一种甘草次酸衍生物及其制备方法与应用
文献发布时间:2023-06-19 09:27:35
技术领域
本发明属于有机合成领域以及医药领域,具体涉及一种甘草次酸衍生物及其制备方法与应用。
背景技术
水凝胶是一类由3D聚合物网络和大量水组成的软物质材料,与生物体软组织在结构上有诸多相似之处。水凝胶是一种新型的理想医用敷料,其在外敷时可以锁住水分,维持创面的湿润环境,有利于伤口的愈合,且伤口不易成痂,减少疤痕并保持皮肤的美观性;作为内创性手术填充料时与创面贴合良好。因此,水凝胶可广泛用于生物医学工程领域,如内创性手术填充料、创伤敷料、药物输送等。
目前水凝胶形成的主要方式为化学交联和物理交联,这两种方式形成的水凝胶需要往体系中引入蛋白质、金属离子、小分子表面活性剂等桥连剂,以通过桥连作用来诱导形成水凝胶。但这两种方式形成的水凝胶大部分纯度不高,不能被人体完全吸收,影响人体的正常代谢。且传统敷料更换时容易造成二次损伤的问题,给病人带来痛苦。目前水凝胶敷料抗菌效果一般来自加入的抗菌药物或者是引入金属离子抗菌,而水凝胶往往只是起到提供湿润环境的效果,功能和效果单一,早已经不能满足临床医学的使用要求,且生物敷料的原材料来源有限,制备过程较为繁杂,不适于大规模推广。另外,目前大部分水凝胶敷料的制备难免会使用到一些有毒有害的化学品,且生产制备过程会产生有毒有害物质,水凝胶敷料使用之后若不及时回收会污染环境和散播病毒。
甘草是典型的药食同源的中药,其主要成分甘草次酸的药理应用价值极高,对健康人和多种动物,有抗利尿、抗炎、镇咳、抗病毒等作用,但甘草次酸因抗菌活性不强、亲水性差、生物活性低等而使应用范围受限,且以甘草次酸开发出来的产品功能性也较为单一。
发明内容
为解决上述现有技术中存在的缺点和不足,本发明的目的在于提供一种甘草次酸衍生物及其制备方法与应用,以解决甘草次酸抗菌活性不强、亲水性差、生物活性低、应用范围窄、制得的产品功效单一等问题以及传统水凝胶敷料功能性单一、不能完全被人体吸收、对人体存在潜在危害、制备过程繁杂不环保、使用后易污染环境和散播病毒等问题。
为了达到上述目的,第一方面,本发明提供了一种甘草次酸衍生物,其结构式如下所示:
其中,R为含氟基团。
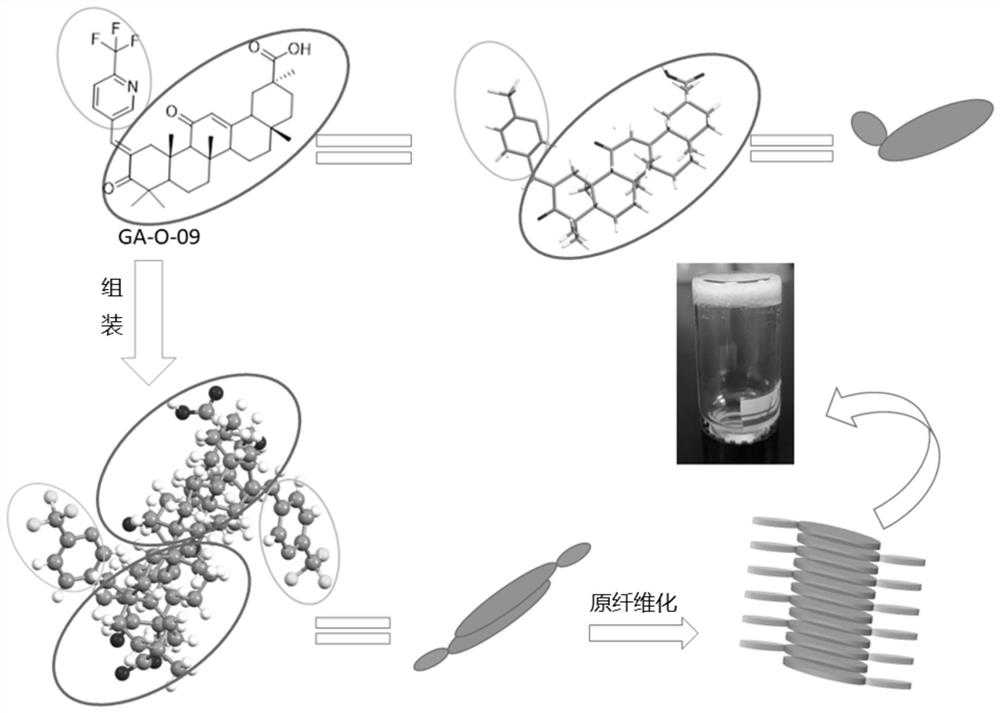
相比甘草次酸,上述甘草次酸衍生物结构中的C3位上的羟基被氧化为羰基,C2位上引入含氟基团,具有更优异的抗菌活性、亲水性以及生物活性;该甘草次酸衍生物与水以及有机溶剂混合后,可通过药物小分子(即该甘草次酸衍生物分子)之间非共价键作用自组装形成三维棒状结构的水凝胶,不需要外界提供能量,且该水凝胶融合了抗菌抗炎与凝胶的特点,实现了抗菌抗炎凝胶一体化,该水凝胶敷用后能被人体完全吸收,没有有害残留,并且对人体安全,具有生物可降解性,不会对环境造成污染。
作为本发明甘草次酸衍生物的优选实施方式,所述R为6-三氟甲基吡啶基。发明人针对构效关系进行结构优化,发现R为6-三氟甲基吡啶基时,所得甘草次酸衍生物的抑菌效果更好。
第二方面,本发明提供了一种上述甘草次酸衍生物的制备方法,包括以下步骤:
(1)选择性氧化:将甘草次酸(GA)于有机溶剂中与氧化剂进行氧化反应,然后淬灭反应,得到甘草次酸氧化物,所述甘草次酸氧化物的结构式如下所示:
(2)修饰处理:在保护气保护下,将所述甘草次酸氧化物、氢氧化物以及含氟基团的化合物于有机溶剂中反应,然后淬灭反应,即得所述甘草次酸衍生物。
上述制备方法,先使甘草次酸发生选择性氧化,以使其结构中的C3位上的羟基被氧化为羰基,得到所述甘草次酸氧化物,再使所述甘草次酸氧化物发生反应,以在其结构中的C2位上引入含氟基团,即得所述甘草次酸衍生物,其中所述甘草次酸氧化物的产率能达到85%-91%,所述甘草次酸衍生物的产率能达到75%-85%;上述制备方法操作简单,可选择无毒性或低毒性的原料,制备过程更加环保。
上述制备方法中,步骤(1)中所采用的有机溶剂为任意能溶解甘草次酸的有机溶剂,优选为丙酮。
作为本发明甘草次酸衍生物的制备方法的优选实施方式,所述步骤(1)中,所述氧化剂为琼斯试剂,所述淬灭所用试剂为甲醇。
作为本发明甘草次酸衍生物的制备方法的优选实施方式,所述步骤(1)中,所述氧化反应在0℃下进行;所述氧化剂被滴加至所述甘草次酸中以进行氧化反应,且所述氧化剂用量过量;在所述氧化反应进行过程中,采用TLC板确认是否反应完全,如果反应完全,则进行淬灭反应;所述甘草次酸氧化物在用于所述步骤(2)之前,还经过纯化处理。其中氧化剂用量过量是指氧化剂用量超过理论用量,即按化学反应方程式计算的用量。
作为本发明甘草次酸衍生物的制备方法的优选实施方式,所述氧化剂被滴加至反应体系呈淡黄色并保持不变,即停止滴加。
上述制备方法中,步骤(2)中所采用的有机溶剂为任意能溶解所述甘草次酸氧化物、所述氢氧化物以及所述含氟基团的化合物的有机溶剂,优选为乙醇。
作为本发明甘草次酸衍生物的制备方法的优选实施方式,所述步骤(2)中,所述氢氧化物为氢氧化钾、氢氧化钠中的至少一种,所述含氟基团的化合物为6-三氟甲基吡啶-3-甲醛,所述淬灭所用试剂为酸溶液。优选所述酸溶液为盐酸。
作为本发明甘草次酸衍生物的制备方法的优选实施方式,所述步骤(2)中,所述反应在室温下进行,所述反应的时间为1-12h;所述甘草次酸氧化物、所述氢氧化物以及所述含氟基团的化合物的摩尔比为甘草次酸氧化物:氢氧化物:含氟基团的化合物=1:1:1-1:2.5:2.5;在所述反应进行过程中,采用TLC板确认是否反应完全,如果反应完全,则进行淬灭反应;所述淬灭反应后,还进行纯化处理。
第三方面,本发明提供了一种水凝胶,所述水凝胶由上述甘草次酸衍生物制备而成。
第四方面,本发明提供了一种上述水凝胶的制备方法,包括以下步骤:将上述甘草次酸衍生物完全溶解于有机溶剂中,然后加入水,即得所述水凝胶。所述水凝胶通过甘草次酸衍生物分子之间的非共价键作用自组装而成,其T
作为本发明水凝胶的制备方法的优选实施方式,所述水和所述有机溶剂的体积比为1-3:1。
作为本发明水凝胶的制备方法的优选实施方式,所述有机溶剂为丙酮、乙醇、乙酸乙酯、二甲基亚砜中的至少一种。
作为本发明水凝胶的制备方法的优选实施方式,所述甘草次酸衍生物完全溶解于有机溶剂后,边被加入水边被摇晃或搅拌。
作为本发明水凝胶的制备方法的优选实施方式,所述水被滴加至所述甘草次酸衍生物中。
作为本发明水凝胶的制备方法的优选实施方式,所述甘草次酸通过超声辅助以完全溶解于有机溶剂中。
作为本发明水凝胶的制备方法的优选实施方式,所述超声辅助的温度为35-50℃,处理时间为60-110s。
第五方面,本发明还提供了一种医用水凝胶敷料,其包括上述水凝胶。
与现有技术相比,本发明具有如下优点:
(1)本发明对甘草次酸进行改性,将其结构中的C3位上的羟基氧化为羰基,C2位上引入含氟基团,提高了其抗菌活性、亲水性以及生物生化;
(2)本发明含氟的甘草次酸衍生物与水以及有机溶剂混合后,可通过药物小分子之间的非共价键作用自组装形成三维棒状结构的水凝胶,不需要外界提供能量,且所得水凝胶融合了抗菌抗炎与凝胶的特点,实现了抗菌抗炎凝胶一体化,不仅对人体安全,能被人体完全吸收,还具有生物可降解性,不会对环境造成污染,制备方法简单,适合大批量生产,具有非常高的商业价值。
附图说明
图1为各实施例中由GA-O-09制备水凝胶的流程图;
图2为各实施例中GA-O-09的的合成路线图;
图3为本发明所得抗菌抗炎水凝胶实物图;
图4为GA-O-09凝胶小分子SEM下的干凝胶样貌。
具体实施方式
为更好的说明本发明的目的、技术方案和优点,下面将结合具体实施例对本发明作进一步说明。
实施例1
本实施例提供了一种基于甘草次酸衍生物制成医用水凝胶的制备方法,包括以下步骤:
1、甘草次酸衍生物的合成
(1)称取18β-甘草次酸(1g,2.12mmol)于干燥的圆底烧瓶中,加入5-10mL丙酮,在冰浴(0℃)条件下搅拌至甘草次酸完全溶解于丙酮中;
(2)将26.72g三氧化铬以少量水溶解烧杯中,然后缓慢滴入2-3mL浓硫酸(质量浓度98%),再用水稀释至100mL即得琼斯试剂(Jones Regent);
(3)往上述圆底烧瓶的反应溶液中滴加配制好的琼斯试剂,待溶液呈现淡黄色并保持30s以上不变时,即琼斯试剂过量,停止滴加琼斯试剂,通过TLC板确认反应完全后,加入甲醇以淬灭反应,得到含有GA-O(即18β-甘草次酸氧化物)的溶液;
(4)把含有GA-O的溶液旋蒸至干燥,再用乙酸乙酯和水进行萃取,收集有机相溶液并用无水硫酸镁干燥,然后旋蒸至干燥,得到GA-O粗品;
(5)将GA-O粗品进行柱层析分离(固定相为硅胶,流动相为石油醚和乙酸乙酯的混合液,其中石油醚和乙酸乙酯的体积比为石油醚:乙酸乙酯=4:1),得到产物GA-O(白色固体,909mg,产率91%);
(6)将GA-O(200mg,0.43mmol,1eq)、6-三氟甲基吡啶-3-甲醛(149.4mg,0.99mmol,2.5eq)和氢氧化钾(47.9mg,0.85mmol,2eq)置于反应器中,并溶解于10-20mL乙醇中;
(7)在氮气保护下,于室温条件下反应1h,并通过TLC板确定反应是否进行彻底;
(8)通过TLC板确认反应完全后,往反应器中加入2-3mL盐酸溶液以对反应进行猝灭,得到含有GA-O-09(即目标甘草次酸衍生物)的溶液;
(9)把含有GA-O-09的溶液旋蒸至干燥,用乙酸乙酯和水进行萃取,收集有机相溶液并用无水硫酸镁干燥,然后旋蒸至干燥,得到GA-O-09粗品;
(10)将GA-O-09粗品进行柱层析分离(固定相为硅胶,流动相为石油醚和乙酸乙酯的混合液,其中石油醚和乙酸乙酯的体积比为石油醚:乙酸乙酯=4:1),得到产物GA-O-09(白色固体,185mg,产率76.8%);
2、水凝胶的制备
(11)称取步骤(10)得到的GA-O-09 15mg于直径1cm、容积4mL的样品瓶中,用1000μL移液枪向其中加入1mL无水乙醇,然后在40℃下超声100s使其充分溶解;
(12)用1000μL移液枪向其中加1mL去离子水,边缓慢滴加去离子水边缓慢晃动样品瓶,等待30-60s,即可得到能稳定存在的水凝胶(如图3所示);
3、创伤医用敷料的制备
(13)将上述制得的水凝胶结合医用胶带,根据伤口的受伤程度,既可以将水凝胶涂抹在医用胶带的粘胶面上,也可以将水凝胶制成块状敷在伤口并用医用胶带固定。
实施例2
本实施例提供了一种基于甘草次酸衍生物制成医用水凝胶的制备方法,包括以下步骤:
1、甘草次酸衍生物的合成
(1)称取18β-甘草次酸(1g,2.12mmol)于干燥的圆底烧瓶中,加入5-10mL的丙酮,在冰浴(0℃)条件下搅拌至甘草次酸完全溶解于丙酮中;
(2)将26.72g三氧化铬以少量水溶解烧杯中,然后缓慢滴入2-3mL浓硫酸(质量浓度98%),再用水稀释至100mL即得琼斯试剂(Jones Regent);
(3)往上述圆底烧瓶的反应溶液中滴加配制好的琼斯试剂,待溶液呈现淡黄色并保持不变时,即琼斯试剂过量,停止滴加琼斯试剂,通过TLC板确认反应完全后,加入甲醇以淬灭反应,得到含有GA-O(即18β-甘草次酸氧化物)的溶液;
(4)把含有GA-O的溶液旋蒸至干燥,再用乙酸乙酯和水进行萃取,收集有机相溶液并用无水硫酸镁干燥,然后旋蒸至干燥,得到GA-O粗品;
(5)将GA-O粗品进行柱层析分离(固定相为硅胶,流动相为石油醚和乙酸乙酯的混合液,其中石油醚和乙酸乙酯的体积比为石油醚:乙酸乙酯=13:3),得到产物GA-O(白色固体,948mg,产率93.1%);
(6)将GA-O(200mg,0.43mmol,1eq)、6-三氟甲基吡啶-3-甲醛(132.4mg,0.88mmol,2eq)和氢氧化钾(47.9mg,0.85mmol,2eq)置于反应器中,并溶解于12-20mL乙醇溶液中;
(7)在氮气保护下,于室温条件下反应5h,并通过TLC板确定反应是否进行彻底;
(8)通过TLC板确认反应完全后,往反应器中加入mL盐酸溶液以对反应进行猝灭,得到含有GA-O-09(即目标甘草次酸衍生物)的溶液;
(9)把含有GA-O-09的溶液旋蒸至干燥,用乙酸乙酯和水进行萃取,收集有机相溶液并用无水硫酸镁干燥,然后旋蒸至干燥,得到GA-O-09粗品;
(10)将GA-O-09粗品进行柱层析分离(固定相为硅胶,流动相为石油醚和乙酸乙酯的混合液,其中石油醚和乙酸乙酯的体积比为石油醚:乙酸乙酯=4:1),得到产物GA-O-09(白色固体,202mg,产率83.8%);
2、水凝胶的制备
(11)称取步骤(10)得到的GA-O-09 15mg于直径1cm、容积4mL的样品瓶中,用1000μL移液枪向其中加入1mL丙酮,然后在30℃下超声90s使其充分溶解;
(12)用1000μL移液枪向其中加1.5mL去离子水,边缓慢滴加去离子水边缓慢晃动样品瓶,等待30-60s,即可得到能稳定存在的水凝胶(如图3所示);
3、创伤医用敷料的制备
(13)将上述制得的水凝胶结合医用胶带,根据伤口的受伤程度,既可以将水凝胶涂抹在医用胶带的粘胶面上,也可以将水凝胶制成块状敷在伤口并用医用胶带固定。
实施例3
本实施例提供了一种基于甘草次酸衍生物制成医用水凝胶的制备方法,包括以下步骤:
1、甘草次酸衍生物的合成
(1)称取18β-甘草次酸(1g,2.12mmol)于干燥的圆底烧瓶中,加入7-11mL的丙酮,在冰浴(0℃)条件下搅拌至甘草次酸完全溶解于丙酮中;
(2)将26.72g三氧化铬以少量水溶解烧杯中,然后缓慢滴入2-3mL浓硫酸(质量浓度98%),再用水稀释至100mL即得琼斯试剂(Jones Regent);
(3)往上述圆底烧瓶的反应溶液中滴加配制好的琼斯试剂,待溶液呈现淡黄色并保持不变时,即琼斯试剂过量,停止滴加琼斯试剂,通过TLC板确认反应完全后,加入甲醇以淬灭反应,得到含有GA-O(即18β-甘草次酸氧化物)的溶液;
(4)把含有GA-O的溶液旋蒸至干燥,再用乙酸乙酯和水进行萃取,收集有机相溶液并用无水硫酸镁干燥,然后旋蒸至干燥,得到GA-O粗品;
(5)将GA-O粗品进行柱层析分离(固定相为硅胶,流动相为石油醚和乙酸乙酯的混合液,其中石油醚和乙酸乙酯的体积比为石油醚:乙酸乙酯=7:2),得到产物GA-O(白色固体,885mg,产率87.8%);
(6)将GA-O(200mg,0.43mmol,1eq)、6-三氟甲基吡啶-3-甲醛(149.4mg,0.99mmol,2.5eq)和氢氧化钾(40.2mg,0.78mmol,1.8eq)置于反应器中,并溶解于10-16mL乙醇溶液中;
(7)在氮气保护下,于室温条件下反应12h,并通过TLC板确定反应是否进行彻底;
(8)通过TLC板确认反应完全后,往反应器中加入mL盐酸溶液以对反应进行猝灭,得到含有GA-O-09(即目标甘草次酸衍生物)的溶液;
(9)把含有GA-O-09的溶液旋蒸至干燥,用乙酸乙酯和水进行萃取,收集有机相溶液并用无水硫酸镁干燥,然后旋蒸至干燥,得到GA-O-09粗品;
(10)将GA-O-09粗品进行柱层析分离(固定相为硅胶,流动相为石油醚和乙酸乙酯的混合液,其中石油醚和乙酸乙酯的体积比为石油醚:乙酸乙酯=4:1),得到产物GA-O-09(白色固体,193mg,产率81.7%);
2、水凝胶的制备
(11)称取步骤(10)得到的GA-O-09 15mg于直径1cm、容积4mL的样品瓶中,用1000μL移液枪向其中加入1mL乙酸乙酯,然后在50℃下超声110s使其充分溶解;
(12)用1000μL移液枪向其中加3mL去离子水,边缓慢滴加去离子水边缓慢晃动样品瓶,等待30-60s,即可得到能稳定存在的水凝胶(如图3所示);
3、创伤医用敷料的制备
(13)将上述制得的水凝胶结合医用胶带,根据伤口的受伤程度,既可以将水凝胶涂抹在医用胶带的粘胶面上,也可以将水凝胶制成块状敷在伤口并用医用胶带固定。
实施例1-3所得水凝胶的T
对比例1
本对比例提供了一种基于甘草次酸制成医用水凝胶的制备方法,包括如下步骤:
(1)称取18β-甘草次酸18mg于直径1cm、容积4mL的样品瓶中,用250μL移液枪往瓶子中加入0.25mL无水乙醇,再在40℃下超声100s使其充分溶解,然后置于室温环境中;
(2)用250μL移液枪向其中加入0.25mL去离子水,边缓慢滴加去离子水边缓慢旋摇样品瓶,然后静置2小时,无法形成水凝胶。
对比例2
本对比例提供了一种基于甘草次酸氧化物制成医用水凝胶的制备方法,包括如下步骤:
(1)称取实施例1所得甘草次酸氧化物(GA-O)18mg于直径1cm、容积4mL的样品瓶中,用250μL移液枪往瓶子中加入0.25mL无水乙醇,再在40℃下超声100s使其充分溶解,然后置于室温环境中;
(2)用250μL移液枪向其中加入0.25mL去离子水,边缓慢滴加去离子水边缓慢旋摇样品瓶,然后静置2小时,无法形成水凝胶。
效果例1:MIC(最小抑菌浓度)和MBC(最小杀菌浓度)测试
实验方法:
(1)将通过二倍稀释法得到的系列梯度待测天然药物医用水凝胶用10μL移液器量取5μL加入到无菌96孔板中,其中阳性对照为加替沙星,阴性对照为DMSO,每孔重复四次;
(2)将提前准备好用于MIC测定浓度为1.5×10
(3)把微孔板放入恒温箱中,在37℃培养24h后,将微孔板取出;
(4)通过多功能酶标仪在610nm波长下测定每孔的OD值或者用肉眼观察微孔是否变浑浊,我们将微孔板中未变浑浊的临界孔所对应的样品浓度定义为该待测样品对该待测茵种的最小抑菌浓度;
(5)测得本发明的天然药物医用水凝胶前体对金黄色葡萄球菌(ATCC6538)的最小抑菌浓度为6.25μmol/L,对表皮葡萄球茵(ATCC 12228)的最小抑菌浓度为3.125μmol/L,对白色葡萄球菌(ATCC 29213)的最小抑菌浓度为3.125μmol/L,结果如表1所示;
(6)测得本发明干燥后的天然药物医用水凝胶(即干凝胶)对金黄色葡萄球菌(ATCC 6538)的最小抑菌浓度为6.25μmol/L,对表皮葡萄球茵(ATCC12228)的最小抑菌浓度为3.125μmol/L,对白色葡萄球菌(ATCC 29213)的最小抑菌浓度为3.125μmol/L,结果如表1所示;
(7)从做MIC的96孔板中取其中微孔的50μL试液均匀涂布在固体培养基上(取肉眼观察不长菌的和比它大的几个浓度),培养皿倒置放在37℃恒培养温箱中培养24h;
(8)取出培养皿并观察其中的菌落个数。根据MBC定义(杀死99.9%的供试微生物所需的最低药物浓度)计算,长出的菌落个数<8个所对应的浓度即为相应的MBC浓度。
表1:最小抑菌浓度和最小杀菌浓度测试结果
由表1可知,本发明制备的甘草次酸小分子及以其为前体自组装制备的水凝胶均具有抑菌功效,能同时抑制金黄色葡萄球菌、表皮葡萄球茵、白色葡萄球菌,且与GA小分子相比,抑菌效果有了极大的提升,因此,本发明制备所得的水凝胶在抗菌方面具有十分优良的效果。
效果例2:抑菌圈测试
实验方法:
(1)抑菌圈的测定采用的是经典的滤纸片-琼脂扩散法进行完成的。将准备好的米勒-海顿琼脂培养基在50℃左右的时候,倒出约45mL的营养琼脂培养基于120mm的培养皿中,冷却、凝固后备用;
(2)用1000mL移液器量取400μL己经配制好浓度为1.5x10
(3)用无菌摄子夹取直径为6mm的无菌滤纸片压在琼脂平板上,用10μL的移液器量取稀释好的各待测样品5μL轻轻滴加在滤纸片上,保证待测样品完全被滤纸片吸附;
(4)以加替沙星为阳性对照组,以DMSO为阴性对照组,每组重复三次;
(5)静置片刻,将培养皿反过来放置于恒温箱中培养24h后,将培养基取出,用游标卡尺测定抑菌圈直径的大小并做统计;
(5)测得本发明的天然药物医用水凝胶小分子对金黄色葡萄球菌(ATCC6538)的抑制区为8.22±0.08mm,对白色葡萄球菌(ATCC 29213)的最小抑菌浓度为8.31±0.21mm,对表皮葡萄球茵(ATCC 12228)的抑制区为8.80±0.03mm,结果如表2所示;
(6)测得本发明干燥后的天然药物医用水凝胶对金黄色葡萄球菌(ATCC6538)的抑制区为7.82±0.12mm,对白色葡萄球菌(ATCC 29213)的最小抑菌浓度为8.73±0.10mm,对表皮葡萄球茵(ATCC 12228)的抑制区为8.89±0.13mm,结果如表2所示。
表2抑菌圈测试结果
效果例3:SEM测试
通过SEM测试发现本发明制备的天然药物医用水凝胶是一种纳米棒状结构。(见附图4)
最后所应当说明的是,以上实施例仅用以说明本发明的技术方案而非对本发明保护范围的限制,尽管参照较佳实施例对本发明作了详细说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的实质和范围。