一种氟唑菌苯胺代谢物的制备方法
文献发布时间:2023-06-19 09:27:35
技术领域
本发明涉及药物代谢技术领域,尤其涉及一种氟唑菌苯胺代谢物的制备方法。
背景技术
氟唑菌苯胺是拜耳公司开发的琥珀酸脱氢酶抑制剂(SDHI)类杀菌剂,2011年在英国获得首次登记,兼具内吸、预防和治疗作用,持效期长,防治纹枯病、根腐病、黑穗病等,化学名称为N-[2-(1,3-二甲基丁基)苯基]-5-氟-1,3-二甲基-1H-吡唑-4-甲酰胺,CAS登录号为494793-67-8,相对分子质量317.41,分子式为C
氟唑菌苯胺在植株、土壤、沉积物、水中的主要代谢物产物为3-羟基丁基-氟唑菌苯胺,其化学名称为5-氟-N-(2-(4-羟基-4-甲基戊烷-2-基)苯基)-1,3-二甲基-1H-吡唑-4-甲酰胺,目前缺少氟唑菌苯胺代谢物的环境行为及毒性等数据,其合成方法未见报道,为氟唑菌苯胺代谢物的系统性研究带来困难,影响氟唑菌苯胺风险评估的准确性。
发明内容
有鉴于此,本发明的目的在于提供一种氟唑菌苯胺代谢物的制备方法。本发明提供的制备方法可操作性强,得率高,可实现工业化生产氟唑菌苯胺代谢物。
为了实现上述发明目的,本发明提供以下技术方案:
本发明提供了一种氟唑菌苯胺代谢物(5-氟-N-(2-(4-羟基-4-甲基戊烷-2-基)苯基)-1,3-二甲基-1H-吡唑-4-甲酰胺)的制备方法,包括以下步骤;
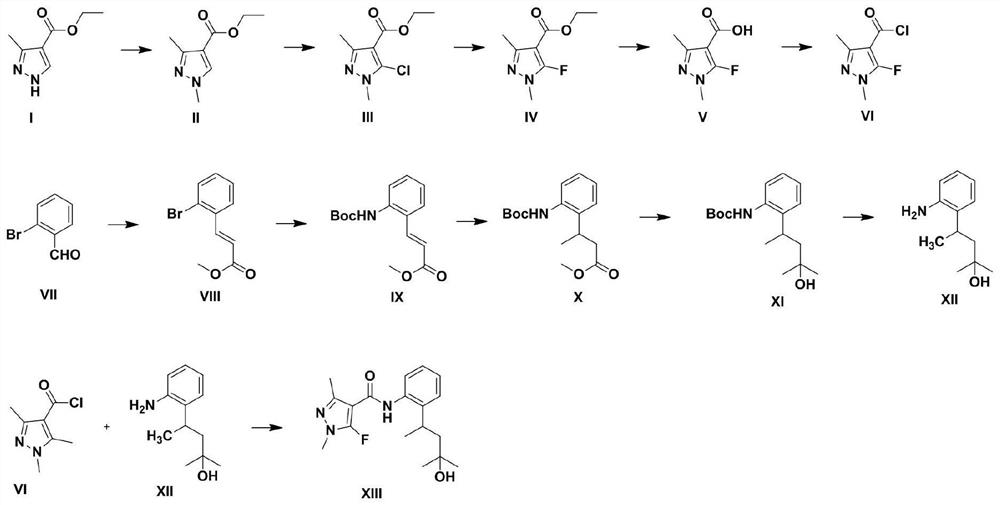
将具有式I所示结构的化合物溶解在极性溶剂中,与强碱和碘甲烷混合进行甲基化反应,得到中间产物II;
将所述中间产物II溶解在极性溶剂中,与氯化剂混合进行氯化反应,得到中间产物III;
将所述中间产物III溶解在极性溶剂中,与氟化剂混合进行氟化反应,得到中间产物IV;
将所述中间产物IV溶解在极性溶剂中,与碱性试剂混合进行水解,得到中间产物V;
将所述中间产物V与酰氯化剂混合进行酰氯化反应,得到酰氯中间产物VI;
将具有式VII所示结构的化合物溶解在稀释剂中,与甲氧甲酰基亚甲基三苯基膦混合进行Wittig反应,得到中间产物VIII;
将所述中间产物VIII、钯类催化剂、膦配体、碳酸铯和氨基甲酸叔丁酯在非极性溶剂和惰性气体的存在下混合进行偶联反应,得到中间产物IX;
将所述中间产物IX、铜试剂和甲基溴化镁在非极性溶剂和惰性气体的存在下混合进行1,4-共轭加成反应,得到中间产物X;
将所述中间产物X和甲基溴化镁在非极性溶剂和惰性气体的存在下混合进行亲核加成反应,得到中间产物XI;
将所述中间产物XI溶解在稀释剂中,与酸性试剂混合进行脱保护,得到中间产物XII;
将所述中间产物XII溶解在稀释剂中,与有机碱试剂和所述中间产物VI混合进行缩合反应,得到氟唑菌苯胺代谢物。
优选地,所述强碱为碳酸钾、碳酸钠、氢氧化钾或氢氧化钠,所述强碱与具有式I所示结构的化合物的当量比为1:1~2:1,所述碘甲烷与具有式I所示结构的化合物的当量比为1:1~2:1,所述甲基化反应的温度为10~40℃,时间为8~24h。
优选地,所述氯化剂为磺酰氯、氯化亚砜或N-氯代丁二酰亚胺,所述氯化剂与中间产物II的当量比为1:1~20:1,所述氯化反应的温度为30~120℃,时间为8~24h。
优选地,所述氟化剂为氟化钠、氟化钾、氟化铯或氟化锂,所述氟化剂与中间产物III的当量比为0.8:1~2:1,所述氟化反应的温度为100~200℃,时间为6~24h。
优选地,所述酰氯化剂为氯化亚砜、草酰氯、三光气或三氯氧磷,所述酰氯化剂与中间产物V的当量比为1:1~5:1,所述酰氯化反应的温度为20~79℃,时间为1~24h。
优选地,所述甲氧甲酰基亚甲基三苯基膦与具有式VII所示结构的化合物的当量比为0.8:1~1.5:1,所述Wittig反应的温度为20~110℃,时间为6~24h。
优选地,所述中间产物VIII、钯类催化剂、膦配体、碳酸铯和氨基甲酸叔丁酯的当量比为1:0.05:0.2:1.2:1.1~1:0.1:0.3:1.5:2.0,所述钯类催化剂为三(二亚苄基丙酮)二钯、四(三苯基膦)钯、[1,1'-双(二苯基膦基)二茂铁]二氯化钯或醋酸钯,所述膦配体为2-二环己基磷-2,4,6-三异丙基联苯、2-二环己膦基-2'-(N,N-二甲胺)-联苯、2-(二环己基膦)3,6-二甲氧基-2′,4′,6′-三异丙基-1,1′-联苯或2-(二叔丁基膦)联苯,所述偶联反应的温度为35~120℃,时间为6~24h。
优选地,所述中间产物IX、铜试剂和甲基溴化镁的当量比为1:0.01:1.2~1:1.5:3.0,所述铜试剂为碘化亚铜、氯化亚铜、三氟甲烷磺酸铜或2-噻吩甲酸铜,所述1,4-共轭加成反应为先将所述铜试剂和甲基溴化镁混合后进行第一反应,然后再加入所述中间产物IX进行第二反应,所述第一反应的温度为-78~0℃,时间为0.5~6h,所述第二反应的温度为-78~0℃,时间为2~24h。
优选地,所述中间产物IX与甲基溴化镁的当量比为1:3.0~1:5.0,所述亲核加成反应的温度为-78~30℃,时间为2~24h。
优选地,所述缩合反应中,有机碱试剂与中间产物XII的当量比为2.0:1~10.0:1,所述中间产物VI与中间产物XII的当量比为0.8:1~1.5:1,所述缩合反应的温度为0~30℃,时间为3~24h。
本发明提供了一种氟唑菌苯胺代谢物(5-氟-N-(2-(4-羟基-4-甲基戊烷-2-基)苯基)-1,3-二甲基-1H-吡唑-4-甲酰胺)的制备方法,所述制备方法工艺设计合理,可操作性强,得率高,可实现工业化生产氟唑菌苯胺代谢物。本发明制备得到的氟唑菌苯胺代谢物可为氟唑菌苯胺的代谢机理研究提供对照品,可用于研究改药物在环境中的代谢过程,对氟唑菌苯胺及其代谢物的系统性研究及风险评估具有极大的价值。
进一步地,本发明通过具体限定了制备步骤和反应条件,整个工艺设计合理,可操作性强,制备得到的氟唑菌苯胺代谢物纯度在99%以上,取得了非常好的技术效果。
附图说明
图1为本发明制备氟唑菌苯胺代谢物的制备流程图;
图2为本发明实施例1制得的氟唑菌苯胺代谢物的液相色谱图。
具体实施方式
本发明提供了一种氟唑菌苯胺代谢物的制备方法,包括以下步骤;
将具有式I所示结构的化合物溶解在极性溶剂中,与强碱和碘甲烷混合进行甲基化反应,得到中间产物II;
将所述中间产物II溶解在极性溶剂中,与氯化剂混合进行氯化反应,得到中间产物III;
将所述中间产物III溶解在极性溶剂中,与氟化剂混合进行氟化反应,得到中间产物IV;
将所述中间产物IV溶解在极性溶剂中,与碱性试剂混合进行水解,得到中间产物V;
将所述中间产物V与酰氯化剂混合进行酰氯化反应,得到酰氯中间产物VI;
将具有式VII所示结构的化合物溶解在稀释剂中,与甲氧甲酰基亚甲基三苯基膦混合进行Wittig反应,得到中间产物VIII;
将所述中间产物VIII、钯类催化剂、膦配体、碳酸铯和氨基甲酸叔丁酯在非极性溶剂和惰性气体的存在下混合进行偶联反应,得到中间产物IX;
将所述中间产物IX、铜试剂和甲基溴化镁在非极性溶剂和惰性气体的存在下混合进行1,4-共轭加成反应,得到中间产物X;
将所述中间产物X和甲基溴化镁在非极性溶剂和惰性气体的存在下混合进行亲核加成反应,得到中间产物XI;
将所述中间产物XI溶解在稀释剂中,与酸性试剂混合进行脱保护,得到中间产物XII;
将所述中间产物XII溶解在稀释剂中,与有机碱试剂和所述中间产物VI混合进行缩合反应,得到氟唑菌苯胺代谢物。
图1为本发明制备氟唑菌苯胺代谢物的制备流程图。
本发明将具有式I所示结构的化合物(3-甲基-1H-吡唑-4-甲酸乙酯)溶解在极性溶剂中,与强碱和碘甲烷混合进行甲基化反应,得到中间产物II;
在本发明中,所述强碱优选为碳酸钾、碳酸钠、氢氧化钾或氢氧化钠,更优选为碳酸钾,所述强碱与3-甲基-1H-吡唑-4-甲酸乙酯的当量比优选为1:1~2:1,更优选为1.2:1,所述碘甲烷与3-甲基-1H-吡唑-4-甲酸乙酯的当量比优选为1:1~2:1,更优选1.2:1。
在本发明中,所述极性溶剂优选为N,N-二甲基甲酰胺、N,N-二甲基乙酰胺或二氯甲烷,更优选为N,N-二甲基甲酰胺,本发明对所述极性溶剂的用量没有特殊的限定,能够使原料完全混合即可。
在本发明中,所述甲基化反应的温度优选为10~40℃,更优选25℃,时间优选为8~24h,更优选为12h。
所述甲基化反应完成后,本发明优选将所得甲基化反应产物用水稀释,再依次用乙酸乙酯萃取、饱和食盐水洗、无水硫酸钠干燥和减压蒸除溶剂,得到所述中间产物II。本发明对所述水稀释、乙酸乙酯萃取、饱和食盐水洗、无水硫酸钠干燥和减压蒸除溶剂的具体操作没有特殊的限定,采用本领域技术人员熟知的方式即可。
得到中间产物II后,本发明将所述中间产物II溶解在极性溶剂中,与氯化剂混合进行氯化反应,得到中间产物III。
在本发明中,所述氯化剂优选为磺酰氯、氯化亚砜或N-氯代丁二酰亚胺,更优选为N-氯代丁二酰亚胺,所述氯化剂与中间产物II的当量比优选为1:1~20:1,更优选为1.0:1。
在本发明中,所述极性溶剂优选为乙酸或二氯甲烷,更优选乙酸,本发明对所述极性溶剂的用量没有特殊的限定,能够使所述中间产物II完全混合即可。
在本发明中,所述氯化反应的温度优选为30~120℃,更优选为70℃,时间优选为8~24h,更优选为12h。
所述氯化反应完成后,本发明优选将所得氯化反应产物用水稀释,再依次用乙酸乙酯萃取、饱和食盐水洗、无水硫酸钠干燥和减压蒸除溶剂,将所得残余物用flash柱层析分离纯化,得到所述中间产物III。本发明对所述水稀释、乙酸乙酯萃取、饱和食盐水洗、无水硫酸钠干燥和减压蒸除溶剂的具体操作没有特殊的限定,采用本领域技术人员熟知的方式即可。在本发明中,所述flash柱层析分离纯化使用的洗脱剂为乙酸乙酯/石油醚混合物,所述乙酸乙酯/石油醚混合物中乙酸乙酯与石油醚的体积比优选为1:10。
得到中间产物III后,本发明将所述中间产物III溶解在极性溶剂中,与氟化剂混合进行氟化反应,得到中间产物IV。
在本发明中,所述氟化剂优选为氟化钠、氟化钾、氟化铯或氟化锂,更优选为氟化钾,所述氟化剂与中间产物III的当量比优选为0.8:1~2:1,更优选为1.7:1。
在本发明中,所述极性溶剂优选为二甲基亚砜、环丁砜、N,N-二甲基甲酰胺或N,N-二甲基乙酰胺,更优选为环丁砜,本发明对所述极性溶剂的用量没有特殊的限定,能够使所述中间产物III和氟化剂完全混合即可。
在本发明中,所述氟化反应的温度优选为100~200℃,更优选为190℃,时间优选为6~24h,更优选为8h。
所述氟化反应完成后,本发明优选将所得氟化反应液自然降温至室温,然后加水稀释,再依次用乙酸乙酯萃取、饱和食盐水洗、无水硫酸钠干燥和减压蒸除溶剂,得到所述中间产物IV。本发明对所述加水稀释、乙酸乙酯萃取、饱和食盐水洗、无水硫酸钠干燥和减压蒸除溶剂的具体操作没有特殊的限定,采用本领域技术人员熟知的方式即可。
得到中间产物IV后,本发明将所述中间产物IV溶解在极性溶剂中,与碱性试剂混合进行水解,得到中间产物V。
在本发明中,所述碱性试剂优选为氢氧化锂、氢氧化钠或氢氧化钾,更优选为氢氧化锂,所述碱性试剂与中间产物IV的当量比优选为1:1~10:1,更优选为10:1。
在本发明中,所述极性溶剂优选包括四氢呋喃、甲醇、乙醇和水中的一种或多种,更优选为四氢呋喃和水的混合物,所述混合物中四氢呋喃和水的体积比优选为4:1,本发明对所述极性溶剂的用量没有特殊的限定,能够使所述中间产物IV和碱性试剂完全溶解即可。
在本发明中,所述水解的温度优选为0~70℃,更优选为25℃,时间优选为6~24h,更优选为12h。
所述水解完成后,本发明优选将所得水解产物的pH值调节至1,然后依次用乙酸乙酯萃取、饱和食盐水洗、无水硫酸钠干燥和减压蒸除溶剂,得到所述中间产物V。本发明对调节pH值的调节剂没有特殊的限定,本发明对所述用乙酸乙酯萃取、饱和食盐水洗、无水硫酸钠干燥和减压蒸除溶剂的具体操作没有特殊的限定,采用本领域技术人员熟知的方式即可。
得到中间产物V后,本发明将所述中间产物V与酰氯化剂混合进行酰氯化反应,得到酰氯中间产物VI。
在本发明中,所述酰氯化剂优选为氯化亚砜、草酰氯、三光气或三氯氧磷,更优选为氯化亚砜,所述酰氯化剂与中间产物V的当量比优选为1:1~5:1,更优选为2:1。
在本发明中,所述酰氯化反应优选在无或有稀释剂的存在下进行,所述稀释剂优选为氯甲苯、二氯甲烷、氯仿或四氯化碳,更优选为二氯甲烷。本发明对所述稀释剂的用量没有特殊的限定。
在本发明中,所述酰氯化反应的温度优选为20~79℃,更优选为40℃,时间优选为1~24h,更优选为2h。
所述酰氯化反应完成后,本发明优选将所得酰氯化产物自然降温至室温,然后减压蒸除溶剂,得到酰氯中间产物VI。
本发明将具有式VII所示结构的化合物(2-溴苯甲醛)溶解在稀释剂中,与甲氧甲酰基亚甲基三苯基膦混合进行Wittig反应,得到中间产物VIII。
在本发明中,所述甲氧甲酰基亚甲基三苯基膦与2-溴苯甲醛的当量比优选为0.8:1~1.5:1,更优选为1.2:1。
在本发明中,所述稀释剂优选为乙醚、四氢呋喃、甲苯、N,N-二甲基甲酰胺或二氯甲烷,更优选为二氯甲烷。
在本发明中,所述Wittig反应的温度优选为20~110℃,更优选为25℃,时间优选为6~24h,更优选为12h。
所述Wittig反应完成后,本发明优选将所得Wittig反应物用水稀释,然后依次进行乙酸乙酯萃取、饱和食盐水洗、无水硫酸钠干燥、减压蒸除溶剂,将所得残余物用flash柱层析分离纯化,得到中间产物VIII。本发明对所述水稀释、乙酸乙酯萃取、饱和食盐水洗、无水硫酸钠干燥和减压蒸除溶剂的具体操作没有特殊的限定,采用本领域技术人员熟知的方式即可。在本发明中,所述flash柱层析分离纯化使用的洗脱剂为乙酸乙酯/石油醚混合物,所述乙酸乙酯/石油醚混合物中乙酸乙酯与石油醚的体积比优选为1:10。
得到中间产物VIII后,本发明将所述中间产物VIII、钯类催化剂、膦配体、碳酸铯和氨基甲酸叔丁酯在非极性溶剂和惰性气体的存在下混合进行偶联反应,得到中间产物IX。
在本发明中,所述中间产物VIII、钯类催化剂、膦配体、碳酸铯和氨基甲酸叔丁酯的当量比优选为1:0.05:0.2:1.2:1.1~1:0.1:0.3:1.5:2.0,更优选为1:0.07:0.28:1.5:1.5。
在本发明中,所述钯类催化剂优选为三(二亚苄基丙酮)二钯、四(三苯基膦)钯、[1,1'-双(二苯基膦基)二茂铁]二氯化钯或醋酸钯,更优选为三(二亚苄基丙酮)二钯,所述膦配体优选为2-二环己基磷-2,4,6-三异丙基联苯、2-二环己膦基-2'-(N,N-二甲胺)-联苯、2-(二环己基膦)3,6-二甲氧基-2′,4′,6′-三异丙基-1,1′-联苯或2-(二叔丁基膦)联苯,更优选为2-二环己基磷-2,4,6-三异丙基联苯,所述非极性溶剂优选为乙醚、四氢呋喃、甲苯或二氧六环,更优选为甲苯。
在本发明中,所述惰性气体优选为氮气或氩气,更优选为氮气。
在本发明中,所述偶联反应的温度优选为35~120℃,更优选为110℃,时间优选为6~24h,更优选为12h。
所述偶联反应完成后,本发明优选将所得偶联反应产物用水稀释,然后依次进行乙酸乙酯萃取、饱和食盐水洗、无水硫酸钠干燥、减压蒸除溶剂,将所得残余物用flash柱层析分离纯化,得到中间产物IX。本发明对所述水稀释、乙酸乙酯萃取、饱和食盐水洗、无水硫酸钠干燥和减压蒸除溶剂的具体操作没有特殊的限定,采用本领域技术人员熟知的方式即可。在本发明中,所述flash柱层析分离纯化使用的洗脱剂为乙酸乙酯/石油醚混合物,所述乙酸乙酯/石油醚混合物中乙酸乙酯与石油醚的体积比优选为1:10。
得到中间产物IX后,本发明将所述中间产物IX、铜试剂和甲基溴化镁在非极性溶剂和惰性气体的存在下混合进行1,4-共轭加成反应,得到中间产物X。
在本发明中,所述中间产物IX、铜试剂和甲基溴化镁的当量比优选为1:0.01:1.2~1:1.5:3.0,更优选为1:1:2.5。
在本发明中,所述铜试剂优选为碘化亚铜、氯化亚铜、三氟甲烷磺酸铜或2-噻吩甲酸铜,更优选为碘化亚铜,所述非极性溶剂优选为乙醚、四氢呋喃、甲苯或二氧六环,更优选为四氢呋喃。
在本发明中,所述惰性气体优选为氮气或氩气,更优选为氮气。
在本发明中,所述1,4-共轭加成反应优选为先将所述铜试剂和甲基溴化镁混合后进行第一反应,然后再加入所述中间产物IX进行第二反应,所述第一反应的温度优选为-78~0℃,更优选为-20℃,时间优选为0.5~6h,更优选为0.5h,所述第二反应的温度优选为-78~0℃,更优选为-15℃,时间优选为2~24h,更优选为12h。
所述1,4-共轭加成反应完成后,本发明优选将所得1,4-共轭加成反应产物与饱和氯化铵的水溶液混合后,升至室温,然后依次进行乙酸乙酯萃取、饱和食盐水洗、无水硫酸钠干燥和减压蒸除溶剂,得到所述中间产物X。本发明对所述混合、升至室温、乙酸乙酯萃取、饱和食盐水洗、无水硫酸钠干燥和减压蒸除溶剂的具体方式没有特殊的限定,采用本领域技术人员熟知的方式即可。
得到中间产物X后,本发明将所述中间产物X和甲基溴化镁在非极性溶剂和惰性气体的存在下混合进行亲核加成反应,得到中间产物XI。
在本发明中,所述中间产物IX与甲基溴化镁的当量比优选为1:3.0~1:5.0,更优选为1:3.3。
在本发明中,所述非极性溶剂优选为乙醚、四氢呋喃、甲苯或二氧六环,更优选为四氢呋喃,所述惰性气体优选为氮气或氩气,更优选为氮气。
在本发明中,所述亲核加成反应的温度优选为-78~30℃,更优选为25℃,时间优选为2~24h,更优选为12h。
所述亲核加成反应完成后后,本发明优选将所得亲核加成反应产物与饱和氯化铵的水溶液混合后,然后依次进行乙酸乙酯萃取、饱和食盐水洗、无水硫酸钠干燥和减压蒸除溶剂,将所得残余物用flash柱层析分离纯化,得到中间产物XI。本发明对所述与饱和氯化铵的水溶液混合、乙酸乙酯萃取、饱和食盐水洗、无水硫酸钠干燥和减压蒸除溶剂的具体操作没有特殊的限定,采用本领域技术人员熟知的方式即可。在本发明中,所述flash柱层析分离纯化使用的洗脱剂为乙酸乙酯/石油醚混合物,所述乙酸乙酯/石油醚混合物中乙酸乙酯与石油醚的体积比优选为1:10。
得到中间产物XI后,本发明将所述中间产物XI溶解在稀释剂中,加入酸性试剂进行脱保护,得到中间产物XII。
在本发明中,所述酸性试剂优选为盐酸或三氟乙酸,更优选为三氟乙酸,所述稀释剂优选为乙醚、二氧六环或二氯甲烷,更优选为二氯甲烷,所述酸性试剂与稀释剂的体积比优选为1:1~1:20,更优选为1:10。
在本发明中,所述脱保护的温度优选为0~30℃,更优选为25℃,时间优选为1~24h,更优选为2h。
所述脱保护完成后,本发明优选将所得脱保护反应物减压蒸除溶剂,将所得残余物用flash柱层析分离纯化,得到中间产物XII。在本发明中,所述flash柱层析分离纯化使用的洗脱剂为乙酸乙酯/石油醚混合物,所述乙酸乙酯/石油醚混合物中乙酸乙酯与石油醚的体积比优选为1:1。
得到中间产物XII后,本发明将所述中间产物XII溶解在稀释剂中,与有机碱试剂和所述中间产物VI混合进行缩合反应,得到氟唑菌苯胺代谢物。
在本发明中,所述有机碱试剂优选为吡啶、三乙胺、N-甲基吗啉或N,N-二异丙基乙胺,更优选为吡啶,所述有机碱试剂与中间产物XII的当量比优选为2.0:1~10.0:1,更优选为6.5:1,所述稀释剂优选为乙醚、二氧六环、四氢呋喃、N,N-二甲基甲酰胺或二氯甲烷,更优选为二氯甲烷,本发明对所述稀释剂的用量没有特殊的限定。
在本发明中,所述中间产物VI与中间产物XII的当量比优选为0.8:1~1.5:1,更优选为0.9:1。
在本发明中,所述缩合反应温度优选为0~30℃,更优选为25℃,时间优选为3~24h,更优选为12h。
所述缩合反应完成后,本发明优选将所得缩合反应产物用水稀释,然后依次进行乙酸乙酯萃取、饱和食盐水洗、无水硫酸钠干燥和减压蒸除溶剂,将所得残余物用flash柱层析分离纯化,得到氟唑菌苯胺代谢物(具有式XIII所示结构的化合物,5-氟-N-(2-(4-羟基-4-甲基戊烷-2-基)苯基)-1,3-二甲基-1H-吡唑-4-甲酰胺)。在本发明中,所述flash柱层析分离纯化使用的洗脱剂为乙酸乙酯/石油醚混合物,所述乙酸乙酯/石油醚混合物中乙酸乙酯与石油醚的体积比优选为1:1。
为了进一步说明本发明,下面结合实例对本发明提供的氟唑菌苯胺代谢物的制备方法进行详细地描述,但不能将它们理解为对本发明保护范围的限定。
实施例1
一种氟唑菌苯胺代谢物的合成方法,包括以下步骤:
(1)将3-甲基-1H-吡唑-4-甲酸乙酯(式I)(1.00g,6.16mmol)溶于N,N-二甲基甲酰胺(10mL)后,加入碳酸钾(1.03g,7.39mmol)和碘甲烷(1.05g,7.39mmol),室温反应12小时。反应完毕后加水(20mL)稀释,乙酸乙酯(20mL×3)萃取,饱和食盐水洗,无水硫酸钠干燥后,减压蒸除溶剂,得到白色固体II(1,3-二甲基-1H-吡唑-4-甲酸乙酯,0.99g,收率96%),白色固体II无须进一步纯化直接投入下一步反应。
(2)将白色固体II(0.99g,5.91mmol)溶于乙酸(20mL)后,加入N-氯代丁二酰亚胺(0.79g,5.91mmol),加热至70℃反应12小时。反应完毕后加水(20mL)稀释,乙酸乙酯(20mL×3)萃取,饱和食盐水洗,无水硫酸钠干燥后,减压蒸除溶剂,残余物用flash柱层析分离纯化(乙酸乙酯/石油醚体积比1:10),得到白色固体III(5-氯-1,3-二甲基-1H-吡唑-4-甲酸乙酯,1.12g,收率94%)。
(3)将白色固体III(1.12g,5.55mmol)溶于环丁砜(20mL)后,加入氟化钾(0.53g,9.28mmol),加热至190℃反应8小时。反应完毕后将反应液降温至室温,加水(30mL)稀释,乙酸乙酯(20mL×3)萃取,饱和食盐水洗,无水硫酸钠干燥后,减压蒸除溶剂,得到棕色固体IV(5-氟-1,3-二甲基-1H-吡唑-4-甲酸乙酯,0.35g,收率34%),棕色固体IV无须进一步纯化直接投入下一步反应。
(4)将棕色固体IV(0.35g,1.89mmol)溶于四氢呋喃(20mL)后,依次加入水(5mL),氢氧化锂一水合物(0.79g,18.9mmol),室温反应12小时。反应完毕后调节pH值至1,乙酸乙酯(10mL×3)萃取,饱和食盐水洗,无水硫酸钠干燥后,减压蒸除溶剂得到白色固体V(5-氟-1,3-二甲基-1H-吡唑-4-甲酸,0.28g,收率94%),白色固体V无须进一步纯化直接投入下一步反应。
(5)将白色固体V(0.28g,1.77mmol)溶于二氯甲烷(10mL)后,加入二氯亚砜(0.44g,3.54mmol),加热回流反应2小时。反应完毕后将反应液降温至室温,减压蒸除溶剂,得到黄色固体VI(5-氟-1,3-二甲基-1H-吡唑-4-甲酰氯,0.30g,收率95%),黄色固体VI无须进一步纯化直接投入下一步反应。
(6)将2-溴苯甲醛(式VII,1.00g,5.40mmol)溶于二氯甲烷(20mL)后,加入甲氧甲酰基亚甲基三苯基膦(2.18g,6.37mmol),室温反应12小时。反应完毕后加水(20mL)稀释,乙酸乙酯(20mL×3)萃取,饱和食盐水洗,无水硫酸钠干燥后,减压蒸除溶剂,残余物用flash柱层析分离纯化(乙酸乙酯/石油醚体积比1:10),得到橙色固体VIII((E)-3-(2-溴苯基)丙烯酸甲酯,1.18g,收率91%)。
(7)氮气保护下,250mL三口反应瓶依次加入橙色固体VIII(1.18g,4.91mmol)、氨基甲酸叔丁酯(0.86g,7.37mmol)、三(二亚苄基丙酮)二钯(0.31g,0.34mmol)、2-二环己基磷-2,4,6-三异丙基联苯(0.66g,1.37mmol)、碳酸铯(2.40g,7.37mmol)和甲苯(30mL)后,加热回流反应12小时。反应完毕后加水(20mL)稀释,乙酸乙酯(20mL×3)萃取,饱和食盐水洗,无水硫酸钠干燥后,减压蒸除溶剂,残余物用flash柱层析分离纯化(乙酸乙酯/石油醚体积比1:10),得到白色固体IX((E)-3-(2-(叔丁氧羰基氨基)苯基)丙烯酸甲酯,1.06g,收率78%)。
(8)氮气保护下100mLSchlenk瓶加入碘化亚铜(0.07g,3.83mmol)和四氢呋喃(10mL),降温至-20℃后加入甲基溴化镁的四氢呋喃溶液(3.20mL,9.58mmol,3M),保持温度反应0.5小时。然后加入白色固体IX(1.06g,3.83mmol),并升温至-15℃反应12小时。反应完毕后加饱和氯化铵的水溶液(10mL),将反应缓慢升至室温。然后乙酸乙酯(10mL×3)萃取,饱和食盐水洗,无水硫酸钠干燥后,减压蒸除溶剂得到淡黄色固体X(3-(2-(叔丁氧羰基氨基)苯基)丁酸甲酯,0.78g,收率69%),淡黄色固体X无须进一步纯化直接投入下一步反应。
(9)氮气保护下100mLSchlenk瓶加入淡黄色固体X(0.78g,2.64mmol)和四氢呋喃(10mL),降温至-78℃后加入甲基溴化镁的四氢呋喃溶液(2.91mL,8.72mmol,3M)。然后将反应液升温至室温反应12小时。反应完毕后加饱和氯化铵的水溶液(10mL),然后乙酸乙酯(10mL×3)萃取,饱和食盐水洗,无水硫酸钠干燥后,减压蒸除溶剂,残余物用flash柱层析分离纯化(乙酸乙酯/石油醚体积比1:10),得到白色固体XI(4-(2-(叔丁氧羰基氨基)苯基)-2-甲基戊烷-2-醇,0.65g,收率83%)。
(10)将白色固体XI(0.65g,2.19mmol)溶于二氯甲烷(10mL)后,加入三氟乙酸(1mL),室温反应2小时。反应完毕后减压蒸除溶剂,残余物用flash柱层析分离纯化(乙酸乙酯/石油醚体积比1:1),得到黄色油状液体XII(4-(2-氨基苯基)-2-甲基戊烷-2-醇,0.37g,收率87%)。
(11)将黄色油状液体XII(0.37g,1.91mmol)和吡啶(1mL)溶于二氯甲烷(10mL)后,加入黄色固体VI(0.30g,1.68mmol),室温反应12小时。反应完毕后加水(20mL)稀释,乙酸乙酯(20mL×3)萃取,饱和食盐水洗,无水硫酸钠干燥后,减压蒸除溶剂,残余物用flash柱层析分离纯化(乙酸乙酯/石油醚体积比1:1),得到白色固体XIII(氟唑菌苯胺代谢物,5-氟-N-(2-(4-羟基-4-甲基戊烷-2-基)苯基)-1,3-二甲基-1H-吡唑-4-甲酰胺,0.22g,收率39%)。
对实施例1制得的白色固体XIII进行液相色谱检测,色谱条件如下:
色谱柱:Shim-pack GIST C18(250×4.6mm×5μm);
流动相:0.05%三氟乙酸甲醇(A)和0.05%三氟乙酸水溶液(B);
流速:1.0mL/min;
进样量:5μL;
柱温:30℃;
紫外检测波长:254nm
梯度洗脱条件如表1所示:
表1梯度洗脱条件
得到的色谱图如图2所示,出峰时间和峰面积如表2所示,可知,白色固体XIII的纯度为99.7%。
表2色谱图的出峰时间和峰面积
实施例2
一种氟唑菌苯胺代谢物的合成方法,包括以下步骤:
(1)将3-甲基-1H-吡唑-4-甲酸乙酯(式I)(1.00g,6.16mmol)溶于N,N-二甲基甲酰胺(10mL)后,加入碳酸钾(0.86g,6.16mmol)和碘甲烷(0.88g,6.16mmol),室温反应12小时。反应完毕后加水(20mL)稀释,乙酸乙酯(20mL×3)萃取,饱和食盐水洗,无水硫酸钠干燥后,减压蒸除溶剂,得到白色固体II(1,3-二甲基-1H-吡唑-4-甲酸乙酯,0.75g,收率73%),白色固体II无须进一步纯化直接投入下一步反应。
(2)将白色固体II(0.99g,5.91mmol)溶于乙酸(20mL)后,加入N-氯代丁二酰亚胺(1.58g,11.82mmol),25℃反应24小时。反应完毕后加水(20mL)稀释,乙酸乙酯(20mL×3)萃取,饱和食盐水洗,无水硫酸钠干燥后,减压蒸除溶剂,残余物用flash柱层析分离纯化(乙酸乙酯/石油醚体积比1:10),得到白色固体III(5-氯-1,3-二甲基-1H-吡唑-4-甲酸乙酯,0.75g,收率63%)。
(3)将白色固体III(1.12g,5.55mmol)溶于环丁砜(20mL)后,加入氟化钾(0.25g,4.36mmol),加热至150℃反应24小时。反应完毕后将反应液降温至室温,加水(30mL)稀释,乙酸乙酯(20mL×3)萃取,饱和食盐水洗,无水硫酸钠干燥后,减压蒸除溶剂,得到棕色固体IV(5-氟-1,3-二甲基-1H-吡唑-4-甲酸乙酯,0.14g,收率14%),IV无须进一步纯化直接投入下一步反应。
(4)将棕色固体IV(0.35g,1.89mmol)溶于四氢呋喃(20mL)后,依次加入水(5mL),氢氧化锂一水合物(0.79g,18.9mmol),40℃反应6小时。反应完毕后调节pH值至1,乙酸乙酯(10mL×3)萃取,饱和食盐水洗,无水硫酸钠干燥后,减压蒸除溶剂得到白色固体V(5-氟-1,3-二甲基-1H-吡唑-4-甲酸,0.24g,收率82%),白色固体V无须进一步纯化直接投入下一步反应。
(5)将白色固体V(0.28g,1.77mmol)溶于氯仿(10mL)后,加入二氯亚砜(0.22g,1.77mmol),20℃反应24小时。反应完毕后,减压蒸除溶剂,得到黄色固体VI(5-氟-1,3-二甲基-1H-吡唑-4-甲酰氯,0.20g,收率64%),黄色固体VI无须进一步纯化直接投入下一步反应。
(6)将2-溴苯甲醛(式VII,1.00g,5.40mmol)溶于乙醚(20mL)后,加入甲氧甲酰基亚甲基三苯基膦(1.75g,4.32mmol),室温反应12小时。反应完毕后加水(20mL)稀释,乙酸乙酯(20mL×3)萃取,饱和食盐水洗,无水硫酸钠干燥后,减压蒸除溶剂,残余物用flash柱层析分离纯化(乙酸乙酯/石油醚体积比1:10),得到橙色固体VIII((E)-3-(2-溴苯基)丙烯酸甲酯,0.80g,收率62%)。
(7)氮气保护下,250mL三口反应瓶依次加入橙色固体VIII(1.18g,4.91mmol)、氨基甲酸叔丁酯(0.86g,7.37mmol)、三(二亚苄基丙酮)二钯(0.31g,0.34mmol)、2-二环己膦基-2'-(N,N-二甲胺)-联苯(0.54g,1.37mmol)、碳酸铯(1.92g,5.90mmol)和四氢呋喃(30mL)后,加热回流反应12小时。反应完毕后加水(20mL)稀释,乙酸乙酯(20mL×3)萃取,饱和食盐水洗,无水硫酸钠干燥后,减压蒸除溶剂,残余物用flash柱层析分离纯化(乙酸乙酯/石油醚体积比1:10),得到白色固体IX((E)-3-(2-(叔丁氧羰基氨基)苯基)丙烯酸甲酯,0.87g,收率64%)。
(8)氮气保护下100mLSchlenk瓶加入碘化亚铜(0.01g,0.38mmol)和四氢呋喃(10mL),降温至-20℃后加入甲基溴化镁的四氢呋喃溶液(2.55mL,7.66mmol,3M),保持温度反应6小时。然后加入白色固体IX(1.06g,3.83mmol),并升温至-15℃反应24小时。反应完毕后加饱和氯化铵的水溶液(10mL),将反应缓慢升至室温。然后乙酸乙酯(10mL×3)萃取,饱和食盐水洗,无水硫酸钠干燥后,减压蒸除溶剂得到淡黄色固体X(3-(2-(叔丁氧羰基氨基)苯基)丁酸甲酯,0.28g,收率25%),淡黄色固体X无须进一步纯化直接投入下一步反应。
(9)氮气保护下100mLSchlenk瓶加入淡黄色固体X(0.78g,2.64mmol)和四氢呋喃(10mL),降温至-78℃后加入甲基溴化镁的四氢呋喃溶液(4.40mL,13.20mmol,3M)。然后将反应液升温至室温反应24小时。反应完毕后加饱和氯化铵的水溶液(10mL),然后乙酸乙酯(10mL×3)萃取,饱和食盐水洗,无水硫酸钠干燥后,减压蒸除溶剂,残余物用flash柱层析分离纯化(乙酸乙酯/石油醚体积比1:10),得到白色固体XI(4-(2-(叔丁氧羰基氨基)苯基)-2-甲基戊烷-2-醇,0.63g,收率81%)。
(10)将白色固体XI(0.65g,2.19mmol)溶于乙醚(10mL)后,加入盐酸(1mL),室温反应12小时。反应完毕后减压蒸除溶剂,残余物用flash柱层析分离纯化(乙酸乙酯/石油醚体积比1:1),得到黄色油状液体XII(4-(2-氨基苯基)-2-甲基戊烷-2-醇,0.28g,收率67%)。
(11)将黄色油状液体XII(0.37g,1.91mmol)和三乙胺(1mL)溶于乙醚(10mL)后,加入黄色固体VI(0.34g,1.91mmol),室温反应24小时。反应完毕后加水(20mL)稀释,乙酸乙酯(20mL×3)萃取,饱和食盐水洗,无水硫酸钠干燥后,减压蒸除溶剂,残余物用flash柱层析分离纯化(乙酸乙酯/石油醚体积比1:1),得到白色固体XIII(氟唑菌苯胺代谢物,5-氟-N-(2-(4-羟基-4-甲基戊烷-2-基)苯基)-1,3-二甲基-1H-吡唑-4-甲酰胺,0.21g,收率38%)。
对实施例2制得的白色固体XIII进行
实施例3
一种氟唑菌苯胺代谢物的合成方法,包括以下步骤:
(1)将3-甲基-1H-吡唑-4-甲酸乙酯(式I)(1.00g,6.16mmol)溶于N,N-二甲基甲酰胺(10mL)后,加入碳酸钠(0.79g,7.39mmol)和碘甲烷(1.05g,7.39mmol),室温反应12小时。反应完毕后加水(20mL)稀释,乙酸乙酯(20mL×3)萃取,饱和食盐水洗,无水硫酸钠干燥后,减压蒸除溶剂,得到白色固体II(1,3-二甲基-1H-吡唑-4-甲酸乙酯,0.78g,收率76%),白色固体II无须进一步纯化直接投入下一步反应。
(2)将白色固体II(0.99g,5.91mmol)溶于二氯甲烷(20mL)后,加入磺酰氯(8.00g,59.10mmol),40℃反应8小时。反应完毕后加水(20mL)稀释,乙酸乙酯(20mL×3)萃取,饱和食盐水洗,无水硫酸钠干燥后,减压蒸除溶剂,残余物用flash柱层析分离纯化(乙酸乙酯/石油醚体积比1:10),得到白色固体III(5-氯-1,3-二甲基-1H-吡唑-4-甲酸乙酯,0.54g,收率45%)。
(3)将白色固体III(1.12g,5.55mmol)溶于环丁砜(20mL)后,加入氟化钠(0.38g,9.28mmol),加热至190℃反应8小时。反应完毕后将反应液降温至室温,加水(30mL)稀释,乙酸乙酯(20mL×3)萃取,饱和食盐水洗,无水硫酸钠干燥后,减压蒸除溶剂,得到棕色固体IV(5-氟-1,3-二甲基-1H-吡唑-4-甲酸乙酯,0.28g,收率28%),棕色固体IV无须进一步纯化直接投入下一步反应。
(4)将棕色固体IV(0.35g,1.89mmol)溶于四氢呋喃(20mL)后,依次加入水(5mL),氢氧化钠(0.75g,18.9mmol),室温反应5小时。反应完毕后调节pH值至1,乙酸乙酯(10mL×3)萃取,饱和食盐水洗,无水硫酸钠干燥后,减压蒸除溶剂得到白色固体V(5-氟-1,3-二甲基-1H-吡唑-4-甲酸,0.19g,收率65%),白色固体V无须进一步纯化直接投入下一步反应。
(5)将白色固体V(0.28g,1.77mmol)溶于二氯甲烷(10mL)后,加入草酰氯(0.22g,1.77mmol),25℃反应2小时。反应完毕后,减压蒸除溶剂,得到黄色固体VI(5-氟-1,3-二甲基-1H-吡唑-4-甲酰氯,0.28g,收率90%),黄色固体VI无须进一步纯化直接投入下一步反应。
(6)将2-溴苯甲醛(VII,1.00g,5.40mmol)溶于四氢呋喃(20mL)后,加入甲氧甲酰基亚甲基三苯基膦(1.85g,5.40mmol),室温反应24小时。反应完毕后加水(20mL)稀释,乙酸乙酯(20mL×3)萃取,饱和食盐水洗,无水硫酸钠干燥后,减压蒸除溶剂,残余物用flash柱层析分离纯化(乙酸乙酯/石油醚体积比1:10),得到橙色固体VIII((E)-3-(2-溴苯基)丙烯酸甲酯,1.06g,收率82%)。
(7)氮气保护下,250mL三口反应瓶依次加入橙色固体VIII(1.18g,4.91mmol)、氨基甲酸叔丁酯(0.86g,7.37mmol)、四(三苯基膦)钯(0.39g,0.34mmol)、2-(二环己基膦)-3,6-二甲氧基-2′,4′,6′-三异丙基-1,1′-联苯(0.74g,1.37mmol)、碳酸铯(2.40g,7.37mmol)和四氢呋喃(30mL)后,加热回流反应24小时。反应完毕后加水(20mL)稀释,乙酸乙酯(20mL×3)萃取,饱和食盐水洗,无水硫酸钠干燥后,减压蒸除溶剂,残余物用flash柱层析分离纯化(乙酸乙酯/石油醚体积比1:10),得到白色固体IX((E)-3-(2-(叔丁氧羰基氨基)苯基)丙烯酸甲酯,0.65g,收率48%)。
(8)氮气保护下100mLSchlenk瓶加入氯化亚铜(0.04g,3.83mmol)和乙醚(10mL),降温至-78℃后加入甲基溴化镁的四氢呋喃溶液(3.20mL,9.58mmol,3M),保持温度反应6小时。然后加入白色固体IX(1.06g,3.83mmol),并升温至-20℃反应24小时。反应完毕后加饱和氯化铵的水溶液(10mL),将反应缓慢升至室温。然后乙酸乙酯(10mL×3)萃取,饱和食盐水洗,无水硫酸钠干燥后,减压蒸除溶剂得到淡黄色固体X(3-(2-(叔丁氧羰基氨基)苯基)丁酸甲酯,0.34g,收率30%),淡黄色固体X无须进一步纯化直接投入下一步反应。
(9)氮气保护下100mLSchlenk瓶加入淡黄色固体X(0.78g,2.64mmol)和乙醚(10mL),降温至-50℃后加入甲基溴化镁的四氢呋喃溶液(2.91mL,8.72mmol,3M)。然后将反应液升温至室温反应12小时。反应完毕后加饱和氯化铵的水溶液(10mL),然后乙酸乙酯(10mL×3)萃取,饱和食盐水洗,无水硫酸钠干燥后,减压蒸除溶剂,残余物用flash柱层析分离纯化(乙酸乙酯/石油醚体积比1:10),得到白色固体XI(4-(2-(叔丁氧羰基氨基)苯基)-2-甲基戊烷-2-醇,0.58g,收率74%)。
(10)将白色固体XI(0.65g,2.19mmol)溶于二氧六环(10mL)后,加入盐酸(1mL),室温反应24小时。反应完毕后减压蒸除溶剂,残余物用flash柱层析分离纯化(乙酸乙酯/石油醚体积比1:1),得到黄色油状液体XII(4-(2-氨基苯基)-2-甲基戊烷-2-醇,0.29g,收率68%)。
(11)将黄色油状液体XII(0.37g,1.91mmol)和三乙胺(1mL)溶于二氧六环(10mL)后,加入黄色固体VI(0.30g,1.68mmol),0℃反应24小时。反应完毕后加水(20mL)稀释,乙酸乙酯(20mL×3)萃取,饱和食盐水洗,无水硫酸钠干燥后,减压蒸除溶剂,残余物用flash柱层析分离纯化(乙酸乙酯/石油醚体积比1:1),得到白色固体XIII(氟唑菌苯胺代谢物,5-氟-N-(2-(4-羟基-4-甲基戊烷-2-基)苯基)-1,3-二甲基-1H-吡唑-4-甲酰胺,0.06g,收率11%)。
对实施例3制得的白色固体XIII进行
实施例4
一种氟唑菌苯胺代谢物的合成方法,包括以下步骤:
(1)将3-甲基-1H-吡唑-4-甲酸乙酯(式I)(1.00g,6.16mmol)溶于N,N-二甲基乙酰胺(10mL)后,加入氢氧化钾(0.42g,7.39mmol)和碘甲烷(1.05g,7.39mmol),40℃反应8小时。反应完毕后加水(20mL)稀释,乙酸乙酯(20mL×3)萃取,饱和食盐水洗,无水硫酸钠干燥后,减压蒸除溶剂,得到白色固体II(1,3-二甲基-1H-吡唑-4-甲酸乙酯,0.64g,收率62%),白色固体II无须进一步纯化直接投入下一步反应。
(2)将白色固体II(0.99g,5.91mmol)溶于二氯甲烷(20mL)后,加入氯化亚砜(6.90g,59.10mmol),加热至40℃反应8小时。反应完毕后加水(20mL)稀释,乙酸乙酯(20mL×3)萃取,饱和食盐水洗,无水硫酸钠干燥后,减压蒸除溶剂,残余物用flash柱层析分离纯化(乙酸乙酯/石油醚体积比1:10),得到白色固体III(5-氯-1,3-二甲基-1H-吡唑-4-甲酸乙酯,0.69g,收率58%)。
(3)将白色固体III(1.12g,5.55mmol)溶于环丁砜(20mL)后,加入氟化钾(1.38g,9.28mmol),加热至190℃反应8小时。反应完毕后将反应液降温至室温,加水(30mL)稀释,乙酸乙酯(20mL×3)萃取,饱和食盐水洗,无水硫酸钠干燥后,减压蒸除溶剂,得到棕色固体IV(5-氟-1,3-二甲基-1H-吡唑-4-甲酸乙酯,0.22g,收率21%),棕色固体IV无须进一步纯化直接投入下一步反应。
(4)将棕色固体IV(0.35g,1.89mmol)溶于甲醇(20mL)后,依次加入水(5mL),氢氧化钠(0.75g,18.9mmol),室温反应5小时。反应完毕后调节pH值至1,乙酸乙酯(10mL×3)萃取,饱和食盐水洗,无水硫酸钠干燥后,减压蒸除溶剂得到白色固体V(5-氟-1,3-二甲基-1H-吡唑-4-甲酸,0.13g,收率43%),白色固体V无须进一步纯化直接投入下一步反应。
(5)将白色固体V(0.28g,1.77mmol)溶于四氯化碳(10mL)后,加入三光气(4.41g,1.77mmol),50℃反应8小时。反应完毕后将反应液降温至室温,减压蒸除溶剂,得到黄色固体VI(5-氟-1,3-二甲基-1H-吡唑-4-甲酰氯,0.23g,收率74%),黄色固体VI无须进一步纯化直接投入下一步反应。
(6)将2-溴苯甲醛(VII,1.00g,5.40mmol)溶于N,N-二甲基甲酰胺(20mL)后,加入甲氧甲酰基亚甲基三苯基膦(1.85g,5.40mmol),80℃反应6小时。反应完毕后加水(20mL)稀释,乙酸乙酯(20mL×3)萃取,饱和食盐水洗,无水硫酸钠干燥后,减压蒸除溶剂,残余物用flash柱层析分离纯化(乙酸乙酯/石油醚体积比1:10),得到橙色固体VIII((E)-3-(2-溴苯基)丙烯酸甲酯,1.00g,收率77%)。
(7)氮气保护下,250mL三口反应瓶依次加入橙色固体VIII(1.18g,4.91mmol)、氨基甲酸叔丁酯(0.86g,7.37mmol)、[1,1'-双(二苯基膦基)二茂铁]二氯化钯(0.25g,0.34mmol)、2-(二环己基膦)-3,6-二甲氧基-2′,4′,6′-三异丙基-1,1′-联苯(0.74g,1.37mmol)、碳酸铯(2.40g,7.37mmol)和乙醚(30mL)后,加热回流反应6小时。反应完毕后加水(20mL)稀释,乙酸乙酯(20mL×3)萃取,饱和食盐水洗,无水硫酸钠干燥后,减压蒸除溶剂,残余物用flash柱层析分离纯化(乙酸乙酯/石油醚体积比1:10),得到白色固体IX((E)-3-(2-(叔丁氧羰基氨基)苯基)丙烯酸甲酯,0.60g,收率44%)。
(8)氮气保护下100mLSchlenk瓶加入三氟甲烷磺酸铜(1.90g,3.83mmol)和甲苯(10mL),降温至-78℃后加入甲基溴化镁的四氢呋喃溶液(3.20mL,9.58mmol,3M),保持温度反应2小时。然后加入IX(1.06g,3.83mmol),并升温至-0℃反应24小时。反应完毕后加饱和氯化铵的水溶液(10mL),将反应缓慢升至室温。然后乙酸乙酯(10mL×3)萃取,饱和食盐水洗,无水硫酸钠干燥后,减压蒸除溶剂得到淡黄色固体X(3-(2-(叔丁氧羰基氨基)苯基)丁酸甲酯,0.14g,收率12%),X无须进一步纯化直接投入下一步反应。
(9)氮气保护下100mLSchlenk瓶加入淡黄色固体X(0.78g,2.64mmol)和甲苯(10mL),降温至-30℃后加入甲基溴化镁的四氢呋喃溶液(2.91mL,8.72mmol,3M)。然后将反应液升温至室温反应4小时。反应完毕后加饱和氯化铵的水溶液(10mL),然后乙酸乙酯(10mL×3)萃取,饱和食盐水洗,无水硫酸钠干燥后,减压蒸除溶剂,残余物用flash柱层析分离纯化(乙酸乙酯/石油醚体积比1:10),得到白色固体XI(4-(2-(叔丁氧羰基氨基)苯基)-2-甲基戊烷-2-醇,0.52g,收率66%)。
(10)将白色固体XI(0.65g,2.19mmol)溶于二氯甲烷(10mL)后,加入三氟乙酸(1mL),0℃反应24小时。反应完毕后减压蒸除溶剂,残余物用flash柱层析分离纯化(乙酸乙酯/石油醚体积比1:1),得到黄色油状液体XII(4-(2-氨基苯基)-2-甲基戊烷-2-醇,0.36g,收率85%)。
(11)将黄色油状液体XII(0.37g,1.91mmol)和N-甲基吗啉(1mL)溶于四氢呋喃(10mL)后,加入黄色固体VI(0.34g,1.91mmol),室温反应24小时。反应完毕后加水(20mL)稀释,乙酸乙酯(20mL×3)萃取,饱和食盐水洗,无水硫酸钠干燥后,减压蒸除溶剂,残余物用flash柱层析分离纯化(乙酸乙酯/石油醚体积比1:1),得到白色固体XIII(氟唑菌苯胺代谢物,5-氟-N-(2-(4-羟基-4-甲基戊烷-2-基)苯基)-1,3-二甲基-1H-吡唑-4-甲酰胺,0.19g,收率33%)。
对实施例4制得的白色固体XIII进行
实施例5
一种氟唑菌苯胺代谢物的合成方法,包括以下步骤:
(1)将3-甲基-1H-吡唑-4-甲酸乙酯(式I)(1.00g,6.16mmol)溶于二氯甲烷(10mL)后,加入氢氧化钠(0.30g,7.39mmol)和碘甲烷(1.05g,7.39mmol),室温反应8小时。反应完毕后加水(20mL)稀释,乙酸乙酯(20mL×3)萃取,饱和食盐水洗,无水硫酸钠干燥后,减压蒸除溶剂,得到白色固体II(1,3-二甲基-1H-吡唑-4-甲酸乙酯,0.62g,收率60%),白色固体II无须进一步纯化直接投入下一步反应。
(2)将白色固体II(0.99g,5.91mmol)溶于二氯甲烷(20mL)后,加入氯化亚砜(15.80g,118.20mmol),30℃反应24小时。反应完毕后加水(20mL)稀释,乙酸乙酯(20mL×3)萃取,饱和食盐水洗,无水硫酸钠干燥后,减压蒸除溶剂,残余物用flash柱层析分离纯化(乙酸乙酯/石油醚体积比1:10),得到白色固体III(5-氯-1,3-二甲基-1H-吡唑-4-甲酸乙酯,0.39g,收率33%)。
(3)将白色固体III(1.12g,5.55mmol)溶于环丁砜(20mL)后,加入氟化锂(0.24g,9.28mmol),加热至190℃反应8小时。反应完毕后将反应液降温至室温,加水(30mL)稀释,乙酸乙酯(20mL×3)萃取,饱和食盐水洗,无水硫酸钠干燥后,减压蒸除溶剂,得到棕色固体IV(5-氟-1,3-二甲基-1H-吡唑-4-甲酸乙酯,0.20g,收率19%),棕色固体IV无须进一步纯化直接投入下一步反应。
(4)将棕色固体IV(0.35g,1.89mmol)溶于乙醇(20mL)后,依次加入水(5mL),氢氧化钾(1.05g,18.9mmol),室温反应5小时。反应完毕后调节PH至1,乙酸乙酯(10mL×3)萃取,饱和食盐水洗,无水硫酸钠干燥后,减压蒸除溶剂得到白色固体V(5-氟-1,3-二甲基-1H-吡唑-4-甲酸,0.07g,收率24%),白色固体V无须进一步纯化直接投入下一步反应。
(5)将白色固体V(0.28g,1.77mmol)溶于氯甲苯(10mL)后,加入三氯氧磷(0.57g,3.54mmol),60℃反应4小时。反应完毕后将反应液降温至室温,减压蒸除溶剂,得到黄色固体VI(5-氟-1,3-二甲基-1H-吡唑-4-甲酰氯,0.18g,收率56%),黄色固体VI无须进一步纯化直接投入下一步反应。
(6)将2-溴苯甲醛(式VII,1.00g,5.40mmol)溶于甲苯(20mL)后,加入甲氧甲酰基亚甲基三苯基膦(2.18g,6.37mmol),110℃反应12小时。反应完毕后加水(20mL)稀释,乙酸乙酯(20mL×3)萃取,饱和食盐水洗,无水硫酸钠干燥后,减压蒸除溶剂,残余物用flash柱层析分离纯化(乙酸乙酯/石油醚体积比1:10),得到橙色固体VIII((E)-3-(2-溴苯基)丙烯酸甲酯,1.17g,收率90%)。
(7)氮气保护下,250mL三口反应瓶依次加入橙色固体VIII(1.18g,4.91mmol)、氨基甲酸叔丁酯(0.86g,7.37mmol)、醋酸钯(0.08g,0.34mmol)、2-(二叔丁基膦)联苯(0.41g,1.37mmol)、碳酸铯(2.40g,7.37mmol)和二氧六环(30mL)后,加热回流反应24小时。反应完毕后加水(20mL)稀释,乙酸乙酯(20mL×3)萃取,饱和食盐水洗,无水硫酸钠干燥后,减压蒸除溶剂,残余物用flash柱层析分离纯化(乙酸乙酯/石油醚体积比1:10),得到白色固体IX((E)-3-(2-(叔丁氧羰基氨基)苯基)丙烯酸甲酯,0.71g,收率52%)。
(8)氮气保护下100mLSchlenk瓶加入2-噻吩甲酸铜(0.07g,3.83mmol)和二氧六环(10mL),降温至-78℃后加入甲基溴化镁的四氢呋喃溶液(3.20mL,9.58mmol,3M),保持温度反应4小时。然后加入白色固体IX(1.06g,3.83mmol),并升温至-0℃反应24小时。反应完毕后加饱和氯化铵的水溶液(10mL),将反应缓慢升至室温。然后乙酸乙酯(10mL×3)萃取,饱和食盐水洗,无水硫酸钠干燥后,减压蒸除溶剂得到淡黄色固体X(3-(2-(叔丁氧羰基氨基)苯基)丁酸甲酯,0.25g,收率22%),淡黄色固体X无须进一步纯化直接投入下一步反应。
(9)氮气保护下100mLSchlenk瓶加入淡黄色固体X(0.78g,2.64mmol)和二氧六环(10mL),降温至-30℃后加入甲基溴化镁的四氢呋喃溶液(2.91mL,8.72mmol,3M)。然后将反应液升温至室温反应2小时。反应完毕后加饱和氯化铵的水溶液(10mL),然后乙酸乙酯(10mL×3)萃取,饱和食盐水洗,无水硫酸钠干燥后,减压蒸除溶剂,残余物用flash柱层析分离纯化(乙酸乙酯/石油醚体积比1:10),得到白色固体XI(4-(2-(叔丁氧羰基氨基)苯基)-2-甲基戊烷-2-醇,0.26g,收率33%)。
(10)将白色固体XI(0.65g,2.19mmol)溶于二氯甲烷(10mL)后,加入三氟乙酸(1mL),30℃反应4小时。反应完毕后减压蒸除溶剂,残余物用flash柱层析分离纯化(乙酸乙酯/石油醚体积比1:1),得到黄色油状液体XII(4-(2-氨基苯基)-2-甲基戊烷-2-醇,0.24g,收率57%)。
(11)将黄色油状液体XII(0.37g,1.91mmol)和N,N-二异丙基乙胺(1mL)溶于N,N-二甲基甲酰胺(10mL)后,加入黄色固体VI(0.51g,2.87mmol),室温反应24小时。反应完毕后加水(20mL)稀释,乙酸乙酯(20mL×3)萃取,饱和食盐水洗,无水硫酸钠干燥后,减压蒸除溶剂,残余物用flash柱层析分离纯化(乙酸乙酯/石油醚体积比1:1),得到白色固体XIII(氟唑菌苯胺代谢物,5-氟-N-(2-(4-羟基-4-甲基戊烷-2-基)苯基)-1,3-二甲基-1H-吡唑-4-甲酰胺,0.21g,收率38%)。
对实施例5制得的白色固体XIII进行
以上所述仅是本发明的优选实施方式,并非对本发明作任何形式上的限制。应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。