基于肠型分型的微生物组合物、过敏预测模型的构建方法及其模型和应用
文献发布时间:2024-01-17 01:26:37
技术领域
本发明涉及微生物分子诊断技术领域,具体涉及基于肠型分型的微生物组合物、过敏预测模型的构建方法及其模型和应用。
背景技术
过敏性疾病是一种常见的免疫系统疾病,是机体免疫系统受到特定抗原持续刺激,或同一抗原再次刺激的病理性过度反应。常见的过敏性疾病包括过敏性鼻炎、特应性皮炎、过敏性哮喘和食物过敏等。全球约有30%~40%的人口患有过敏性疾病,已经成为一种普遍的公共健康问题。常见的过敏反应属于Ⅰ型超敏反应,其原始机制是进入体内的过敏原与肥大细胞等表面的IgE抗体结合,会引起细胞膜一系列生物化学反应,导致肥大细胞脱颗粒而释放过敏反应介质如组胺、白三烯、5-羟色胺、前列腺素(PG)、激肽等,从而引起一系列过敏性反应症状。
过敏多发生儿童身上,根据流行病学调查的研究发现,导致过敏症状越来越多的原因除了遗传易感性,生活方式的改变也起到了重要的影响,包括剖腹产的增加、抗生素的使用和饮食生活习惯的发展等等。儿童在成长期间并未充分接触周围环境的微生物,因此缺乏相应抗原的刺激,导致后期发生过敏性疾病的概率大大增加。这些元素都直接或间接影响了肠道微生物的发展,而微生物在形成免疫反应方面发挥着主导作用,特别是在生命早期的时候。
肠道微生物是人体组成不可缺少的部分,不仅帮助人体从食物中吸收营养,还在包括物质代谢、生物屏障、免疫调控和宿主防御等功能上发挥重要的作用。肠道微生物可以间接的影响个体对免疫治疗的反应,定植在肠道黏膜表面的菌群对于宿主免疫系统的成熟起着关键作用,表现在肠道菌群及其代谢产物在维持上皮细胞的完整性和刺激免疫耐受性方面。如短链脂肪酸是由肠道菌群发酵降解一些膳食纤维所产生的代谢产物,而这些产物则参与调控机体的健康和疾病的发生,其中如丁酸作为组蛋白脱乙酰基酶抑制剂,有利于FOXP3的表达,因此增强了诱导的Treg细胞的抑制功能,有效进行免疫调控。反过来,过敏性疾病的发生以及药物的使用也会导致肠道微生物菌群的失调改变。在一项利用抗组胺药物治疗慢性自发性荨麻疹的研究中发现,荨麻疹患者肠道中的Prevotella,Megamonas和Escherichia显著的富集,而耐抗组胺药患者肠道中的Blautia,Alistipes,Anaerostipes和Lachnospira则明显的降低。肠道微生物菌群与人体存在的共生和协同进化的关系,可以促进宿主免疫系统的发育及调节机体免疫系统平衡。
目前报道的关于无创方式判断过敏性疾病的方法是通过检测尿液中的相关蛋白的表达量进行判断,还有报道通过肠道中的菌群进行诊断食物过敏的方法,但是该方法是停留在小鼠实验阶段,并没有直接进行人群中进行验证,更没有针对儿童过敏进行验证,其准确度还是有待提高的,目前暂时还没有通过粪便样本中的特定菌群来直接判断儿童过敏性疾病的方法和手段,因此,亟需一种能够通过粪便样本中的特定菌群直接对儿童的过敏性疾病进行判断。
发明内容:
发明目的:本发明所要解决的技术问题时提供了基于肠型分型的过敏人群的微生物标志物组合物。
本发明还要解决的技术问题是提供了基于肠型分型的过敏人群的微生物标志物组合在制备鉴定过敏性疾病药物的试剂或试剂盒中的应用。
本发明还要解决的技术问题是提供了一种鉴定过敏性疾病的试剂盒。
本发明还要解决的技术问题是提供了所述的基于肠型分型的过敏预测模型的构建方法。
技术方案:为了解决上述技术问题,本发明提供了基于肠型分型的过敏人群的微生物标志物组合物,其包括Incertae Sedis、Bombiscardovia、Eggerthella、Erysipelatoclostridium、Lachnospira、[Eubacterium]eligens group、Ligilactobacillus、[Ruminococcus]gauvreauii group、Lachnospiraceae NK4A136group、Bifidobacterium、Veillonella、Collinsella、Faecalibacterium、[Clostridium]innocuum group和Enterococcus中的一种或几种。
本发明内容还包括所述的基于肠型分型的过敏人群的微生物标志物组合物在制备鉴定过敏性疾病的试剂或试剂盒中的应用。
本发明内容还包括一种鉴定过敏性疾病的试剂盒,所述试剂盒包含检测试剂,所述检测试剂用于检测包括所述受试者的肠道微生物菌群的样品中包括Incertae Sedis、Bombiscardovia、Eggerthella、Erysipelatoclostridium、Lachnospira、[Eubacterium]eligens group、Ligilactobacillus、[Ruminococcus]gauvreauii group、Lachnospiraceae NK4A136 group、Bifidobacterium、Veillonella、Collinsella、Faecalibacterium、[Clostridium]innocuum group和Enterococcus中的一种或几种。
其中,所述样品包括但不仅限于粪便样品。
本发明还包括所述的基于肠型分型的过敏预测模型的构建方法,包括以下步骤:
1)在NCBI或SRA公开数据中筛选适合年龄段的样本;
2)下载第一步中筛选出的原始16S测序数据和对应的样本信息数据,年龄,过敏与否;
3)通过16S分析流程鉴定每个样本的菌群组成结构数据;
4)肠型模型训练:利用结构主题模型来训练上述处理好的样本的菌群组成结构数据,得到最优的肠型种类数,各肠型的组成结构以及肠型模型参数;
5)运用肠型模型对用来训练的样本进行肠型预测、统计过敏在各肠型中的例数以及各样本的肠型分布;
6)特征筛选:将步骤5)中的数据拆分为训练集和测试集,进而通过随机森林方法在训练集中评估微生物特征在模型中的重要性,最终选取15个重要性最高的微生物特征作为标志物;
7)模型训练:以步骤6)的15个微生物标志物为特征,根据逻辑斯蒂回归、支持向量机或随机森林算法训练出对正常儿童和过敏儿童进行区分的二分类模型即得。
其中,步骤1)中的年龄段为1-2岁的儿童。
其中,步骤5)中的肠型分布包括以Blautia为代表的E1,以Bacteroides为代表的E2,以Bifidobacterium为代表的E3。
其中,步骤6)中的15个微生物标志物包括Incertae Sedis、Bombiscardovia、Eggerthella、Erysipelatoclostridium、Lachnospira、[Eubacterium]eligens group、Ligilactobacillus、[Ruminococcus]gauvreauii group、Lachnospiraceae NK4A136group、Bifidobacterium、Veillonella、Collinsella、Faecalibacterium、[Clostridium]innocuum group和Enterococcus。
本发明内容还包括所述的构建方法得到的过敏预测模型。
本发明内容还包括所述的过敏预测模型在制备判断样本过敏风险的系统中的应用。
本发明内容还包括一种基于肠型和机器学习的过敏诊断和预测的方法。
本发明内容包括过敏判断方法其包括以下步骤:
1)基于大规模公开的16S测序数据进行肠型分类判断,训练出肠型分类模型;
2)基于上述的肠型分类结果,应用机器学习方法建模寻找过敏儿童相关的微生物标志物;
3)正常儿童粪便样本和过敏儿童粪便样本,提取其中的微生物基因组DNA;
4)PCR扩增16S核糖体rRNA序列的V3-V4区(或其他可变区),构建文库并上机进行测序;
4)对测序数据进行生物信息分析,鉴定微生物菌群结构;
6)针对上述菌群鉴定结果,应用第一步中的肠型分类模型计算出该样本的肠型;
7)对于上述肠型分类结果,应用肠型相对应的机器模型和微生物标志物来判断该样本的过敏风险。
综上,本发明基于大规模公开数据的研究,收集大批量公开微生物测序数据,对不同年龄段的儿童进行肠道菌群类型分型,继而通过机器学习的方法寻找不同肠型中的过敏疾病标志物,并通过微生物标志物预测婴幼儿过敏发病风险。本发明通过采取儿童粪便,在实验室环境下提取粪便中微生物的基因组DNA,扩增后利用二代测序技术检测粪便中菌群结构,从菌群结构判断该样本的肠型,进行通过机器学习方法预测和判断该样本的过敏风险。
有益效果:与现有技术相比,本发明具备以下优点:
1)本发明首次提出了基于肠型分层的过敏预测模型,先通过肠型进行分类,进而利用对应机器学习模型预测该样本的过敏风险。
2)本发明通过超过2000例的数据训练和学习,在以Bifidibacterium为核心的肠型寻找出15种微生物标志物,可有效预测该肠型种儿童的过敏风险。
3)本发明通过一种无创非侵入的方式,仅需要儿童的粪便即可检测出该儿童的过敏风险。本发明为未来儿童过敏的研究提供新的技术基础,为儿童过敏检测提供新的无创非侵入的新方法。
附图说明
图1肠型在2310份样本中的分布;
图2各肠型菌属组成(肠型1代表E1,肠型2代表E2,肠型3代表E3);
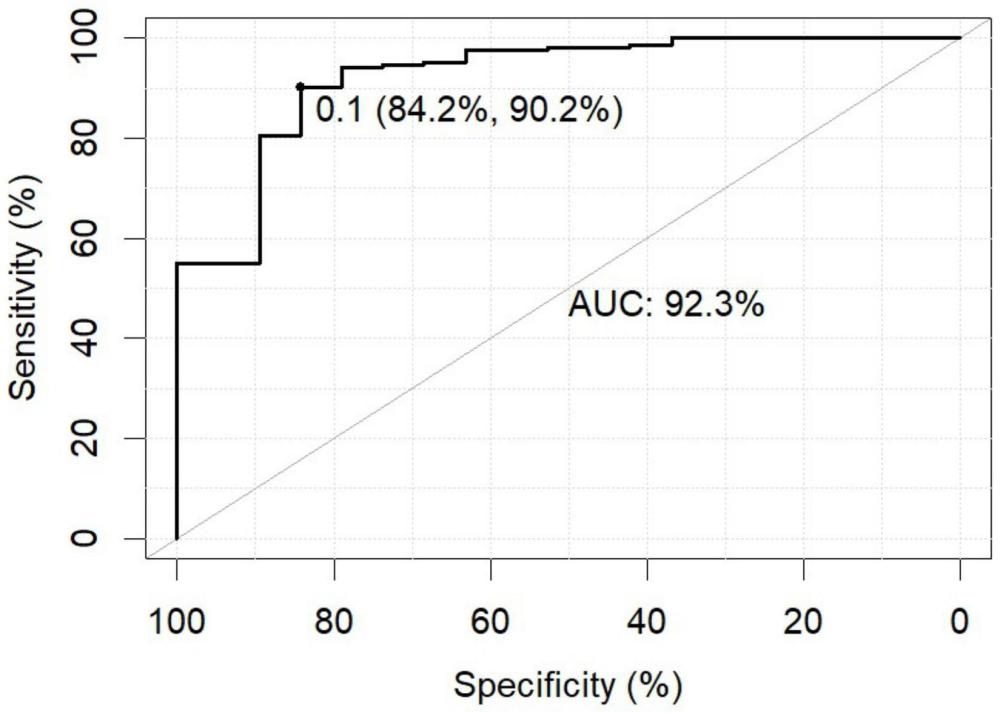
图3机器学习过敏模型在测试集上的表现。
具体实施方式
实施例1 1~2岁儿童肠型分类模型学习
本实施例主要包括以下步骤,即数据查找筛选、数据下载、数据分析、模型训练和模型预测。
1、通过查找阅读儿童粪便肠道菌群文献,筛选适合年龄段(1~2岁)的文献,再对文献进行分析,选取利用16S测序且数据已公开的研究,具体选择了来源于NCBI或ENA,总共2311份样本;
2、在公开数据库中下载第一步中筛选出的原始测序数据和对应的样本信息(如年龄,过敏与否等);
表1
3、测序数据分析:
基于DADA2(https://benjjneb.github.io/dada2/tutorial.html)的16S扩增子分析流程对测序下机数据进行处理。操作步骤如下:
(1)首先,下机序列经过去接头和拆分得到每个样本的16S rRNA序列,然后经过质量过滤、去噪音、合并以及去除嵌合体过程,得到100%一致性的ASV(Amplicon SequenceVariant)序列。
(2)将ASV序列比对到Silva数据库(v138)得到序列的物种命名。
(3)整合不同参考序列的命名,将注释到同一物种的序列丰度合并为同一物种的丰度,获得所用样本的菌群组成数据;
4、肠型模型训练:利用结构主题模型(R语言stm包)来训练上述处理好的样本菌群组成数据,得到最优的肠型种类数,各肠型的组成结构以及肠型模型参数;
5、运用肠型模型对用来训练的样本进行肠型预测、统计过敏在各肠型中的例数(见表2,三种肠型的组成结构)以及各样本的肠型分布(见图1),明显可以看出1~2岁婴幼儿中,拟杆菌属肠型占比最高,其次为双岐杆菌属肠型,这与婴儿早期成长阶段的母乳喂养和辅食添加有密切相关。而从过敏发生的比率来看,双岐杆菌属肠型的婴幼儿中,过敏比率仅有8.59%,远远低于其他两种肠型(都在14%以上)。
表2不同肠型中过敏和正常婴幼儿分布
实施例2 1~2岁Bifidobacterium肠型儿童过敏相关微生物标志物的寻找
本实施例主要包括以下步骤,即数据整理、特征筛选、模型训练、模型评估。
1、数据整理:从实施例1中的2310多样本中选取肠型为Bifidobacterium型的样本;
2、特征筛选:以8:2为比例将第一步中的数据拆分为训练集和测试集,进而通过随机森林方法在训练集中评估微生物特征在模型中的重要性,最终选取15个重要性最高的微生物特征作为标志物(见下表3,以Bifidobacterium为核心的肠型中找到的15种微生物标志物);
表3 15种微生物标志物
3、模型训练:以第二步的15个标志物为特征,训练出对正常儿童和过敏儿童进行区分的二分类模型,模型训练选择的是随机森林算法;根据模型输出的概率,选择大于50%为过敏,小于等于50%为正常;
4、模型评估:在测试集上,对上述训练出的模型进行评估,模型在测试集上的AUC如图3。模型在测试集上预测的混淆矩阵如表4。从表4中可以看出,模型预测出的19例过敏中有7例是真正过敏的,而模型预测出的正常的样本中,实际中均为正常,证明了模型较好特异性。模型在测试集上的表现参数如表5,从标准可以看出,模型的准确率能达到94.62%,特异性能达到100%。
表4模型预测的混淆矩阵
表5模型在预测集上的参数表现
实施例3 1~2岁儿童过敏风险判断
本实施例主要包括以下步骤,即样本收集、提取建库和测序、数据处理、肠型确定和过敏风险判断。
1、样本收集:按照样本采集流程收集表4中的223例的粪便样本2g(如1~2岁儿童样本),冷冻保存;
2、提取建库和测序:
a)提取:使用土壤微生物DNA抽提试剂盒(OMEGASoil DNA Kit,M5635-02)用于提取微生物的基因组DNA。
b)PCR扩增:微生物RNA含有多个保守区和可变区,这里利用引物338F(5'-ACTCCTACGGGAGGCAGCA-3')和806R(5'-GGACTACHVGGGTWTCTAAT-3')对样本的16S rRNA基因V3-V4区进行PCR扩增。
PCR采用NEB Q5 DNA高保真聚合酶,体系见表6:
表6
操作过程如下:
将PCR反应所需的成分配置完后,在PCR仪上于98℃预变性30s,使模板DNA充分变性,然后进入扩增循环。在每一个循环中,先于98℃保持15s使模板变性,然后将温度降到50℃,保持30秒钟,使引物于模板充分退火;在72℃保持30秒钟,使引物在模板上延伸,合成DNA,完成一个循环。重复这样的循环25~27次,使扩增的DNA片段大量累积。最后,在72℃保持5分钟,使产物延伸完整,4℃保存。
扩增结果进行2%琼脂糖凝胶电泳,用Axygen凝胶回收试剂盒回收目的片段,片段大小为480bp左右。
c)建库:利用Illumina公司的TruSeq Nano DNA LT Library Prep Kit进行建库。首先进行的末端修复过程是利用试剂盒中的End Repair Mix2将DNA5’端突出的碱基切除,3’端缺失的碱基补齐,同时在5’端加上一个磷酸基团;
具体步骤如下:
第一步是DNA5’端突出的碱基切除:
(1)取30ng混好的上述步骤得到的DNA片段补水至60μL,加入40μL End RepairMix2;
(2)用枪吹打混匀,并置于PCR仪上30℃孵育30min;
(3)利用BECKMAN AMPure XP beads纯化末端修复体系(BECKMAN公司购买),最终用17.5μL Resuspension buffer洗脱。
第二步是3’端加A,在这一过程中,DNA的3’端会单独加上一个A碱基,以防止DNA片段的自连,同时保证DNA与3’端有一个突出T碱基的测序接头相连,具体步骤如下:
(1)向片段选择后的DNA中加入12.5μL A-Tailing Mix;
(2)用枪吹打混匀,并置于PCR仪上孵育,程序如下:37℃,30min;70℃,5min;4℃,5min;4℃,∞。
第三步是加一个有特异性标签的接头,此过程是为了让DNA最终杂交到Flow Cell上,具体步骤如下:
(1)在第二步得到的产物的体系中,加入2.5μL Resuspension buffer,2.5μLLigation Mix和2.5μLDNA adapter Index;
(2)用枪吹打混匀,并置于PCR仪上,30℃孵育10min;
(3)加入5μL Stop Ligation buffer;
(4)利用BECKMAN AMPure XP beads纯化已加接头的体系。
第四步是通过PCR扩增已经加上接头的DNA片段,然后利用BECKMAN AMPure XPbeads纯化PCR体系。
第五步是通过2%琼脂糖凝胶电泳来对文库做最终的片段选择与纯化。
d)上机测序:首先对文库进行质量检验,待质检合格后,进行上机测序。首先将需要上机的文库(Index不可重复)梯度稀释到2nM,然后按所需数据量比例混样。混好的文库经0.1N NaOH变性成单链进行上机测序,具体在Illumina NovaSeq机器上利用NovaSeq6000SP Reagent Kit(500cycles)进行2×250bp的双端测序。
3、数据处理:基于DADA2(https://benjjneb.github.io/dada2/tutorial.html)的16S扩增子分析流程对测序下机数据进行处理。操作步骤如下:
a)首先,下机序列经过去接头和拆分得到每个样本的16S rRNA序列,然后经过质量过滤、去噪音、合并以及去除嵌合体过程,得到100%一致性的ASV序列(AmpliconSequence Variant)。
b)将ASV序列比对到Silva数据库(v138)得到序列的物种命名。
c)整合不同参考序列的命名,将注释到同一物种的序列丰度合并为同一物种的丰度;
d)单个样本中,将物种的丰度转化为相对丰度,即每个物种所占的序列(Read)数量除以该样本总的序列(Read)数量;最终得到每个菌的相对丰度,即样本的菌群组成结构。4、肠型确定:根据上述样本的菌群组成结构,应用实施例1中的肠型分类算法计算出该样本的肠型类型;
5、过敏风险:选择不同肠型对应的机器学习模型及微生物标志物,机器模型输出结果即为该样本过敏风险判断结果。最终从以上的223个样本中检测得到过敏的有7例,正常的有216例,其结论与实施例2的模型验证的结果一致。