在氮气保护下进行。
本发明提供的制备抗体药物偶联物中间体(具体为连接基团-SN38共价偶联物)的方法,除制备方法步骤简单,降低了由于重金属残留导致的安全性问题,且使用该抗体药物偶联物中间体制备的抗体药物偶联物在体内将具有更高的稳定性。
同时,发明人惊奇的发现,本发明中的连接子与SN38组合,所制备的抗体药物偶联物能够产生显著的肿瘤抑制效果。
附图说明
图1为小鼠体重变化情况;
图2为肿瘤体积变化情况;
图3相对肿瘤体积情况。
具体实施方式
[缩写]
除非另有说明,本发明使用的所有缩写具有本领域普通技术人员所理解的相同含义。如本发明所用,常用的缩写及其定义如下所示:
/>
[定义]
与说明书的各方面相关的各种术语在说明书和权利要求书中通篇使用。除非另外指明,否则此类术语被赋予本领域的普通含义。其它具体定义的术语应按照与本文所提供的定义相符的方式理解。
如本文所用,术语“一个”和“一种”和“所述”是按照标准惯例使用的并且意指一个或多个,除非上下文另有指示。因此例如,对“一种抗体药物偶联物”的提及包括两个或更多个抗体药物偶联物的组合等等。
应当理解,无论何处在本文中用语言“包含”描述方面,除此之外还提供了以“由......组成”和/或“基本上由......组成”描述的类似方面。
本发明所使用的术语“抗体药物偶联物”表示抗体/抗体功能性片段、连接子、药物部分经过化学反应连接在一起的化合物,其结构通常由三部分组成:抗体或抗体类配体、药物部分、以及将抗体或抗体类配体及药物偶联起来的连接子(linker)。目前抗体药物偶联物的制备通常分为两步:第一步是将连接子与药物部分通过化学反应形成“连接子-药物”偶联物,第二步是将“连接子-药物”偶联物中的连接子部分与抗体/抗体功能性片段通过巯基或者氨基共价偶合在一起。本发明所使用的术语“抗体药物偶联物中间体”是指上述所述的“连接子-药物”偶联物,进一步的,本发明涉及的“抗体药物偶联物中间体”泛指“连接基团”和SN38共价偶联物。
本发明所用的“抗体药物偶联物”采用本领域通用制备方法制备,示例性的,本发明所用的抗体药物偶联物的制备方法为:用pH=7.4的PBS缓冲液,把抗体配制为10mg/mL的溶液,加入适量当量的TCEP,振荡混匀1小时,再加入5.0摩尔当量的连接子-毒素(即式1-式16所示的化合物),振荡混匀,反应1h,反应结束后,超滤除去残留的小分子,加载至疏水色谱(HIC-HPLC)进行DAR、药物分布、裸抗比例检测。
可以理解的是,本发明旨在提供一种新的药物偶联物中间体,并示例性的以Heceptin制备多例ADC对技术效果进行验证,抗体的选择不对本专利造成限制,示例性地给出具体ADC的结构,其中,ADC-1中的连接子和毒素对应于所用的式(1)化合物与Heceptin利用上述本领域制备“抗体药物偶联物”的通用制备方法制备而成(下同,ADC-2中的连接子和毒素对应于式(2)化合物;ADC-3中的连接子和毒素对应于式(3)化合物;ADC-4中的连接子和毒素对应于式(4)化合物;ADC-5中的连接子和毒素对应于式(5)化合物;ADC-6中的连接子和毒素对应于式(6)化合物;ADC-7中的连接子和毒素对应于式(7)化合物;ADC-8中的连接子和毒素对应于式(8);化合物ADC-9中的连接子和毒素对应于式(9);化合物ADC-10中的连接子和毒素对应于式(10)化合物;ADC-11中的连接子和毒素对应于式(11)化合物;ADC-12中的连接子和毒素对应于式(12)化合物;ADC-13中的连接子和毒素对应于式(13)化合物;ADC-14中的连接子和毒素对应于式(14)化合物;ADC-15中的连接子和毒素对应于式(15)化合物;ADC-16中的连接子和毒素对应于式(16)化合物;ADC-17中的连接子和毒素对应于CL2A-SN38)与:其中,q可选自1、2、3、4、5、6、7、8、9、10。
/>
/>
/>
/>
本发明中的术语“连接基团”是指一种具有双官能团或多官能团的分子,可分别与蛋白/抗体分子和SN38反应,因此作为一种“桥梁”将蛋白/抗体与SN38连接起来。本发明涉及的连接基团特指那些结构中含有酰基结构的基团。
[具体实施例]
下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的实验方法,通常按照常规条件或按照制造厂商建议的条件进行,未注明具体来源的试剂,为市场购买的常规试剂。除非另外说明,否则所有的百分数、比率、比例、或份数按重量计。
除非另行定义,文中所使用的所有专业与科学用于与本领域熟悉人员所熟悉的意义相同。此外,任何与所记载内容相似或均等的方法及材料皆可应用于本发明办法中。文中所述的较佳实施方式与材料仅作示范之用。
实施例1式(12)化合物的制备方法
1)化合物c(2-Boc-N-甲基乙基乙二醇)的合成方法
将化合物a(氨基乙二醇)0.6g和化合物b(N-Boc-乙醛)1.0g溶于20mL甲醇中,室温搅拌6h。将反应置于冰水浴,加入三乙酰氧基硼氢化钠,冰水浴搅拌1h,恢复至室温搅拌过夜。通过LC-MS法检测,产物没有紫外吸收。将溶剂旋干,用乙酸乙酯萃取2次,无水Na
2)SN38-PNP(10-对硝基苯基碳酸酯-喜树碱)的合成方法
将SN38(喜树碱)1.0g和DNPC(二(对硝基苯)碳酸酯)1.6g溶于100mL四氢呋喃中,加三乙胺2mL,室温搅拌1.5h。通过LC-MS法检测,反应结束。反应完成后,将溶剂旋干,用乙酸乙酯:正己烷=20mL:100mL打浆,过滤得固体,用二氯甲烷20mL打浆,过滤,LC-MS监测收集产品得SN38-PNP固体700mg,产率49.3%。LC-MS:(M+H)
3)化合物d(Boc-N-甲基乙基二醇-喜树碱)的合成方法
将SN38-PNP 250mg溶于30mL N,N-二甲基甲酰胺中,加入235mg Boc-DMEDA-PEG和233uL N,N-二异丙基乙胺,室温搅拌16h。通过LC-MS法检测,反应结束。反应完成后,将溶剂旋干,柱层析法纯化,洗脱剂为二氯甲烷:甲醇=20:1,冲洗。LC-MS监测收集产品得Boc-DMEDA-PEG-SN38固体213mg。收率70%。LC-MS:(M+H)
4)化合物g(10-(2-N-甲基乙基乙二醇)-喜树碱三氟乙酸盐)的合成方法
将化合物d 73.0mg溶于3mL二氯甲烷和3mL三氟乙酸中,置于冰水浴中,恒温搅拌1h。通过LC-MS法检测,反应结束。反应完成后,将溶剂旋干,再加入甲苯2mL和二氯甲烷2mL,旋干,带两次。LC-MS监测收集产品得SN38-DMEDA·TFA,直接投入下一步反应。LC-MS:(M+H)
5)式(12)化合物(马来酰亚胺二乙氧基-L-缬氨酰基-L-瓜氨酸-对氨基汴基-(2-N-甲基乙基乙二醇)-喜树碱)的合成方法
将MP2-VC-PAB-PNP 50mg溶于4mL N,N-二甲基甲酰胺中,放入冰水浴恒温,加氮气保护。加入化合物g 38mg,再加入三乙胺42uL,恒温搅拌0.5h后,恢复至室温反应0.5h。LC-MS法检测,反应完成。反应结束后,将溶剂低温旋出,制备液相法纯化,制备柱:Sun
实施例2式(4)化合物的制备方法
/>
按照实施例1的合成路线制备化合物g(10-(2-N-甲基乙基乙二醇)-喜树碱三氟乙酸盐):将化合物d(73.0mg)溶于3mL二氯甲烷和3mL三氟乙酸中,置于冰水浴中,恒温搅拌1h。通过LC-MS法检测,反应结束。反应完成后,将溶剂旋干,再加入二氯甲烷2mL,旋干,带两次。LC-MS监测收集产品得化合物g(70mg),直接投入下一步反应。LC-MS:(M+H)
按照实施例1的合成路线制备式(4)化合物(马来酰亚胺己酸-L-缬氨酰基-L-瓜氨酸-对氨基汴基-(2-N-甲基乙基乙二醇)-喜树碱):将Mc-VC-PAB-PNP(马来酰亚胺己酸-L-缬氨酰基-L-瓜氨酸-对氨基汴基-对硝基苯基碳酸酯)(58mg),化合物g(42mg)溶于2.5mLN,N-二甲基甲酰胺中后,冰水浴搅拌30mins,滴加DIPEA (42mg),冰水浴搅拌1h。通过LC-MS法检测,反应未完成。继续加入加三乙胺(10mg),室温搅拌1h,反应溶液变黄,通过LC-MS法检测,反应完成。反应完成后,将溶剂低温旋干,制备液相法纯化,制备柱:Sun
实施例3式(1)化合物的制备方法
1)化合物c(2-Boc-N-甲基乙基乙二醇)的合成方法
将化合物a(氨基乙二醇)0.6g和化合物b(N-Boc-(甲胺基)乙醛)1.0g溶于20mL甲醇中,室温搅拌6h。将反应体系置于冰水浴,加入三乙酰氧基硼氢化钠,冰水浴搅拌1h,恢复至室温搅拌过夜。通过LC-MS法检测,产物没有紫外吸收。将溶剂旋干,用乙酸乙酯萃取2次,加入无水Na
2)SN38-PNP(10-对硝基苯基碳酸酯-喜树碱)的合成方法
将SN38 1.0g和DNPC(二(对硝基苯)碳酸酯)1.6g溶于100mL四氢呋喃中,加三乙胺2mL,室温搅拌1.5h。通过LC-MS法检测,反应结束。反应完成后,将溶剂旋干,用乙酸乙酯:正己烷=20mL:100mL打浆,过滤得固体,用二氯甲烷20mL打浆,过滤,LC-MS监测收集产品得SN38-PNP固体700mg,产率49.3%。LC-MS:(M+H)
3)化合物d(Boc-N-甲基乙基二醇-喜树碱)的合成方法
将500mg SN38-PNP溶于30mL N,N-二甲基甲酰胺中,加入370mg化合物c和233uLN,N-二异丙基乙胺,室温搅拌3h。通过LC-MS法检测,反应结束。反应完成后,将溶剂旋干,柱层析法纯化,洗脱剂为二氯甲烷:甲醇=20:1,冲洗。LC-MS监测收集产品得化合物d固体250mg。收率45%。LC-MS:(M+H)
4)化合物h(Boc-N-甲基乙基二乙氧基对硝基活性酯-喜树碱)的合成方法
将化合物d 220.0mg溶于5mL二氯甲烷中,加入二(对硝基苯)碳酸酯150mg、DIPEA86mg,恒温搅拌16h。通过LC-MS法检测,反应结束。反应完成后,将溶剂旋干,柱层析法纯化,洗脱剂为二氯甲烷:甲醇=50:1。收集产品得化合物h固体220mg,收率68.5%直接投入下一步反应。LC-MS:(M+H)
5)化合物i(Boc-N-甲基乙基二乙氧基-N,N,N-三甲基乙二胺-喜树碱)的合成方法
将化合物h(Boc-N-甲基乙基二乙氧基对硝基活性酯-喜树碱)220mg,N,N,N-三甲基乙二胺50mg溶于5mL N,N-二甲基甲酰胺中后,滴加DIPEA(65mg),冰水浴搅拌1h。通过LC-MS法检测,反应完成后,将溶剂旋干,柱层析法纯化,洗脱剂为二氯甲烷:甲醇=20:1。收集产品得化合物i固体157mg,收率75%直接投入下一步反应。LC-MS:(M+H)
6)化合物j(N-甲基乙基二乙氧基-N,N,N-三甲基乙二胺-喜树碱-三氟乙酸盐)的合成方法
将化合物i(Boc-N-甲基乙基二乙氧基-N,N,N-三甲基乙二胺-喜树碱)157.0mg溶于3mL二氯甲烷和1.3mL三氟乙酸中,置于冰水浴中,恒温搅拌1h。通过LC-MS法检测,反应结束。反应完成后,将溶剂旋干,再加入二氯甲烷2mL,旋干,带两次。得化合物j 120mg,直接投入下一步反应。LC-MS:(M+H)
7)式(1)化合物(马来酰亚胺己酸-L-缬氨酰基-L-瓜氨酸-对氨基汴基-(N-甲基乙基二乙氧基-N,N,N-三甲基乙二胺)-喜树碱)的合成方法
将Mc-VC-PAB-PNP(马来酰亚胺己酸-L-缬氨酰基-L-瓜氨酸-对氨基汴基-对硝基苯基碳酸酯)52mg,化合物j 35mg溶于2.5mL N,N-二甲基甲酰胺中后,冰水浴搅拌30mins,滴加DIPEA129mg,冰水浴搅拌0.5h。通过LC-MS法检测,反应未完成。继续加入加DIPEA129mg,室温搅拌0.5h,反应溶液变黄,通过LC-MS法检测,反应完成。反应完成后,将溶剂低温旋干,制备液相法纯化,制备柱:Sun
实施例4式(9)化合物的制备方法
按照实施例3的合成路线制备式(9)化合物(马来酰亚胺二乙氧基-L-缬氨酰基-L-瓜氨酸-对氨基汴基-(N-甲基乙基二乙氧基-N,N,N-三甲基乙二胺)-喜树碱):将MP2-VC-PAB-PNP(马来酰亚胺二乙氧基-L-缬氨酰基-L-瓜氨酸-对氨基汴基-对硝基苯基碳酸酯)(52mg),化合物j(34mg)溶于2.5mL N,N-二甲基甲酰胺中后,冰水浴搅拌30mins,滴加DIPEA(36mg),冰水浴搅拌0.5h。通过LC-MS法检测,反应未完成。继续加入加DIPEA(8mg),室温搅拌1.5h,反应溶液变黄,通过LC-MS法检测,反应完成。反应完成后,将溶剂低温旋干,制备液相法纯化,制备柱:Sun
实施例5ADC-1对NCI-N87细胞的抑制作用
将式(1)化合物和CL2A-SN38中间体分别通过本领域常规方法与Heceptin进行巯基偶联(例如参见中国专利申请公开号CN102448494A中的实施例12),制备得到平均DAR值为8的ADC-1和HER2-CL2A-SN38(ADC-17)抗体药物偶联物。将NCI-N87细胞胰酶消化后调整细胞密度为50000个/ml,100μL/孔加入细胞培养板中,37℃,5% CO
表1供试品成分
表2对NCI-N87细胞的抑制作用
实验结果表明,ADC-1组与ADC-17组对NCI-N87细胞的抑制效果相当。
实施例6稳定性实验研究
1)精密称取化合物式(1)化合物(2.1mg)、CL2A-SN38(2.1mg)分别溶于DMSO 700μL;
2)分别配制pH为6、7、8的PB(0.2M)缓冲液;
3)取50μL式(1)化合物的DMSO溶液于离心管,加入pH=6的PB(0.2M)缓冲液950μL,化合物浓度为60μg/mL。放置于37℃恒温体系中;取50μL CL2A-SN38的DMSO溶液于离心管,加入pH=6的PB(0.2M)缓冲液950μL,化合物浓度为60μg/mL。放置于37℃恒温体系中;
4)分别在时间为0h、2h、4h、6h、24h、48h取样40μL,加入乙腈120μL,离心,取上清液送LC-MS定量检测。
5)结果处理:
以T=0h的LC-MS浓度为基准,数据=T/T
表3式(1)化合物
在pH为6时,式(1)化合物48小时后分解了21.2%;
在pH为7时,式(1)化合物48小时后分解了87.8%;
在pH为8时,式(1)化合物48小时后分解了98.7%。
表4 CL2A-SN38
在pH为6时,CL2A-SN38 48小时后分解了98.0%;
在pH为7时,CL2A-SN38 48小时后分解了98.1%;
在pH为8时,CL2A-SN38 48小时后分解了98.1%。
由表3-表4中的结果,可以看出:
1)式(1)化合物在弱酸性条件下比CL2A-SN38更稳定。
2)式(1)化合物在中性条件下,6h分解了18%,24h分解了75%,CL2A-SN38在中性条件下,6h分解了43%,24h分解了98%。
3)式(1)化合物在碱性条件下,24h分解了97%,CL2A-SN3824h分解了98%。
因此,采用本发明制备的式(1)化合物与CL2A-SN38相比,在体内弱酸性或中性条件下具有更高的稳定性,预计将显著提高ADC药物安全性。
实施例7评估抗Her2-药物偶联物对原位乳腺癌发展的抑制作用
1.实验方法
BT474细胞株复苏后传1-2代,待细胞生长状态稳定后扩大培养,制备肿瘤细胞悬液,裸鼠乳脂垫接种0.2*10
2.试验结果及分析
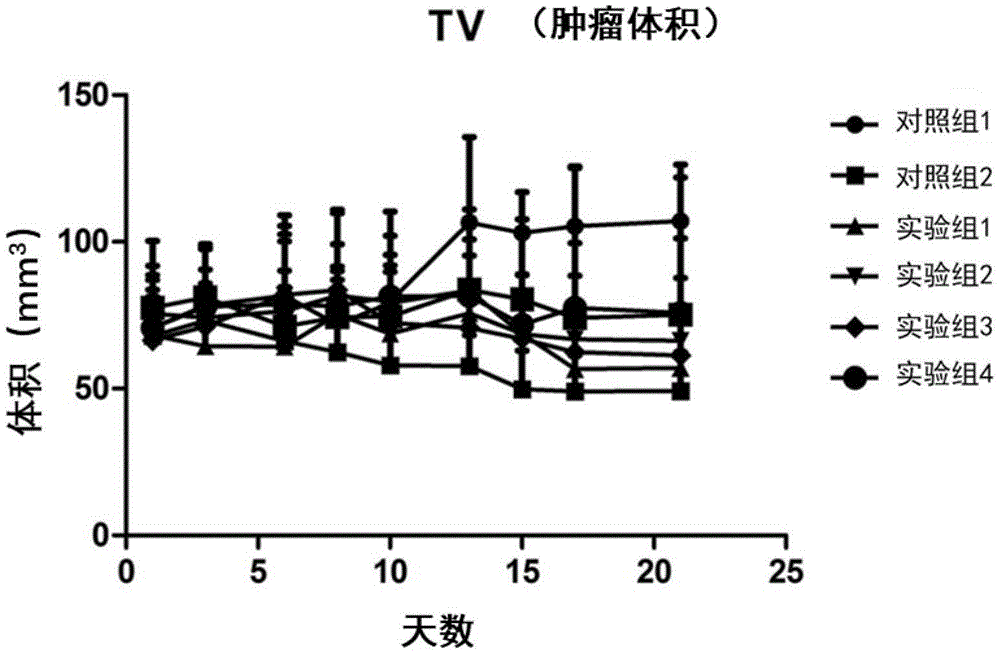
1)小鼠体重变化情况:
表5小鼠的体重情况
实验过程无小鼠死亡,实验结束时,各组与对照组1相比,对照组2
(ADC-17组)、实验组1(ADC-4)、实验组2(ADC-12)、实验组3
(ADC-1)、实验组4(ADC-9)体重未出现明显差异,具体见表5和图1。
2)小鼠肿瘤体积情况:
表6小鼠肿瘤体积情况
注:各组与对照组1生理盐水组相比,“*”表示P<0.05;“**”表示P<0.01;
各组与对照组2相比,“#”表示P<0.05,“##”表示P<0.01
在给药后21天对照组1的肿瘤体积达到107±19.30mm
综上所述,利用本发明中药物偶联物中间体构建的ADC对乳腺癌BT474细胞模型显示出显著的抗肿瘤活性,优选的ADC-1、ADC-4、ADC-12具有明显优势,预计在体内条件下会显著提高安全性的情况下,还能起到与ADC-17组效果相当的肿瘤抑制效果。
本发明已通过各个具体实施例作了举例说明。但是,本领域普通技术人员能够理解,本发明并不限于各个具体实施方式,普通技术人员在本发明的范围内可以作出各种改动或变型,并且在本说明书中各处提及的各个技术特征可以相互组合,而仍不背离本发明的精神和范围。这样的改动和变型均在本发明的范围之内。
- 一种抗体-蒜酶的偶联物及其制备方法和应用
- 一种叶酸受体介导、光激发光敏药物偶联物及其制备方法
- 一种用于合成抗肿瘤药物niraparib的中间体的制备方法及中间体
- 一种用于肿瘤光热治疗联合免疫治疗的抗体偶联硒化铋纳米颗粒及其制备方法
- 一种用于抗体药物偶联物的药物-连接子MC-MMAF的制备方法及其中间体
- 一种用于抗体药物偶联物的药物毒素PNU-159682的制备方法及其中间体