高纯度氟比洛芬酯的制备方法
文献发布时间:2024-04-18 19:58:21
技术领域
本发明属于药物合成技术领域,具体涉及高纯度氟比洛芬酯的制备方法。
背景技术
氟比洛芬酯是一种具有抗炎镇痛功能的药物,为无色至微黄色透明油状物,结构式如下:
目前,氟比洛芬酯一般采用氟比洛芬和乙酸-1-溴乙酯反应制得,例如国大亮等在《氟比洛芬酯的合成》(齐鲁药事,2007,Vol.26,No.9,557-558)中公开的氟比洛芬酯制备方法,其采用摩尔比1:1.6的氟比洛芬和乙酸-1-溴乙酯作为原料,采用丙酮作为溶剂进行反应,并在反应后加乙酸乙酯稀释,然后常压蒸去溶剂,再高真空(0.8mmHg)减压蒸馏产品。
再例如专利CN102381970A中公开的氟比洛芬酯制备方法,其采用摩尔比1:(1.5-1.7)的氟比洛芬和乙酸-1-溴乙酯作为原料,在二甲基甲酰胺(DMF)溶剂的作用下进行反应,反应结束后加入乙酸乙酯和水后,分出有机相,水洗涤,饱和碳酸钠溶液洗涤,再水洗涤,最后用饱和食盐水洗涤,有机相旋干后抽真空去除溶剂,得粗品,然后用醚类和酯类混合有机溶剂溶解粗品,加入硅胶活性炭升温回流20-40min,冷却抽滤,抽真空得到氟比洛芬酯。
但是,上述制备方法中还存在以下缺陷:(1)混合溶剂难以套用,尤其是难以分离的混合溶剂,比如丙酮-乙酸乙酯混合溶剂、DMF-乙酸乙酯混合溶剂;(2)水洗时溶剂进入水中,有机废液增加环保压力;(3)利用高真空减压蒸馏产品的方法对产品进行精制,工业化难度大;(4)乙酸-1-溴乙酯作为基因毒杂质,其限度要求低至50ppm,上述方法难以达到标准;(5)产物收率较低;(6)产物纯度低。此外,专利CN102381970A采用的高沸点溶剂二甲基甲酰胺难以回收套用,尤其在产物氟比洛芬酯高温下不稳定、后处理中与乙酸乙酯、水混合的情况下,工业化价值低。
发明内容
本发明要解决的技术问题是:提供一种高纯度氟比洛芬酯的制备方法,收率高、成本低、纯度高,便于工业化生产,制备的氟比洛芬酯纯度≥99.5%,氟比洛芬含量≤0.4%,其他最大杂质含量≤0.1%,其他总杂质含量≤0.2%,乙酸-1-溴乙酯含量≤50ppm,收率90%以上。
本发明所述的高纯度氟比洛芬酯的制备方法,包括以下步骤:
(1)将氟比洛芬、乙酸-1-溴乙酯、溶剂、碳酸氢钾加入反应瓶中,进行反应;
(2)反应结束后,对反应物进行水洗,得到有机层;
(3)将有机层先在0-50℃低温下减压蒸馏除去溶剂,至溶剂不出后,再升温至60-90℃,在氮气流条件下进行后期减压蒸馏除杂,得到氟比洛芬酯。
本发明中,氟比洛芬酯的合成反应式如下:
步骤(1)中,氟比洛芬和乙酸-1-溴乙酯的摩尔比为1:(1.4-1.6)。
步骤(1)中,氟比洛芬和碳酸氢钾的摩尔比为1:(1.8-2.2)。
步骤(1)中,溶剂为二氯甲烷、1,2-二氯乙烷、三氯甲烷中的一种。氟比洛芬和溶剂的摩尔体积比为1mol:(0.8L-1.2L)。
步骤(1)中,反应温度为40-50℃,反应时间为12-16h。
步骤(2)中,对反应物进行水洗时,控制温度0-20℃,先向反应液中加入水、碳酸钾,搅拌、静置、分层,得有机相,再将有机相水洗3次,得到有机层。
在一种实施方式中,对反应物进行水洗时,控制温度0-20℃,先以水与氟比洛芬的体积摩尔比为0.6L:1mol向反应液中加入水,再以碳酸钾与氟比洛芬的摩尔为(0.4-0.6):1加入碳酸钾,搅拌、静置、分层,得有机相,再将有机相水洗3次,每次水洗时所使用的水与氟比洛芬的体积摩尔比为0.6L:1mol,得到有机层。
步骤(3)中,在0-50℃低温下减压蒸馏时,真空度高于-0.095MPa。
步骤(3)中,在60-90℃下进行后期减压蒸馏除杂时,开通氮气流调节真空度在-0.08MPa到-0.09MPa之间,后期减压蒸馏除杂的目的是除去残留的溶剂和乙酸-1-溴乙酯等杂质。
步骤(3)中,工业生产减压蒸馏时,在减压蒸馏釜内底部设置呈等速螺线分布的氮气流管路,使氮气更加均匀的通入物料体系中。
本发明制备得到的氟比洛芬酯纯度≥99.5%,氟比洛芬含量≤0.4%,其他最大杂质含量≤0.1%,其他总杂质含量≤0.2%,乙酸-1-溴乙酯含量≤50ppm。收率90%以上。
与现有技术相比,本发明的有益效果如下:
(1)本发明采用二氯甲烷等溶剂代替丙酮、DMF、乙酸乙酯混合溶剂,利于溶剂回收套用;
(2)本发明通过减压低温蒸馏除去溶剂,氮气流条件下减压蒸馏带走乙酸-1-溴乙酯,基因毒杂质乙酸-1-溴乙酯易达到标准,乙酸-1-溴乙酯≤50ppm,且通过使用二氯甲烷等溶剂进行反应、低温减压蒸去溶剂等技术,降低了反应液中杂质含量,从而无需进行高真空蒸馏,大大降低了工业化生产难度;
(3)本发明制备的氟比洛芬酯纯度≥99.5%,氟比洛芬含量≤0.4%,其他最大杂质含量≤0.1%,其他总杂质含量≤0.2%,乙酸-1-溴乙酯含量≤50ppm。收率90%以上。
附图说明
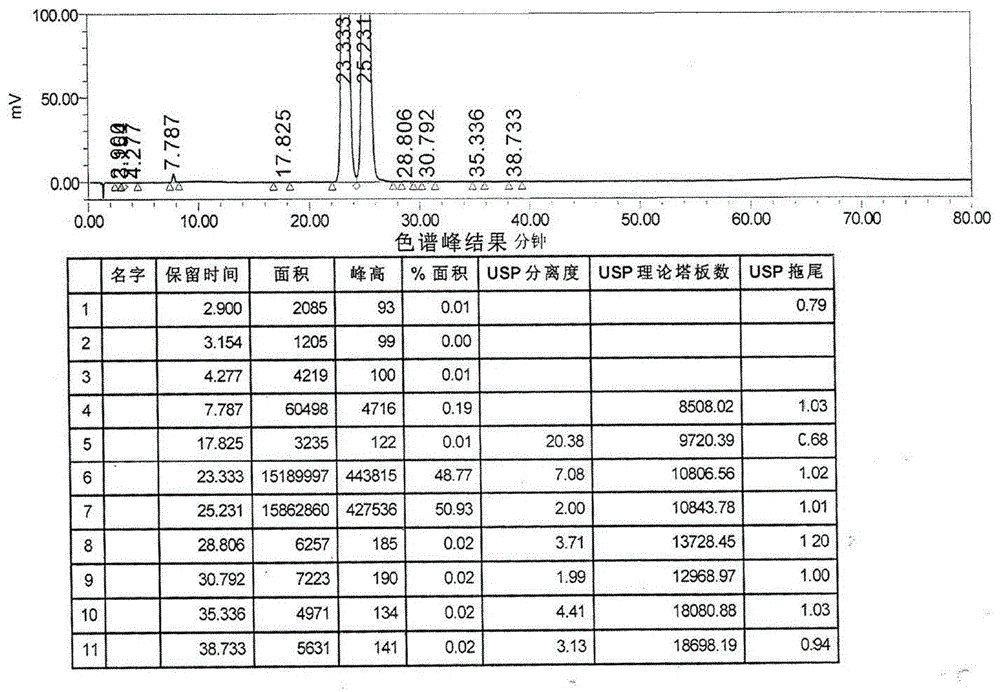
图1为本发明制备的氟比洛芬酯HPLC谱图;
图2为乙酸-1-溴乙酯对照液GC谱图
图3为本发明制备的氟比洛芬酯的GC分析谱图。
具体实施方式
下面结合实施例对本发明做出进一步说明。实施例中所使用的原料,如无特别说明,均为市售常规原料;实施例中所使用的工艺方法,如无特别说明,均为本领域常规方法。
实施例中采用HPLC对制备得到的氟比洛芬酯进行分析,测试氟比洛芬酯的色谱纯度、有关物质含量。参照高效液相色谱法(中国药典2020年版四部通则0512)测定,具体测试方法如下:
溶剂:乙腈-水(体积比45:55)。
供试品溶液:取本品适量,精密称定,加溶剂溶解并定量稀释制成每1ml中约含0.5mg的溶液。
氟比洛芬定位溶液:取氟比洛芬适量,加溶剂溶解并定量稀释制成每1ml中约含25μg的溶液。
色谱条件:用十八烷基硅烷键合硅胶为填充剂(Waters symmetry shield RP18柱,3.9×150mm,5μm);以水-冰醋酸-乙腈混合液为流动相,水和冰醋酸的体积比为60:5,水和冰醋酸的总体积与乙腈的体积比为55:45;流速为每分钟1.0ml;检测波长为254nm;进样体积20ul。
系统适用性要求理论板数按氟比洛芬峰计算不低于3000。
测定法:精密量取供试品溶液与氟比洛芬定位溶液,分别注入液相色谱仪,记录色谱图。
限度:供试品溶液色谱图中如有杂质峰,按面积归一化法计算,氟比洛芬不大于0.5%,其他单个杂质不大于0.1%,其他总杂质不大于0.5%。
实施例中采用GC分析方法对制备得到的氟比洛芬酯进行分析,测试氟比洛芬酯中残留乙酸-1-溴乙酯的含量。参照气相色谱法(中国药典2020年版四部通则0521)测定,具体测试方法如下:
供试品溶液:取本品约0.25g,精密称定,置2ml量瓶中,加二氯甲烷溶解并稀释至刻度,摇匀。
对照品溶液:取乙酸-1-溴乙酯对照品约25mg,精密称定,置50ml量瓶中,加二氯甲烷溶解并稀释至刻度,摇匀,精密量取1ml,置20ml量瓶中,用二氯甲烷稀释至刻度,摇匀,再精密量取1ml,置20ml量瓶中,用二氯甲烷稀释至刻度,摇匀。
色谱条件:以6%氰丙基苯基-94%二甲基聚硅氧烷为固定液的毛细管为色谱柱(30m×0.53mm,3.0μm);起始温度80℃,维持12min,再以50℃/min速率升温至180℃,维持10min;流速5.0ml/min,进样口温度为200℃;检测器温度为250℃;载气为氮气,检测器为电子捕获检测器(ECD);进样体积1μl。
系统适用性要求:理论板数按乙酸-1-溴乙酯峰计算不低于3000。
测定法:分别精密量取供试品溶液和对照品溶液,分别注入气相色谱仪,记录色谱图。
限度:供试品溶液色谱图中如有与乙酸-1-溴乙酯保留时间一致的色谱峰,其含量不得大于0.005%。
计算公式:乙酸-1-溴乙酯%=(A
其中,
A
W
V
A
W
V
实施例1
采用本发明的制备方法制备氟比洛芬酯,包括以下步骤:
(1)将12.2g(99%,0.05mol)氟比洛芬、12.8g(97%,0.075mol)乙酸-1-溴乙酯、60ml二氯甲烷、10.0g(99.5%,0.10mol)碳酸氢钾加入反应瓶中,升温至40℃反应16h;
(2)反应结束后,控制温度10℃,向反应液中加30ml水、3.5g(99%,0.025mol)碳酸钾,搅拌、静置、分层,得有机相,再水洗3次,每次加水30ml,得到有机层;
(3)检查反应瓶真空系统-0.095MPa以上,将有机层加入反应瓶,搅拌,减压蒸二氯甲烷,反应瓶内温度由于抽真空自然降温,最低降至0℃,然后逐步升至10℃,真空度逐步升至-0.095MPa,馏分不出后,升温至60℃,开通氮气流调节真空度在-0.08MPa到-0.09MPa之间,进一步蒸去二氯甲烷及乙酸-1-溴乙酯等杂质,3h后取样,得氟比洛芬酯15.63g,收率94.6%。
经HPLC和GC分析,产品氟比洛芬酯纯度99.55%,氟比洛芬含量0.30%,其他最大杂质含量0.04%,其他总杂质含量0.15%,乙酸-1-溴乙酯含量3.3ppm。
实施例2
采用本发明的制备方法制备氟比洛芬酯,包括以下步骤:
(1)将12.2g(99%,0.05mol)氟比洛芬、12.0g(97%,0.07mol)乙酸-1-溴乙酯、40ml1,2二氯乙烷、9.0g(99.5%,0.09mol)碳酸氢钾加入反应瓶中,升温至50℃反应12h;
(2)反应结束后,控制温度20℃,向反应液中加30ml水、2.8g(99%,0.020mol)碳酸钾,搅拌、静置、分层,得有机相,再水洗3次,每次加水30ml,得到有机层;
(3)检查反应瓶真空系统0.099MPa以上,将有机层加入反应瓶,搅拌,减压蒸1,2-二氯乙烷,控制温度20-50℃之间,真空度逐步升至-0.099MPa,馏分不出后,升温至90℃,开通氮气流调节真空度在-0.08MPa到-0.09MPa之间,进一步蒸去1,2-二氯乙烷及乙酸-1-溴乙酯等杂质,3h后取样,得氟比洛芬酯15.17g,收率91.8%。
经HPLC和GC分析,产品氟比洛芬酯纯度99.71%,氟比洛芬含量0.12%,其他最大杂质含量0.04%,其他总杂质含量0.17%,乙酸-1-溴乙酯含量8.2ppm。
实施例3
采用本发明的制备方法制备氟比洛芬酯,包括以下步骤:
(1)将12.2g(99%,0.05mol)氟比洛芬、13.7g(97%,0.08mol)乙酸-1-溴乙酯、50ml氯仿、11.0g(99.5%,0.011mol)碳酸氢钾加入反应瓶中,升温至45℃反应20h;
(2)反应结束后,控制温度5℃,向反应液中加30ml水、4.1g(99%,0.030mol)碳酸钾,搅拌、静置、分层,得有机相,再水洗3次,每次加水30ml,得到有机层;
(3)检查反应瓶真空系统0.098MPa以上,将有机层加入反应瓶,搅拌,减压蒸氯仿,控制温度20-50℃之间,真空度逐步升至-0.098MPa,馏分不出后,升温至70℃,开通气流调节真空度在-0.08MPa到-0.09MPa之间,进一步蒸去氯仿及乙酸-1-溴乙酯等杂质,3h后取样,得氟比洛芬酯15.24g,收率92.2%。
经HPLC和GC分析,产品氟比洛芬酯纯度99.70%,氟比洛芬含量0.19%,其他最大杂质含量0.02%,其他总杂质含量0.11%,盐酸-1-溴乙酯含量18.5ppm。
图1为氟比洛芬酯HPLC谱图;图中23.333′、25.231′为氟比洛芬酯的两个互变异构体,均为氟比洛芬酯的有效成分,二者相加99.70%即为氟比洛芬酯的色谱纯度;图中7.787′峰为氟比洛芬,含量为0.19%;其他最大杂质含量为0.02%;其他总杂质0.11%。
图2为乙酸-1-溴乙酯对照液GC谱图,从图2可以看出,对照品峰面积为86094。
图3为氟比洛芬酯的GC分析谱图;从图3可以看出,样品中乙酸-1-溴乙酯峰面积为151823,乙酸-1-溴乙酯含量=(A
对比例1
与《氟比洛芬酯的合成》(齐鲁药事,2007,Vol.26,No.9,557-558)中公开的氟比洛芬酯制备方法,进行对比,具体如下:
向250mL四颈瓶加入氟比洛芬12.2g(0.05mol),碳酸氢钾8g,搅拌下加入丙酮110mL,滴加乙酸-1-溴乙酯13.4g(0.08mol),室温反应5h,加入200mL乙酸乙酯稀释,将反应液转入分液漏斗中,用3%碳酸钠洗涤(2×200mL),分取有机层,无水硫酸钠干燥,过滤除去干燥剂,加入活性炭回流脱色20min,过滤除去活性炭,将滤液常压浓缩至无液体馏出,将剩余物减压蒸馏,收集173-175℃/0.8mm Hg馏分,得无色液体13.8g,氟比洛芬酯收率83.7%。
对比例2
与专利CN102381970A中公开的氟比洛芬酯制备方法进行对比,其实施例1如下:
向2L反应瓶中,加入2-氟-α-甲基(1,1'-二苯基)-4-乙酸(0.5mol),DMF(1000ml),碳酸钾(0.6mol),搅拌升温70~75℃,反应2h,降至20℃,滴加乙酸-1-溴乙酯(0.75mol),反应5h。然后转入分液漏斗中,加入2L乙酸乙酯,1.5L水,分出有机相继续水洗2L两次,有机相继续使用饱和碳酸钠溶液500ml洗涤一次,然后水洗2L两次,最后使用1.5L饱和食盐水洗涤两次。有机相无水硫酸钠干燥旋干,得到红褐色油状物,使用油泵在80℃抽真空2h。将所得油状物用2400ml乙酸乙酯:石油醚=1:20(体积比)溶解,加入84g硅胶,84g活性炭,升温回流30min,稍微冷却抽滤,再加入42g硅胶,84g活性炭,升温回流30min,稍微冷却后过滤,滤液旋干得到无色透明液体,油泵90℃真空抽1h得到131.3g无色油状物,收率:76.9%,纯度99.2%。
对比例3-5
为考察溶剂对产品氟比洛芬酯收率和纯度的影响,实施例1的预实验和对比例3-5对工业化前景较好的潜在溶剂二氯甲烷、丙酮、甲基叔丁基醚(MEBE)、二氧六环进行对比研究,对比例3-5与实施例1的预实验的不同之处仅在于所采用的溶剂和反应条件不同,且根据不同的溶剂沸点适应性改变反应条件,即反应温度和时间。
具体的,实施例1的预实验和对比例3-5的溶剂类型、反应条件以及反应结果如表1所示。
表1
从表1可以看出,MTBE、二氧六环做溶剂,反应慢,未反应的氟比洛芬含量高,杂质大;丙酮和二氯甲烷做溶剂,反应速度快,杂质低,考虑到二氯甲烷在后续水洗时,容易分层,水洗液中不含有机溶剂,对环境友好,而且二氯甲烷作为溶剂反应副产物(其他总杂质)特别小,可以不经蒸馏即得到高纯度产品所以,选择二氯甲烷作为反应溶剂;二氯甲烷、二氯乙烷、氯仿均为卤代烷,在反应中表现出良好的优势,反应较快,且副产物很小。
- 一种氟比洛芬的制备方法及氟比洛芬酯的制备方法
- 一种氟比洛芬酯与去氟氟比洛芬酯的分离检测方法