使用Cas9碱基编辑器的淋巴造血改造
文献发布时间:2023-06-19 09:41:38
相关申请的交叉引用
本申请要求于2018年3月13日提交的美国临时专利申请号62/642,151的优先权,其公开内容出于所有目的通过引用整体并入本文。
背景技术
原代人细胞的精确调节在免疫治疗、自身免疫和酶病领域中具有多种应用。在遗传水平上调节患者免疫细胞由于治疗的永久性和低患者排斥风险而成为有吸引力的治疗途径。用于免疫细胞基因编辑的一种方法是使用成簇的规律间隔的短回文重复(ClusteredRegularly Interspaced Short Palindromic Repeat,CRISPR)系统在目的基因中诱导双链断裂(double stranded break,DSB),从而导致形成通过经由非同源末端连接(Non-Homologous End Joining,NHEJ)途径进行高度可变修复而产生的小插入或缺失(统称为“indel”)。或者,可以通过引入DSB以及共递送DNA模板用于通过同源指导的修复(homologydirected repair,HDR)进行修复来实现精确的基因组改变。尽管这种方法在通过NHEJ单纯破坏单个基因时是有效且可靠的,但是通过HDR精确改变单个核苷酸的效率非常低。此外,在多重基因编辑程序中诱导多个DSB可导致不期望的遗传毒性和潜在致癌性总染色体易位的形成。因此,在本领域中仍然需要在有限地诱导毒性DSB的情况下对人免疫细胞进行多重基因改造的更受控且更安全的方法。
发明内容
在第一方面,本文提供了用于产生经遗传改造淋巴造血细胞的方法。所述方法可以包括以下或基本上由以下组成:(a)向淋巴造血细胞中引入:(i)编码碱基编辑器融合蛋白的质粒、mRNA或蛋白质,所述碱基编辑器融合蛋白包含与Cas9切口酶结构域融合的脱氨酶结构域,其中所述切口酶结构域包含碱基切除修复抑制剂结构域;和(ii)与待遗传修饰的靶核酸序列具有互补性的一种或更多种剪接受体-剪接供体(splice acceptor-splicedonor,SA-SD)gRNA;以及(b)在促进破坏被一种或更多种SA-SD gRNA靶向的剪接位点的条件下培养经引入的细胞,从而相对于未经转染的淋巴造血细胞,通过碱基编辑器融合蛋白和一种或更多种剪接受体-剪接供体(SA-SD)gRNA来修饰靶核酸序列,并且从而产生经遗传改造淋巴造血细胞。在一些情况下,所述方法还包括向淋巴造血细胞中引入被设计用于产生一个或更多个靶向敲入或错义突变的一种或更多种gRNA,从而经遗传改造淋巴造血细胞包含至少一个基因敲除和一个或更多个基因敲入或错义突变。在一些情况下,所述方法还包括向淋巴造血细胞中引入被设计用于产生一个或更多个靶向敲入和一个或更多个错义突变的一种或更多种gRNA,从而经遗传改造淋巴造血细胞包含至少一个基因敲除、至少一个基因敲入和至少一个错义突变。碱基编辑器融合蛋白可以是BE3、BE4或腺嘌呤碱基编辑器(adenine base editor,ABE)。淋巴造血细胞可以是T细胞、自然杀伤(NK)细胞、B细胞或CD34+造血干祖细胞(hematopoietic stem progenitor cell,HSPC)。一种或更多种SA-SDgRNA可被化学修饰以在每个gRNA的至少一个5’核苷酸和至少一个3’核苷酸上包含2’-O-甲基硫代磷酸酯修饰。碱基编辑器融合蛋白和一种或更多种剪接受体-剪接供体(SA-SD)gRNA可以表现出约50%至约90%的C-T转换效率。一种或更多种SA-SD gRNA可以选自表1中所示的序列。
在另一个方面,本文提供了用于产生经遗传修饰T细胞的方法。所述方法可以包括以下或基本上由以下组成:(a)向人T细胞中引入:(i)编码碱基编辑器融合蛋白的质粒、mRNA或蛋白质,所述碱基编辑器融合蛋白包含与Cas9切口酶结构域融合的脱氨酶结构域,其中所述切口酶结构域包含碱基切除修复抑制剂结构域;(ii)破坏TRAC、B2M和PDCD1中每一种的表达的一种或更多种剪接受体-剪接供体(SA-SD)gRNA;(iii)编码T细胞受体(TCR)和嵌合抗原受体(CAR)的供体DNA模板;和(iv)两种与靶插入位点互补的gRNA;(b)在促进破坏被SA-SD gRNA靶向的剪接位点的条件下培养(a)中的T细胞,从而相对于未经转染的T细胞,TRAC、B2M和PDCD1基因产物的表达降低;以及(c)在促进在靶插入位点靶向敲入供体DNA模板的条件下培养经转染的T细胞。碱基编辑器融合蛋白可以是BE3、BE4或腺嘌呤碱基编辑器(ABE)。一种或更多种SA-SD gRNA可以选自表1中所示的序列。SA-SD gRNA和与靶插入位点互补的gRNA中的一种或更多种可被化学修饰以在每个gRNA的至少一个5’核苷酸和至少一个3’核苷酸上包含2’-O-甲基硫代磷酸酯修饰。供体DNA模板可以以rAAV提供。TCR可以与肿瘤抗原特异性结合。CAR可以包含与肿瘤抗原特异性结合的CAR抗原结合结构域。TCR和CAR可以与不同的抗原结合。
在另一个方面,本文提供了根据本公开内容的方法获得的经遗传修饰的细胞。
附图说明
当考虑本发明以下详细描述时,将更好地理解本发明,并且除了上述那些之外的特征、方面和优点将变得明显。这样的详细描述参照以下附图,其中:
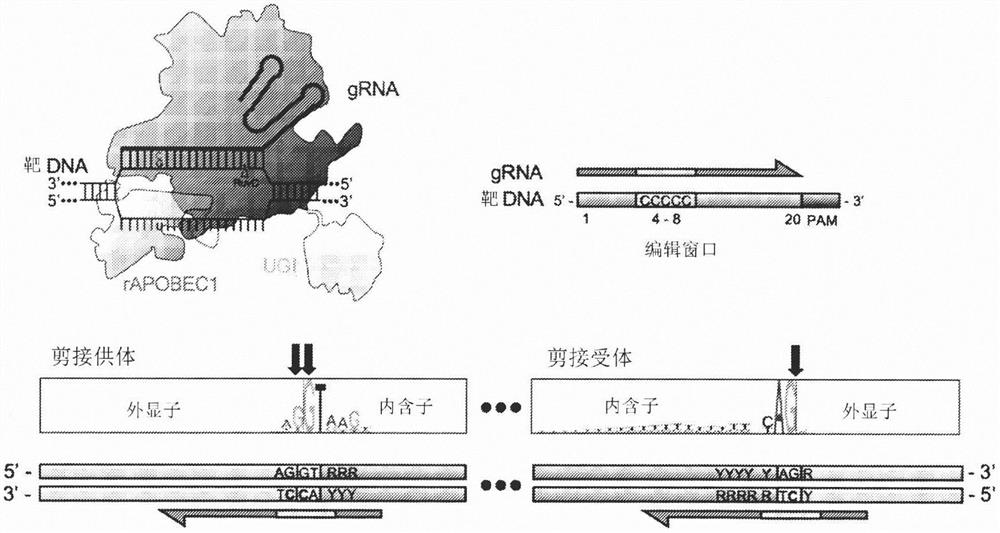
图1示出了描绘与靶DNA结合的Cas9碱基编辑器(BE)(左)和描绘用BE3和BE4实现的碱基编辑窗口的前间隔序列(protospacer)(右)的图。还提供了标识图,其描绘了哺乳动物剪接供体(SD)和剪接受体(SA)元件的共有序列,以及用于通过剪接位点破坏进行BE敲除的前间隔序列的相关取向。
图2示出了通过引入提前终止(pmSTOP)密码子或通过由碱基编辑器进行的剪接位点破坏来进行单基因敲除的工作流程的一个实例。
图3A-3L展示了在PDCD1、B2M和TRAC处用于基因破坏的指导RNA活性的评估。(a)PDCD1基因座的图,指示了每种sgRNA的相对位置。方框的彩色部分表示蛋白质编码区,垂直红线表示终止密码子。(b)如通过Sanger测序迹线的EditR分析确定的与BE3或BE4 mRNA共递送之后每种PDCD1 sgRNA的靶碱基的C-T转换的定量(n=3个独立的T细胞供体)。(c)如通过流式细胞术确定的在递送指定sgRNA与BE3或BE4mRNA之后的PDCD1蛋白敲除频率(n=3个独立的T细胞供体)。(d)如通过Sanger测序迹线的EditR分析确定的与BE3或BE4 mRNA共递送之后PDCD1 Ex1 SD sgRNA的检出编辑窗口中所有C(以红色显示)的C-T/A/G转换的定量(n=3个独立的T细胞供体)。带下划线的C指示对于正确剪接至关重要的靶核苷酸。(e)TRAC基因座的图,指示了每种sgRNA的相对位置。(f)如通过Sanger测序迹线的EditR分析确定的与BE3或BE4 mRNA共递送之后每种TRAC sgRNA的靶碱基的C-T转换的定量(n=3个独立的T细胞供体)。(g)如通过用于CD3损失的流式细胞术确定的在递送指定sgRNA与BE3或BE4mRNA之后的TRAC蛋白敲除频率(n=3个独立的T细胞供体)。(h)如通过Sanger测序迹线的EditR分析确定的与BE3或BE4 mRNA共递送之后TRAC Ex3 SA sgRNA的检出编辑窗口中所有胞嘧啶(以红色显示)的C-T/A/G转换的定量(n=3个独立的T细胞供体)。(i)B2M基因座的图,指示了每种sgRNA的相对位置。(j)如通过Sanger测序迹线的EditR分析确定的在共递送BE3或BE4mRNA之后每种B2MsgRNA的靶碱基的C-T转换的定量(n=3个独立的T细胞供体)。(k)如通过用于B2M损失的流式细胞术确定的在递送指定sgRNA与BE3或BE4 mRNA之后的B2M蛋白敲除频率(n=3个独立的T细胞供体)。(1)如通过Sanger测序迹线的EditR分析确定的与BE3或BE4mRNA共递送之后B2M Ex1 SD sgRNA的检出编辑窗口中所有胞嘧啶(以红色显示)的C-T/A/G转换的定量(数据表示为平均值±SD,n=3个独立的生物T细胞供体)。由最高编辑指导处理与第二高编辑处理之间的Student配对双尾t检验计算的P值(n.s.P>0.05、*P≤0.05、**P≤0.01、***P≤0.001、****P≤0.0001)。
图4A-4F展示了使用最佳sgRNA(TRAC Ex3 SA、B2M Ex1 SD和PDCD1 Ex1 SD)进行的多重编辑优化。(a)如在将三种靶sgRNA与以下共递送之后通过NGS分析的TRAC、PDCD1和B2M的靶胞嘧啶到所有其他碱基的转换效率:以3μg剂量递送的第一代BE3(BE3)或BE4(BE4)mRNA;与sgRNA复合的BE4蛋白(BE4 RNP);或以1.5μg或4μg剂量递送的经密码子优化的BE4mRNA(coBE4)。(b)如在将三种靶sgRNA与以下递送之后通过NGS分析的TRAC、PDCD1和B2M的indel频率:以3μg剂量递送的第一代BE3(BE3)或BE4(BE4)mRNA;与sgRNA复合的BE4蛋白(BE4 RNP);或以1.5μg或4μg剂量递送的经密码子优化的BE4 mRNA(coBE4)。(c)如在将三种靶sgRNA与1.5μg或4μg剂量的SpCas9核酸酶mRNA共递送之后通过NGS分析的TRAC、PDCD1和B2M的indel频率。(d)如在递送三种靶sgRNA和以下之后7天通过流式细胞术测量的TRAC、PDCD1和B2M蛋白损失的频率:以3μg剂量递送的第一代BE3(BE3)和BE4(BE4)mRNA;与sgRNA复合的BE4蛋白(BE4 RNP);以及以1.5μg剂量和4μg剂量递送的经密码子优化的BE4 mRNA(coBE4)。(e)在电穿孔之后7天进行的多重流式细胞术分析的SPICE表示。(f)WT、单基因、双重基因和三重基因KO的分数的定量。数据表示为平均值±SD,n=2两个独立的生物T细胞供体。
图5A-5B展示了多重编辑T细胞中的易位频率。(a)TRAC、B2M、PDCD1和PDCD1 OT位点处由双链断裂诱导引起的可能易位结果的Circos图。(b)易位频率的微滴数字PCR定量。所有测定以n=2个独立生物T细胞供体的技术重复进行。
图6A-6D展示了多重编辑T细胞的功能。(a)编辑和扩增之后记忆标志物CD27和CD45ro的表达。活化之后CD4和CD8 T细胞的细胞因子单独(b)和组合(c)的产生。(d)如在与T细胞共培养之后通过萤光素酶发光测定测量的T细胞杀伤CD19neg K562、CD19pos Raji细胞或CD19pos/PD-L1pos Raji细胞的能力。附图标题示出了E∶T比率。数据表示为平均值±SD,其中测定在两个独立生物T细胞供体中一式三份进行。(n.s.P>0.05、*P≤0.05、**P≤0.01、***P≤0.001、****P≤0.0001)。
图7A-7C展示了图3A-3L中每种sgRNA的非靶编辑。从NGS分析数据。堆叠条的高度表示平均值,误差条±1为标准偏差。n=3个独立供体。
图8A-8C示出了图3A-3L中所有样品的indel。从NGS分析数据。堆叠条的高度表示平均值,误差条±1为标准偏差。n=3个独立供体。
图9展示了使用第一代、低剂量(1.5μg)BE3或BE4 mRNA对T细胞进行的多重碱基编辑。条形图描绘了在蛋白质水平上的碱基编辑器介导的TRAC、B2M和PDCD1敲除。如方法部分描述的,通过流式细胞术评估蛋白质表达。n=2个独立供体。
图10展示了T细胞电穿孔之后的碱基编辑器蛋白水平。评估在两个独立供体中在经刺激T细胞的电穿孔之后24使用编码Cas9、BE3、BE4和经密码子优化BE4的mRNA实现的蛋白质水平的数字western印迹结果。经纯化的BE3蛋白也用作BE蛋白抗体检测的阳性对照。
图11展示了重新刺激之后PDCD1、B2M和TRAC的代表性流式图。电穿孔之后五天,重新刺激T细胞以诱导PDCD1的表达,这允许评估PD-1蛋白的敲除频率。此处显示的是多重co-BE4 mRNA编辑和仅脉冲对照(左列)之后供体匹配T细胞的TRAC、B2M和PDCD1表达的代表性流式细胞术图。
图12A-12B展示了对计算预测的脱靶碱基编辑和indel形成的评估。在T细胞中使用与Cas9或BE4 mRNA组合的靶向TRAC、B2M或PDCD1的最佳sgRNA使用下一代测序评估的在靶和前10个计算预测脱靶sgRNA结合位点的碱基编辑(A)和indel(B)频率。
图13展示了三个靶基因座之间的易位频率。递送三种sgRNA与spCas9蛋白、spCas9mRNA、BE4蛋白或coBE4 mRNA之后TRAC、B2M与PDCD1之间的易位频率的微滴数字PCR定量。n=一式两份测定的2个独立的T细胞供体。
图14展示了靶基因座与PDCD1脱靶位点之间的易位频率。递送三种sgRNA与spCas9蛋白、spCa9 mRNA、BE4蛋白或coBE4 mRNA之后,PDCD1 OT位点与TRAC、B2M和PDCD1之间的易位频率的微滴数字PCR定量。n=一式两份测定的2个独立的T细胞供体。
图15A-15B展示了CAR转导和T细胞扩增效率。(A.)示出了两个独立供体中通过对RQR8染色的使用MND-CD19CAR-RQR8慢病毒载体(MOI为20)的经转导T细胞的频率的条形图。RQR8是包含用于用CD34和CD20特异性抗体染色的结构域的杂合分子,并且用作确定CAR阳性T细胞频率的替代物。(B.)示出了电穿孔和转导之后第5天和第12天活细胞的数目的条形图。n=2个独立供体。
图16示出了相对于Cas9核酸酶和Cas9切口酶使用BE切口酶活性和效率的靶向敲入(KI)。
图17A-17B展示了跨易位连接部的亚克隆PCR产物的测序。(A)如指示的,使用Cas9或BE mRNA或蛋白质在指定靶基因之间进行易位PCR的结果。还进行了AAVS1对照PCR以确定gDNA品质和PCR功能。(B)将来自(A)的PCR产物TA克隆到TOPO质粒中,随后通过Sanger测序进行分析。然后将所得色谱图与指定靶基因gRNA切割位点之间的假设‘完美’连接序列对齐并进行比对。还描绘了用于生成数据的ddPCR探针。
图18A-18C展示了扩增、活化的UCB CD34+HSPC中的高度有效碱基编辑。使用EditR计算C-T转换。
图19示出了CD16中先前鉴定的ADAM17切割区域,其中关键丝氨酸(红色)在突变为脯氨酸时使CD16不可被ADAM17切割。使用C-T碱基编辑器变体,我们能够靶向相邻的缬氨酸(蓝色)。示出了使用C-T碱基编辑器变体可实现的针对该缬氨酸的可能氨基酸变化。
图20A-20B示出了代表性的Sanger测序色谱图。(A)示出了来自对照(单独BE3-VQR)和经编辑(BE3-VQR+CD16gRNA)样品的测序色谱图(上图)。使用EditR计算C-T转换。在条形图(下图)中组合了来自两个单独供体的结果。(B)示出了来自对照(单独ABE)和经编辑(ABE+CD16a gRNA)样品的测序色谱图。使用EditR计算T-C转换。ABE+CD16a gRNA样品显示转换为切割抗性CD16a变体。
图21A-21D显示DNA修复缺陷型范科尼贫血(Fanconi Anemia,FA)成纤维细胞适合于使用BE3和BE4进行碱基编辑。(A)用BE4处理的FA患者来源的成纤维细胞中FANCA Ex.39c.3934+2 T>C pos.4 gRNA靶位点的序列色谱图。用EditR软件对碱基编辑频率进行定量。(B)来自A中但使用ABE和PDCD1 Ex.1 SD gRNA进行的类似实验的结果。(C)使用BE3在对照(MPS1)成纤维细胞和FA患者来源的成纤维细胞中进行碱基编辑的近视图。(D)来自使用TIDE算法分析的Sanger测序数据的成纤维细胞中Cas9核酸酶活性的结果。
尽管本发明可以允许多种修改和替代形式,在附图中以实例的方式示出了并且在本文中详细描述了本发明的一些示例性实施方案。然而,应当理解,示例性实施方案的描述并非旨在将本发明限制于所公开的特定形式,而是相反,意图是涵盖落入由所附权利要求书限定的本发明精神和范围内的所有修改方案、等同方案和替代方案。
具体实施方式
本说明书中引用的所有出版物(包括但不限于专利和专利申请)均通过引用并入本文,就如同在本申请中整体提出一样。
本文所述的方法、系统和组合物至少部分基于发明人开发了用于使用CRISPR-Cas9碱基编辑器对原代人淋巴造血细胞进行基因组改造的方案。为了在不使用同源指导的修复(HDR)的情况下进行点突变,研究人员开发了将Cas9切口酶或dCas9与胞苷脱氨酶(如APOBEC1)融合的CRISPR碱基编辑器。与CRISPR不同,碱基编辑不切割双链DNA,而是使用脱氨酶来精确重排构成DNA或RNA的四种碱基之一中的一些原子,从而转换该碱基而不改变其周围的碱基。碱基编辑器通过指导RNA(gRNA)靶向特定的基因座,并且其可将在前间隔序列相邻基序(PAM)位点附近的小编辑窗口中的胞苷转换为尿苷。尿苷随后通过碱基切除修复转换为胸苷,从而产生C->T变化(或相反链上的G->A)。其中碱基切除修复抑制剂UGI与Cas9切口酶融合的第三代碱基编辑器(BE3系统)对未经修饰的DNA链产生切口,从而促使细胞使用经编辑的链作为模板进行错配修复。结果,细胞使用包含U的链(通过胞苷脱氨引入)作为模板来修复DNA,从而复制了碱基编辑。第四代碱基编辑器(BE4系统)使用两个拷贝的碱基切除修复抑制剂UGI。已开发出腺嘌呤碱基编辑器(ABE),其将基因组DNA中的靶向A·T碱基对有效地转换为G·C(在人细胞中0-100%效率),并且具有高产物纯度(通常至少99.9%)和低indel率(通常不超过0.1%)。参见,例如,Gaudelli et al.,Nature 551:464-471(2017)。
如以下段落和实施例中所述,发明人的进行基因组改造的精简方法采用碱基编辑器(例如,第三代和第四代碱基编辑器、腺嘌呤碱基编辑器)通过敲除和错义突变以及在DNA供体模板的存在下的靶向基因敲入进行靶向基因破坏。这些方法、系统和组合物的优点是多重的。特别地,这些方法可用于在单个方法中进行三重基因编辑:靶向基因敲除、靶向错义突变和靶向基因敲入。本文所述的方法非常适合于研究淋巴造血细胞生物学和基因功能、对疾病(例如原发性免疫缺陷)进行建模、以及校正引起疾病的点突变、以及产生用于治疗应用的新的细胞产物(例如T细胞产物)。不受特定理论或作用机制束缚,认为通过使用预定病毒整合模式以及有限诱导毒性双链断裂,本文所述的方法、系统和组合物允许更安全、受控的细胞改造。
因此,本文提供了用于靶向破坏靶基因的转录或翻译的方法。特别地,所述方法包括通过起始密码子的破坏、提前终止密码子的引入和/或内含子/外显子剪接位点的靶向破坏来靶向破坏靶基因的转录或翻译。在一些情况下,本文提供了一种方法,其包括将编码碱基编辑融合蛋白的核酸序列与指导RNA组合,从而获得意想不到的高碱基编辑率;特别是在原代细胞例如CD34+HSPC、T细胞、自然杀伤细胞和B细胞中。使用本文所述的方法,可以以改善的效率和降低的脱靶indel形成率在原代细胞中敲入和/或敲除一个或更多个目的基因。在一些优选的实施方案中,该方法用于多重碱基编辑,包括基因敲入、基因敲除和错义突变。
在第一方面,本文提供了用于产生经遗传改造淋巴造血细胞的方法。特别地,所述方法包括将碱基编辑组分转染到淋巴造血细胞中,其中所述组分包含:(i)编码碱基编辑器融合蛋白的质粒,所述碱基编辑器融合蛋白包含碱基切除修复抑制剂结构域和与Cas9切口酶结构域融合的脱氨酶结构域,其中Cas9切口酶结构域任选地与碱基切除修复结构域融合;以及(ii)与待遗传修饰的靶核酸序列具有互补性的一种或更多种gRNA。当在促进破坏被一种或更多种gRNA靶向的剪接位点的条件下培养经转染的细胞时,相对于未经转染的细胞,通过碱基编辑器融合蛋白和一种或更多种gRNA来修饰靶核酸序列,并且从而产生经遗传改造淋巴造血细胞。如本文所用,术语“淋巴造血细胞”是指T细胞、自然杀伤(NK)细胞、B细胞、CD34+造血干祖细胞(HSPC),以及参与淋巴细胞以及血液、骨髓、脾、淋巴结和胸腺的细胞的产生的其他细胞。
如本文所用,“碱基编辑器”(也称为“核碱基编辑器”)是Cas9融合蛋白,其包含与脱氨酶融合的Cas9切口酶结构域或死的Cas9(dead Cas9,dCas9)。在一些实施方案中,融合蛋白包含进一步与UGI结构域融合的Cas9切口酶。在一些实施方案中,UGI结构域也提供在系统中,但是不与Cas9结构域融合。在一些情况下,碱基编辑融合蛋白是碱基编辑器3(BE3)或碱基编辑器4(BE4),其中BE3和BE4分别是指第三代碱基编辑器和第四代碱基编辑器。B3和B4可以产生C>G或A或者indel突变。在另一些情况下,碱基编辑融合蛋白是腺嘌呤碱基编辑器(ABE),例如将细菌和人细胞DNA中的A·T转换为G·C碱基对的ABE。参见,例如Gaudelli et al.,Nature 551:464-471(2017)。应当理解,其他碱基编辑器(包括在ATG“起始”密码子处引入无效突变以破坏所靶向基因表达的那些)也适用于根据本文所述的方法。
在某些实施方案中,所述方法包括通过靶向剪接受体-剪接供体(SA-SD)位点或提前终止(pmSTOP)位点来敲除基因。对于这样的方法,将CRISPR gRNA分子设计为破坏靶核苷酸序列中的一个或更多个剪接受体/供体位点。CRISPR指导RNA分子(gRNA)包含具有至少10个连续核苷酸的序列,并且通常具有17-23个连续核苷酸的序列,其与生物体基因组中的靶序列互补并且包含靶碱基对。gRNA包含与gRNA靶位点部分或完全互补的核苷酸序列。gRNA靶位点还包含紧邻靶位点下游存在的前间隔序列相邻基序(PAM)。PAM序列的实例是已知的(参见,例如,Shah et al.,RNA Biology 10(5):891-899,2013)。
SA-SD位点处的破坏是特别有利的,因为可以敲除编码序列和非编码RNA(ncRNA)而不会通读终止密码子。针对与免疫治疗相关的人长非编码RNA和人蛋白质编码基因设计的示例性SA-SD gRNA示于表1中。如以下实施例中所示,在敲除效率方面,使用靶向SA-SD位点的gRNA进行的破坏优于引入提前终止密码子。碱基编辑的效率可以通过Sanger测序迹线的EditR分析或通过下一代测序(NGS)在基因组水平上确定,以及也可以通过流式细胞术在蛋白质水平上确定。靶向剪接供体区和剪接受体区的剪接受体-剪接供体碱基编辑gRNA的碱基转换效率为至少5%,并且在一些情况下为至少80%或更高(例如5%、10%、15%、20%、25%、30%、35%、40%、45%、50%、55%、60%、65%、70%、75%、80%、85%、90%、95%、99%)。在一些情况下,与引入提前终止密码子破坏的gRNA相比,SA-SD gRNA在C-T转换方面的效率显著更高。
可以使用基于R的程序来设计用于靶向SA-SD位点的指导RNA,该程序鉴定靶向所有ncRNA和蛋白质编码基因SA-SD位点的gRNA。在一些情况下,用户提供参考基因组、参考序列的Ensembl转录本ID、前间隔序列相邻基序(PAM)位点以及与外显子-内含子边界上游和下游子集的距离。该程序提取外显子-内含子边界上游20个碱基对+PAM长度和下游15个碱基对的序列、以及剪接位点基序。在一些实施方案中,指导分子的长度可以是20至120个碱基,或更多。在某些实施方案中,指导分子的长度可以是20至60个碱基、或20至50个碱基、或30至50个碱基、或39至46个碱基。
在一些情况下,使用当转染到哺乳动物细胞中时具有提高的稳定性的经化学修饰的gRNA是有利的。例如,可以对gRNA进行化学修饰,以在每个gRNA的至少一个5’核苷酸和至少一个3’核苷酸上包含2’-O-甲基硫代磷酸酯修饰。在一些情况下,对三个末端5’核苷酸和三个末端3’核苷酸进行化学修饰以包含2’-O-甲基硫代磷酸酯修饰。
在某些实施方案中,该方法采用碱基编辑切口酶活性以通过(i)来自模板的同源指导的修复(HDR)或(ii)病毒载体的整合来介导供体序列的插入。对于这样的方法,Cas9切口酶结构域有助于在DNA供体模板的存在下使用gRNA靶向插入位点来进行靶向基因敲入。在一些情况下,将供体序列整合在内源安全港基因座,例如C-C基序趋化因子受体5(CCR5)、腺相关病毒整合位点1(AAVS1)、ROSAβgeo26(Rosa26)、白蛋白(ALB)、T-细胞受体α恒定区(T-Cell Receptor Alpha Constant,TRAC)和/或次黄嘌呤磷酸核糖基转移酶1(HPRT)。例如,指导RNA可以被设计成靶向AAVS1基因座。在这种情况下,指导RNA与DNA靶位点具有互补性。参照示出了靶向敲入(KI)的图16,BE切口酶活性在与rAAV DNA供体递送组合时对刺激HDR非常有效。在一些情况下,相对于使用单一gRNA或使用Cas9核酸酶或Cas9切口酶,使用两种与靶插入位点互补的指导RNA的BE效率得到了极大改善。
一些实施方案包括同时进行的多重基因编辑方法。例如,本文提供了使用碱基编辑在人细胞中进行多重改造的方法。在一些情况下,该方法包括使用碱基编辑器融合蛋白(例如BE3、BE4、ABE)和gRNA进行多重基因编辑,其中破坏一个或更多个基因(用于敲除),并且使用供体DNA模板用于插入来敲入一个或更多个基因。参照图12,碱基编辑器3(BE3)和碱基编辑器4(BE4)与三种SA-SD gRNA一起使用,成功敲除了人T细胞中TRAC、B2M和PDCD1的表达。BE3和BE4也与这三种SA-SD gRNA和两种靶向AAVS1的gRNA一起使用以敲入供体DNA模板。这些数据表明,本文提供的碱基编辑方法可用于在具有或不具有靶DNA模板的敲入或错义突变的情况下进行细胞(例如,T细胞)中与免疫治疗相关的多个基因的多重破坏。
本文还提供了用于在体外、体内或离体的原核或真核细胞中进行基因组改造(例如,用于改变或操纵一种或更多种基因或者一种或更多种基因产物的表达)的方法。特别地,本文提供的方法可用于哺乳动物细胞中的靶向碱基编辑破坏,所述哺乳动物细胞包括人T细胞、自然杀伤(NK)细胞、CD34+造血干祖细胞(HSPC)(例如脐带血HSPC)和成纤维细胞(例如,MPS1成纤维细胞、范科尼贫血成纤维细胞)。重要的是,如图21A-21D中所示,来自范科尼贫血患者(并且因此是DNA修复缺陷型)的成纤维细胞仍然适合使用例如BE3、BE4或ABE进行碱基编辑。因此,本文还提供了经遗传改造淋巴造血细胞,例如已根据本文所述的方法修饰的T细胞。
在一些情况下,所述方法被配置成产生适合作为“普遍可接受的”细胞以用于治疗应用的经遗传改造T细胞。如本文所用,术语“普遍可接受的”是指在免疫学意义上细胞产物的普遍接受,其中不要求患者与细胞的交叉匹配,并且不需要免疫抑制。为了获得这样的细胞,所述方法可以包括例如向人T细胞中转染:(i)编码碱基编辑器融合蛋白的质粒、mRNA或蛋白质,所述碱基编辑器融合蛋白包含与Cas9切口酶结构域融合的脱氨酶结构域,其中所述切口酶结构域包含碱基切除修复抑制剂结构域;(ii)破坏TRAC、B2M和PDCD1中每一种的表达的一种或更多种剪接受体-剪接供体(SA-SD)gRNA;(iii)编码T细胞受体(TCR)和嵌合抗原受体(CAR)的供体DNA模板;以及(iv)两种与靶插入位点互补的gRNA。所述方法还包括在促进破坏被SA-SD gRNA靶向的剪接位点的条件下培养经转染的T细胞,从而相对于未经转染的T细胞,TRAC、B2M和PDCD1基因产物的表达降低;以及在促进在靶插入位点靶向敲入供体DNA模板的条件下培养经转染的T细胞。在该实例中,所得经遗传修饰的T细胞表达CAR/TCR而缺乏TRAC、PDCD1和B2M的表达。在一些情况下,该方法还包括引入被设计用于破坏CTLA-4表达的gRNA。表1中示出了用于编辑基因TRAC、PDCD1和B2M中靶碱基的示例性gRNA的序列。
表1.用于T细胞和CD34+细胞碱基编辑的单指导RNA(sgRNA)
碱基编辑融合蛋白包含与Cas9切口酶结构域或失活Cas9核酸酶结构域(称为死的Cas9或dCas9)融合的脱氨酶结构域,其中,在一些情况下,脱氨酶结构域是载脂蛋白B mRNA编辑复合物(APOBEC)家族脱氨酶。APOBEC胞苷脱氨酶结构域允许进行靶向基因破坏,其中进行胸苷代替胞苷的单碱基替换。
在另一方面,本文提供了用于靶向疾病以进行碱基编辑校正的方法。靶序列可以是本领域中已经确定的任何与疾病相关的多核苷酸或基因。内源基因序列的突变或“校正”的有用应用的实例包括改变疾病相关基因突变、改变编码剪接位点的序列、改变调节序列、改变序列以引起功能获得性突变和/或改变序列以引起功能丧失性突变,以及靶向改变编码蛋白质的结构特征的序列。
在另一方面,本文提供了用于使用碱基编辑来获得切割抗性Fc受体的方法。例如,在一些情况下,碱基编辑酶的诱变结构域被用于引入突变,其产生具有更高亲和力的FCγRIIIa(IgG受体IIIa的Fc片段)基因产物(也称为CD16a)。因此,该方法可以包括向自然杀伤细胞中引入用于碱基编辑的组分(例如BE3、BE4、gRNA、供体模板),以在CD16a中引入增强NK细胞的抗体依赖性细胞介导的细胞毒性(ADCC)的突变。参照图18A-18D,根据本文提供的方法的碱基编辑通过将CD16a修饰成切割抗性形式成功修饰了CD3
在一些情况下,使用本文所述的方法对细胞进行遗传修饰以使细胞表达嵌合抗原受体(CAR)和/或T细胞受体(TCR)将是有利的。“嵌合抗原受体(CAR)”有时称为“嵌合受体”、“T体(T-body)”或“嵌合免疫受体(CIR)”。如本文所用,术语“嵌合抗原受体(CAR)”是指人工构建的杂合蛋白或多肽,其包含与跨膜结构域和至少一个细胞内结构域可操作地连接的抗体的细胞外抗原结合结构域(例如,单链可变片段(scFv))。通常,CAR的抗原结合结构域对在目标靶细胞表面上表达的特定抗原具有特异性。例如,可以对T细胞进行改造以表达对B细胞淋巴瘤上CD19具有特异性的CAR。对于不受供体匹配限制的同种异体抗肿瘤细胞治疗剂,可以对细胞进行改造,以敲入编码CAR的核酸,但也敲除负责供体匹配的基因(TCR和HLA标志物)。
如本文所用,术语“经遗传修饰的”和“经遗传改造的”可互换使用,并且是指包含外源多核苷酸(与用于插入的方法无关)的原核或真核细胞。在一些情况下,效应细胞已被修饰以包含非天然存在的核酸分子,其已人为产生或修饰(例如,使用重组DNA技术)或源自这样的分子(例如,通过转录、翻译等)。包含外源的、重组的、合成的和/或以其他方式修饰的多核苷酸的效应细胞被认为是经改造细胞。
在一些实施方案中,可以将包括碱基编辑器和指导分子的组分体外、离体或体内递送至细胞。在一些情况下,采用病毒或质粒载体系统递送本文所述的碱基编辑组分。优选地,所述载体是病毒载体,例如慢病毒或杆状病毒或优选腺病毒/腺相关病毒(AAV)载体,但是其他递送方式是已知的(例如酵母系统、微囊泡、基因枪/将载体附接到金纳米粒的方式)并且在考虑之中。在某些实施方案中,将编码gRNA和碱基编辑器融合蛋白的核酸包装在一种或更多种病毒递送载体中用于递送至细胞。合适的病毒递送载体包括但不限于腺病毒/腺相关病毒(AAV)载体、慢病毒载体。在一些情况下,可以使用本领域已知的非病毒转移方法将核酸或蛋白质引入哺乳动物细胞中。核酸和蛋白质可以与可药用载剂一起递送,或例如封装在脂质体中。其他递送方式是已知的(例如酵母系统、微囊泡、基因枪/将载体附接到金纳米粒的方式)并且在考虑之中。在一些情况下,对细胞进行电穿孔以摄取gRNA和碱基编辑器(例如BE3、BE4、ABE)。在一些情况下,在通过电穿孔引入gRNA、碱基编辑器和载体之后,通过向培养基添加病毒上清液来将DNA供体模板以腺相关病毒6型(AAV6)载体进行递送。
插入或缺失(indel)形成率可以通过适当的方法确定。例如,Sanger测序或下一代测序(NGS)可用于检测indel形成率。优选地,接触导致在碱基编辑之后少于20%的脱靶indel形成。接触导致在碱基编辑之后至少2∶1的预期产物:意外产物。
可用于本文提供的方法的细胞可以是新鲜分离的原代细胞或获自原代细胞培养物的冷冻等分试样。在一些情况下,对细胞进行电穿孔以摄取gRNA和碱基编辑融合蛋白。如以下实施例中所述,用于一些测定(例如,对于T细胞)的电穿孔条件可以包括1400伏,10毫秒的脉冲宽度,3个脉冲。电穿孔之后,使经电穿孔的T细胞在细胞培养基中恢复,然后在T细胞扩增培养基中培养。在一些情况下,使经电穿孔的细胞在细胞培养基中恢复约5至约30分钟(例如,约5、10、15、20、25、30分钟)。优选地,恢复细胞培养基不含抗生素或其他选择剂。在一些情况下,T细胞扩增培养基是完全CTS OpTmizer T细胞扩增培养基。
如本文所用,术语“核酸”和“核酸分子”是指包含核碱基和酸性部分的化合物,例如核苷、核苷酸或核苷酸的聚合物。通常,聚合核酸(例如包含三个或更多个核苷酸的核酸分子)是线性分子,其中相邻的核苷酸通过磷酸二酯键联彼此连接。在一些实施方案中,“核酸”是指单个核酸残基(例如核苷酸和/或核苷)。在一些实施方案中,“核酸”是指包含三个或更多个单独核苷酸残基的寡核苷酸链。如本文所用,术语“寡核苷酸”和“多核苷酸”可以互换使用,以指核苷酸的聚合物(例如,至少三个核苷酸的串)。在一些实施方案中,“核酸”涵盖RNA以及单链和/或双链DNA。核酸可以是天然存在的,例如在基因组、转录本、mRNA、tRNA、rRNA、siRNA、snRNA、质粒、黏粒、染色体、染色单体或其他天然存在的核酸分子的情况下。另一方面,核酸分子可以是非天然存在的分子,例如重组DNA或RNA、人工染色体、经改造基因组、或其片段,或者合成DNA、RNA、DNA/RNA杂合体,或者包含非天然存在的核苷酸或核苷。此外,术语“核酸”、“DNA”、“RNA”和/或类似术语包括核酸类似物,即除磷酸二酯骨架以外还具有其他的类似物。核酸可以从天然来源中纯化、使用重组表达系统产生并且任选地纯化、化学合成等等。在适当时,例如在化学合成分子的情况下,核酸可以包含核苷类似物,例如具有经化学修饰的碱基或糖、和骨架修饰的类似物。除非另有说明,否则核酸序列以5’至3’方向呈现。在一些实施方案中,核酸是或包含天然核苷(例如腺苷、胸苷、鸟苷、胞苷、尿苷、脱氧腺苷、脱氧胸苷、脱氧鸟苷和脱氧胞苷);核苷类似物(例如2-氨基腺苷、2-硫代胸苷、肌苷、吡咯并嘧啶、3-甲基腺苷、5-甲基胞苷、2-氨基腺苷、C5-溴尿苷、C5-氟尿苷、C5-碘尿苷、C5-丙炔基-尿苷、C5-丙炔基-胞苷、C5-甲基胞苷、2-氨基腺苷、7-脱氮腺苷、7-脱氮鸟苷、8-氧代腺苷、8-氧代鸟苷、O(6)-甲基鸟嘌呤和2-硫代胞苷);经化学修饰的碱基;经生物修饰的碱基(例如甲基化碱基);插入的碱基;经修饰的糖(例如2’-氟核糖、核糖、2’-脱氧核糖、阿拉伯糖和己糖);和/或经修饰的磷酸基团(例如硫代磷酸酯和5’-N-亚磷酰胺键联)。
术语“蛋白质”、“肽”和“多肽”在本文中可互换使用,并且是指通过肽(酰胺)键连接在一起的氨基酸残基的聚合物。该术语是指任何大小、结构或功能的蛋白质、肽或多肽。通常,蛋白质、肽或多肽的长度为至少三个氨基酸。蛋白质、肽或多肽可以指单个蛋白质或蛋白质集合。蛋白质、肽或多肽中的一个或更多个氨基酸可以被修饰,例如,通过添加化学实体,例如碳水化合物基团,羟基,磷酸基团,法呢基,异法呢基,脂肪酸基团,用于缀合、官能化或其他修饰的接头等进行修饰。蛋白质、肽或多肽还可以是单分子或可以是多分子复合物。蛋白质、肽或多肽可以仅仅是天然存在的蛋白质或肽的片段。蛋白质、肽或多肽可以是天然存在的、重组的或合成的,或其任意组合。蛋白质可以包含不同的结构域,例如核酸结合结构域和核酸切割结构域。在一些实施方案中,蛋白质包含蛋白质性部分(例如构成核酸结合结构域的氨基酸序列)和有机化合物(例如可以充当核酸切割剂的化合物)。
在解释本公开内容时,应以与上下文一致的尽可能广泛的方式解释所有术语。应当理解,在本公开内容中描述的本发明的某些改编对于本领域技术人员而言是常规优化问题,并且可以在不脱离本发明的精神或所附权利要求书的范围的情况下实施。
如本文所使用的,术语“合成的”和“经改造的”可互换使用,并且是指已经被人工操纵的方面。
为了使本文提供的组合物和方法可更容易理解,定义了某些术语:
除非上下文另外明确指出,否则本说明书和所附权利要求书中所使用的未用数量词限定的名词包括一个/种和/或更多个/种。除非另有说明,否则本文中对“或”的任何引用旨在涵盖“和/或”。
如本文所用,术语“包含”与“包括”或“含有”同义且是包括性的或开放式的,并且不排除另外的未记载的成员、要素或方法步骤。本文所用的措词和术语是出于描述的目的,并且不应被视为限制。“包括”、“包含”、“具有”、“含有”、“涉及”及其变化形式的使用旨在涵盖其后列出的项目和另外的项目。被提及为“包含”某些要素的实施方案也被构想为“基本上由这些要素组成”和“由这些要素组成”。权利要求书中使用序数术语例如“第一”、“第二”、“第三”等来修饰权利要求要素本身并不表示一个权利要求要素相对于另一个的任何优先级、优先序或顺序或者执行方法的动作的时间顺序。序数术语仅用作标记,以区分具有某名称的一个权利要求要素与具有相同名称(但使用了序数术语)的另一要素,从而区分权利要求要素。
如本文所用,“修饰”一个或更多个靶核酸序列是指改变(一个或更多个)靶核酸序列的全部或一部分,并且包括切割、引入(插入)、替换和/或缺失(去除)靶核酸序列的全部或一部分。可以使用本文提供的方法对靶核酸序列的全部或一部分进行完全或部分修饰。例如,对靶核酸序列进行修饰包括用一个或更多个核苷酸(例如,外源核酸序列)替换靶核酸序列的全部或一部分,或者去除或缺失靶核酸序列的全部或一部分(例如一个或更多个核苷酸)。对一个或更多个靶核酸序列进行修饰还包括将一个或更多个核苷酸(例如外源序列)引入或插入到一个或更多个靶核酸序列中(内)。
术语“约”和“大约”通常是指在考虑到测量的性质或精确度的情况下测量的量的可接受的误差度。通常,示例性误差度在给定值或值范围的10%内,并且优选5%内。或者,并且特别是在生物系统中,术语“约”和“大约”可以意指在给定值的数量级内,优选5倍内,更优选2倍内的值。除非另有说明,否则本文给出的数值量是近似的,这意味着当没有明确说明时,可以推断出术语“约”或“大约”。
除非另有定义,否则本文使用的所有技术术语具有与本发明所属领域的普通技术人员通常所理解的相同含义。除非上下文另外明确指出,否则本文和权利要求书中所使用的未用数量词限定的名词包括一个/种和/或更多个/种。因此,例如,提及“试剂”包括单一试剂和多种这样的试剂。除非另有说明,否则本文中对“或”的任何引用旨在涵盖“和/或”。
现在在以下非限制性实施例中描述根据本发明的组合物和方法的多个示例性实施方案。提供实施例仅用于举例说明目的,而无意以任何方式限制本发明的范围。实际上,本文中示出和描述的那些之外的各种修改根据前述描述和以下实施例对于本领域技术人员而言将明显并且落入所附权利要求书的范围内。
本部分展示了成功使用第三代和第四代碱基编辑器敲除原代人淋巴造血细胞中的四个不同基因,效率高达80%。已证明对于基因敲除,剪接位点破坏比提前终止密码子更有效。通过后代碱基编辑器的Cas9切口酶功能,在施用AAV6载体以用于供体模板递送之后,还以高至70%的效率实现了靶向基因敲入。总体而言,本文所述的测定和结果证明了增强基于经改造T细胞的免疫治疗的安全性和效力二者的改进的多重基因编辑平台。
先前已经使用碱基编辑在小鼠和哺乳动物细胞中诱导提前终止(pmSTOP)密码子用于基因敲除
为了评估pmSTOP引入和剪接位点破坏二者的性能,我们设计了一组单指导RNA(sgRNA)以在PDCD1、TRAC和B2M中将氨基酸密码子转换为pmSTOP或破坏剪接供体(SD)和剪接受体(SA)序列(图2A、2E、2I;表1)。通过电穿孔将单一sgRNA作为经化学修饰的RNA寡核苷酸
首先,我们通过设计八种sgRNA来靶向检查点基因PDCD1(也称为PD-1);预测其中三种将引入pmSTOP密码子,两种靶向破坏SD位点(GT:
根据我们PDCD1结果提供的信息,我们设计了靶向TRAC的sgRNA的聚焦组(图3E)。在此,我们发现在外显子1 SD位点(BE3为47.6±4.6%;BE4为60.0±11.3%)和外显子3 SA位点(BE3为40.3±9.7%;BE4为62.3±11.0%)的C-T转换最高,其中在每个靶标处BE4表现出比BE3更高的编辑率(图3F)。在外显子3中的两个pmSTOP候选位点也观察到有效编辑,但是效率低于任一剪接位点破坏sgRNA(图3F)。外显子1 SD位点和外显子3 SA位点二者以相似的频率编辑,但外显子3 SA位点的破坏导致最高的TCR破坏率,如通过细胞表面CD3表达的损失测量的(BE3为69±15.3%;BE4为83.7±5.8%)(图3G)。
接下来,我们使用类似的策略靶向了B2M(图3I)。与靶向外显子1 SD位点的sgRNA一起递送BE4 mRNA显示出靶碱基的最有效C-T转换(BE3为58.3±2.5%;BE4为70.3±3.2%)(图3J),导致B2M蛋白的有效敲除(BE3为79.1±1.3%;BE4为80.0±3.2%)(图3K)。我们还在外显子2中鉴定了候选pmSTOP位点,其导致相对有效的C-T编辑(BE3为43.3±5.7%;BE4为55.7±5.0%)和蛋白质敲除(BE3为56.2±5.1%;BE4为61.5±1.8%)(图3J,3K)。值得注意的是,靶向非编码外显子3的SA位点产生了有效的C-T编辑,但未导致可检测的蛋白质表达降低(图3J,3K)。
已经报道了BE3的非靶编辑(即C-A或C-G)
为了我们验证可用于产生功能增强的同种异体“现成”T细胞的多重编辑策略的目标,我们将我们用于每种基因的最好sgRNA与第一代BE3或BE4 mRNA共递送。令人惊讶地,当以多重设置递送时,对于BE3和BE4二者而言,在每个靶标处的敲除效率均大幅降低(图9)。为了确定降低的编辑效率是否是由于低蛋白质水平,我们将等剂量的BE3、BE4和核酸酶活性酿脓链球菌(Streptococcus pyogenes)Cas9(SpCas9)mRNA递送至T细胞,并且在电穿孔之后24小时时测量了蛋白质表达。令人惊讶地,尽管在这些时间点可容易检测到SpCas9蛋白表达,但是检测不到BE3和BE4蛋白(图10)。为了解决这个问题,我们首先以在我们初始多重实验中使用的剂量(1.5μg)的2倍高的剂量(3μg)递送BE3和BE4mRNA。该策略改善了每个基因座处的编辑效率,但是效率仍然低于在我们的单基因靶向实验中观察到的效率(图4A)。
在这些实验的过程中,出现了独立的报告,鉴定了与使用第一代BE3和BE4表达载体有关的问题,其严重降低了人细胞中的转录和翻译效率二者
通过流式细胞术分析了每个靶基因的多重蛋白质敲除,并且蛋白质损失的频率与遗传编辑频率很好地相关(图4D,r=0.90,df=28,P=1.3e-11)。BE4 RNP展示出比第一代BE3和BE4 mRNA更有效的蛋白质敲除,而coBE4 mRNA最有效,在低和高mRNA剂量二者下每个基因都具有超过90%的蛋白质损失(图4D;图11)。多重编辑的一个关键考虑因素是携带基因敲除的每种潜在组合的细胞的所得比例。为了在我们的实验中更好地理解这种现象,我们通过流式细胞术同时评价了所有靶基因的蛋白质表达,并且使用SPICE分析确定不具有敲除、具有单基因敲除、具有双基因敲除或具有三基因敲除的单独细胞的比例;以及这些级分中每一个中损失的蛋白质的组合(图4E)。虽然第一代BE4 mRNA产生了具有敲除表型的多样组合的终点细胞群,但三重敲除细胞的频率较低(21.9±1.1%)。使用BE4 RNP(68.6±0.37%),三重敲除细胞的比例明显更高,并且利用1.5μg(86.6±3.75%)和4μg(89.57±4.2%)的coBE4 mRNA时甚至进一步提高(图4F)。
脱靶(OT)DSB诱导是核酸酶平台面临的一个重要挑战
表2计算预测的候选脱靶位点
还已经报道了核酸酶介导的多重编辑在人T细胞中产生不希望的易位(translocation)
接下来,我们试图确定使用我们的碱基编辑策略产生的多重敲除T细胞是否保留细胞因子功能以及在配备有CAR时能够介导靶细胞杀伤。我们通过分析分化标志物
随着我们更好地了解成功的基于细胞的免疫治疗和基因治疗的要求,以及随着对产生通用、同种异体细胞的热情增长,高度多重基因编辑可能会变得更加普遍。然而,有充分的文献证明,DSB是可导致基因组不稳定和细胞死亡的毒性损伤
意想不到的是,我们发现与由转染mRNA表达的coBE4相比在使用BE4 RNP时非靶编辑率和indel形成率均更高。这一观察结果可能是由于与直接BE4蛋白递送相对,当由稳定mRNA以高水平表达时实现了延长BE4保留时间。由于已经表明甚至游离UGI也会在BE3的情况下降低indel频率和非靶编辑二者
在我们当前的研究中,我们利用CD19特异性CAR的慢病毒递送,这是CAR-T治疗中的当前行业标准。但是,这种方法有很多缺点,包括插入诱变、可变CAR表达和基因沉默的风险
使用碱基编辑器和靶向核酸酶之间的一个显著差异是来自编辑事件的潜在结果的数量。核酸酶介导的DSB通过高度可变的非同源末端连接(NHEJ)途径进行修复,导致一系列indel;其中一些不会引入移码突变,并且因此对基因表达和功能的意义未知。作为替代地,即使考虑非靶编辑时,我们的碱基编辑方法的结果数量也有限,所有都导致天然剪接供体或受体的功能损失。然而,重要的是要考虑到天然剪接位点的破坏可并不总是导致无功能的产物,因为选择性剪接或隐蔽剪接可维持基因的生物学功能。
使用横跨sgRNA靶位点的小ddPCR扩增子(>200bp)进行的易位分析表明,利用最佳试剂进行碱基编辑几乎消除了可检测的易位,而Cas9核酸酶产生了许多易位,有些频率高达1.5%。值得注意的是,通过对来自SpCas9处理细胞的亚克隆连接PCR扩增子(约500bp)进行Sanger测序,还在易位位点鉴定出了较大的缺失(图17A-17B)。这些数据表明存在类似于先前报道的基因组重排
尽管我们证明BE4与SpCas9核酸酶相比大幅降低了DSB诱导并且不产生可检测的易位,但仍存在发生不期望事件的可能性。例如,BE4的rAPOBEC1可能在DNA复制过程中非特异性地编辑单链DNA中的胞嘧啶
方法
克隆和病毒产生:通过T2A与RQR8连接的CD19嵌合抗原受体的DNA序列作为gBlock基因片段合成(Integrated DNA Technologies[IDT])。将片段吉布森组装
指导RNA设计:使用碱基编辑剪接位点破坏sgRNA设计程序SpliceR(可从万维网上的z.umn.edu/splicer获得)设计指导RNA(sgRNA)[Kluesner&Lahr et al.,在准备中]。SpliceR用R统计学编程语言(v.3.4.3)编写。简单地说,SpliceR将靶Ensembl转录本ID、碱基编辑器PAM变体和物种作为输入。该程序使用来自Ensembl的外显子和内含子序列基于用户指定的窗口提取每个剪接位点周围的区域。然后将N
CD3+T细胞分离:使用基于氯化铵的红细胞裂解从Trima Accel去白系统(leukoreduction system,LRS)室中分离外周血单个核细胞(PBMC)。使用EasySep人T细胞分离试剂盒(STEMCELL Technologies)通过免疫磁性负选择从PBMC群体中分离出CD3+T细胞。将T细胞以每1mL Cryostor CS10(STEMCELL Technologies)10-20x10
T细胞培养:在37℃和5%CO
T细胞电穿孔:48小时之后,磁力去除Dynabeads,将细胞用PBS洗涤一次,然后重悬于合适的电穿孔缓冲液中。对于单重实验,在以下条件下使用Neon转染系统(ThermoFisher)在10μL尖端中用1μg经化学修饰的sgRNA(Synthego,Menlo Park,CA)和1.5μg SpCas9、BE3或BE4mRNA(TriLink Biotechnologies)对3x10
慢病毒转导:在纤维连接蛋白(Retronectin)(Takara)包被的板上通过旋转转染(spinfection)用MOI为20的pRRL-MND-CAR19-RQR8慢病毒载体(UMN Viral Vector&Cloning Core)转染之后24小时,T细胞被转导。
基因组DNA分析:电穿孔之后5天,通过基于旋转柱的纯化从T细胞分离基因组DNA。通过CRISPR靶向基因座的PCR扩增、PCR扩增子的Sanger测序以及随后使用如前所述的webapp EditR(可在万维网上的baseeditr.com获得)
下一代测序和分析:设计具有Nextera通用引物衔接子(Illumina)的引物,以使用Primer3Plus扩增目标区域周围的375-425bp位点(表2)。使用高保真AccuPrime Taq DNA聚合酶根据制造商的方案(Invitrogen)使用循环[94℃-2:00]-30x[94℃-0:30,55℃-0:30,68℃-0:30]-[68℃-5:00]-[4℃-保持]对基因组DNA进行PCR扩增。使用QIAquick凝胶提取试剂盒(Qiagen)从1%琼脂糖凝胶中纯化扩增子。将样品提交给明尼苏达大学基因组学中心(University of Minnesota Genomics Center)用于随后利用索引引物进行扩增以及在MiSeq 2x300bp运行(Illumina)上进行测序。每个靶上位点产生最少1,000个对齐的读序对(read-pairs),而脱靶位点产生至少10,000个读序对。如先前所述
流式细胞术:在流式细胞术之前,如上所述,使用CD3/CD28Dynabeads将单重PDCD1破坏T细胞重新刺激48小时。在用TRAC敲除进行的多重实验中,将T细胞用佛波醇12-豆蔻酸酯13-乙酸酯(PMA;100ng/mL;Sigma-Aldrich)和离子霉素(250ng/mL;MilliporeSigma)活化24小时。将用PMA/离子霉素处理的T细胞用PBS洗涤,重悬于培养基中,并在流式细胞术之前再孵育24小时。将T细胞用荧光团缀合的抗人CD3(BD Biosciences)、B2M(BioLegend)和CD279(PD-1)(BioLegend)抗体染色。使用抗人CD34单克隆抗体(QBEnd10)(ThermoFisher)检测CD19-T2A-RQR8CAR表达
F’
细胞因子谱分析:简单地说,在不存在或存在K562细胞或Raji细胞或经改造以过表达PDL1的Raji细胞的情况下,将2×10
易位测定:使用2种引物+探针qPCR的设置(表3),使用PrimerQuest软件(Integrated DNA Technologies,Coralville IA)设计了易位PCR测定。每个样品以由内部参考引物+探针组(HEX)和实验引物+探针组(FAM)组成的双重测定运行。引物和探针订购自IDT。使用ddPCR Supermix for Probes(无dUTP)(Biorad,Hercules,CA)和每次测定200ng基因组DNA根据制造商的说明设置反应。使用QX200微滴式数字PCR系统(Bio-Rad)产生并分析微滴。
细胞毒性测定:将表达萤光素酶的K562、Raji或Raji-PDL1细胞接种到96孔圆底板(3x10
免疫印迹测定:在具有蛋白酶和磷酸酶抑制剂(Sigma-Aldrich,COEDTAF-RO、P5726和P0044)的完全RIPA缓冲液中,从1x10
数据分析和可视化:所有统计学分析均在R studio中进行。显著性水平设定为α=0.05。在进行统计学检验之前,对数据进行正态性和同方差性(homeodascity)假设的分析。如文中所述,使用Student配对单尾t检验或双尾t检验。使用Prism 8(Graphpad)或RStudio,使用多种tidyverse(可从万维网上的www.tidyverse.org/获得)和Bioconductor(可从万维网上的www.bioconductor.org/)包可视化数据。
数据可用性:下一代测序读序将在公布前存放在NCBI序列读序存档数据库(NCBISequence Read Archive database)中。
表3.易位ddPCR引物和探针序列
方法
外周血单个核细胞(PBMC)的分离:将外周血用冷却的1X PBS以3∶1稀释。将经稀释的血液滴加到15mL的Lymphoprep(Stem Cell Technologies)之上。在没有制动的情况下将细胞以400×g离心25分钟。移出血沉棕黄层并用冷却的1X PBS洗涤,并以400×g离心10分钟。去除上清液,并将细胞作为PBMC冷冻或立即用于纯化NK细胞。
CD3
CD3-CD56+NK细胞的刺激:对CD3-CD56+NK细胞进行计数并以1.25 x 10
流式细胞术:将细胞用冷却的1X PBS+0.5%FBS洗涤,并用抗人CD3(eBioscience)和抗人CD56(Miltenyi Biotec)染色。使用LSR Fortessa(BD Biosciences)和FlowJo(Treestar)分析细胞。
NK细胞的Neon电穿孔:使用Neon转染试剂盒(Invitrogen)转染经刺激的NK细胞。对细胞进行计数并以3 x 10
材料
表4.CD16指导RNA序列
表5.引物序列
B0培养基:60%mL DMEM 30%mL Ham’s F12
10%mL人AB血清
100U/mL青霉素;
100μg/mL链霉素
20μM 2-巯基乙醇
50μM乙醇胺
10μg/mL抗坏血酸
1.6ng/mL亚硒酸钠
冷冻介质:CryoStor CS10
细胞分离试剂:人NK细胞分离试剂盒(Stem Cell Technologies),人NK细胞富集试剂盒(Stem Cell Technologies)
电穿孔试剂:Neon 10μL转染试剂盒(Invitrogen)
材料和方法
培养基:StemSpan无血清扩增培养基II(Serum Free Expansion Media II,SFEMII)(Stem Cell Technologies目录号#09605);100ng/ml hSCF(Peprotech);100ng/mlhTPO(Peprotech);100ng/ml hFlt-3L(Peprotech);100ng/ml hIL-6(Peprotech);StemRegenin1(0.75μM终浓度)Cayman Chemical
冷冻介质:Cryostor CS10
细胞分离试剂:人UCB CD34+富集试剂盒(Stem Cell Technologies;根据来源,可以有多种变化)。
其他试剂:Neon试剂盒(Thermo Fisher Scientific,根据量和期望的尖端尺寸,有多种选择)。
解冻样品:使用与用于培养的相同类型的培养基,在预热的培养基(37℃)中解冻细胞。将1mL培养基添加到无菌的15mL锥形管。将冷冻的小瓶在37℃水浴中解冻,直到剩下单个冰晶。将小瓶立即移至生物安全柜,喷洒70%乙醇,然后擦拭。小心打开小瓶。将细胞悬液小心地从一个小瓶逐滴吸移到15ml锥形管中。再滴加1ml培养基,并轻轻涡旋。滴加另外1ml培养基,并轻轻涡旋。再添加4ml培养基并轻轻混合。以175g离心10分钟。更高的离心力将导致细胞死亡。吸出上清液,并将细胞沉淀物悬浮在培养基中。对细胞进行计数并测试或放置在培养物中。重要的是不要延迟使细胞进入培养基和培养箱中。
CD34
CD34
对于使用所有mRNA的敲除:100μl尖端:15μg Cas9 mRNA,10-20μg gRNA-RNA;10μl尖端:1.5μg Cas9 mRNA,1-2μg gRNA-RNA。
在转染之后,将细胞以3000个细胞/μl的密度平板接种在含有5%CO
在恢复期之后,将2倍体积的含抗生素培养基添加到孔。将细胞在37℃下在5%CO
CD34+细胞的rAAV转导:将rAAV在冰上解冻并充分混合,然后添加到细胞。在电穿孔之后的以下时间点以指定的MOI添加。
对于Cas9 mRNA编辑的细胞:在电穿孔之后4-6小时添加病毒。
对于Cas9蛋白(RNP):在电穿孔之后15分钟添加病毒。
电穿孔后扩增:电穿孔后观察到的培养基颜色作为培养基添加的指示。时机将根据特定实验/供体基于细胞的健康状况而有所不同。当培养基的颜色开始变成橙色时(在一些情况下早至48小时),使用含有2X浓度细胞因子的培养基(“2X培养基”)将培养基的体积加倍。在整个培养过程中,根据需要继续进行此过程。
在一些情况下,如果细胞生长非常迅速(特别是在第7-9天左右)并且培养基很快耗尽,则将细胞离心并在2-3倍体积的1X培养基中进行重构。
在细胞生长差并且经过3-4天而无需培养基加倍的情况下,通过从顶部移液小心地除去约50%的培养基。小心不要扰乱沉淀在瓶底的细胞。移除的培养基替换为等体积的2X培养基。
方法
细胞培养:将来自健康患者或患有范科尼贫血或MPS1的患者的原代成纤维细胞以每ml 1 x 10
原代成纤维细胞的转染:在第-1天,将90-100%汇合度的成纤维细胞以12孔板每孔3 x 10
基因组分析:基因组DNA分析:通过基于旋转柱的纯化从成纤维细胞中分离基因组DNA。通过CRISPR靶向基因座的PCR扩增、PCR扩增子的Sanger测序以及随后使用如前所述的web app EditR(baseeditr.com)对Sanger测序迹线进行分析,在基因组水平上分析碱基编辑效率。
材料
表6.FANCA sgRNA序列
表7.引物序列
结果:为了确定碱基编辑是否可用于校正引起范科尼贫血的FANCA基因中的致病性突变,我们将BE4maX mRNA和cm-sgRNA引入原代成纤维细胞以试图校正突变。我们发现,我们能够实现将致病性突变19%校正成WT等位基因(图21A)。未来的实验将评估这种校正是否会导致WT表型的恢复,尤其是关于交联DNA的DNA修复,以及重新编程为诱导性多能干细胞(iPSC)的能力。为了确定是否可以在原代范科尼贫血成纤维细胞中进行腺嘌呤脱氨酶碱基编辑,我们将ABE7.10 mRNA和cm-sgRNA一起引入原代成纤维细胞。我们发现,我们能够实现靶位点的49%编辑,这表明对原代细胞进行了高效率碱基编辑(图21B)。在该位点的高效率碱基编辑表明使用碱基编辑作为单核苷酸编辑方法来代替HDR的可行性。未来的工作将使用腺苷脱氨酶和胞苷脱氨酶碱基编辑器来校正来自多种患有遗传疾病的患者的原代成纤维细胞中的致病性突变,目的是产生iPSC分化为造血干细胞以进行自体基因校正和移植。
参考文献
1.Porter,D.L.,Levine,B.L.,Kalos,M.,Bagg,A.&June,C.H.Chimeric antigenreceptor-modified T cells in chronic lymphoid leukemia.N.Engl.J.Med.365,725-733(2011).
2.Kochenderfer,J.N.,Dudley,M.E.,Feldman,S.A.,Wilson,W.H.,Spaner,D.E.,Maric,I.,Stetler-Stevenson,M.,Phan,G.Q.,Hughes,M.S.,Sherry,R.M.,Yang,J.C.,Kammula,U.S.,Devillier,L.,Carpenter,R.,Nathan,D.-A.N.,Morgan,R.A.,Laurencot,C.&Rosenberg,S.A.B-cell depletion and remissions of malignancy along withcytokine-associated toxicity in a clinical trial of anti-CD19 chimeric-antigen-receptor-transduced T cells.Blood blood-2011-10-384388(2011).
3.Qasim,W.,Zhan,H.,Samarasinghe,S.,Adams,S.,Amrolia,P.,Stafford,S.,Butler,K.,Rivat,C.,Wright,G.,Somana,K.,Ghorashian,S.,Pinner,D.,Ahsan,G.,Gilmour,K.,Lucchini,G.,Inglott,S.,Mifsud,W.,Chiesa,R.,Peggs,K.S.,Chan,L.,Farzeneh,F.,Thrasher,A.J.,Vora,A.,Pule,M.&Veys,P.Molecular remission ofinfant B-ALL after infusion of universal TALEN gene-edited CAR Tcells.Sci.Transl.Med.9,(2017).
4.Osborn,M.J.,Webber,B.R.,Knipping,F.,Lonetree,C.L.,Tennis,N.,DeFeo,A.P.,McElroy,A.N.,Starker,C.G.,Lee,C.,Merkel,S.,Lund,T.C.,Kelly-Spratt,K.S.,Jensen,M.C.,Voytas,D.F.,von Kalle,C.,Schmidt,M.,Gabriel,R.,Hippen,K.L.,Miller,J.S.,Scharenberg,A.M.,Tolar,J.&Blazar,B.R.Evaluation of TCR GeneEditing Achieved by TALENs,CRISPR/Cas9,and megaTAL Nucleases.Mol.Ther.24,570-581(2016).
5.Ren,J.,Liu,X.,Fang,C.,Jiang,S.,June,C.H.&Zhao,Y.Multiplex GenomeEditing to Generate Universal CAR T Cells Resistant toPD1Inhibition.Clin.Cancer Res.23,2255-2266(2017).
6.Provasi,E.,Genovese,P.,Lombardo,A.,Magnani,Z.,Liu,P.-Q.,Reik,A.,Chu,V.,Paschon,D.E.,Zhang,L.,Kuball,J.,Camisa,B.,Bondanza,A.,Casorati,G.,Ponzoni,M.,Ciceri,F.,Bordignon,C.,Greenberg,P.D.,Holmes,M.C.,Gregory,P.D.,Naldini,L.&Bonini,C.Editing T cell specificity towards leukemia by zincfinger nucleases and lentiviral gene transfer.Nat.Med.18,807-815(2012).
7.Ihry,R.J.,Worringer,K.A.,Salick,M.R.,Frias,E.,Ho,D.,Theriault,K.,Kommineni,S.,Chen,J.,Sondey,M.,Ye,C.,Randhawa,R.,Kulkarni,T.,Yang,Z.,McAllister,G.,Russ,C.,Reece-Hoyes,J.,Forrester,W.,Hoffman,G.R.,Dolmetsch,R.&Kaykas,A.p53inhibits CRISPR-Cas9 engineering in human pluripotent stemcells.Nat.Med.24,939-946(2018).
8.Haapaniemi,E.,Botla,S.,Persson,J.,Schmierer,B.&Taipale,J.CRISPR-Cas9 genome editing induces a p53-mediated DNA damage response.Nat.Med.24,927-930(2018).
9.Kosicki,M.,Tomberg,K.&Bradley,A.Repair of double-strand breaksinduced by CRISPR-Cas9leads to large deletions and complex rearrangements.Nat.Biotechnol.36,765(2018).
10.Shin,H.Y.,Wang,C.,Lee,H.K.,Yoo,K.H.,Zeng,X.,Kuhns,T.,Yang,C.M.,Mohr,T.,Liu,C.&Hennighausen,L.CRISPR/Cas9 targeting events cause complexdeletions and insertions at 17 sites in the mouse genome.Nat Commun 8:15464.(2017).
11.Chapman,J.R.,Taylor,M.R.G.&Boulton,S.J.Playing the end game:DNAdouble-strand break repair pathway choice.Mol.Cell 47,497-510(2012).
12.Khanna,K.K.&Jackson,S.P.DNA double-strand breaks:signaling,repairand the cancer connection.Nat.Genet.27,247-254(2001).
13.Komor,A.C.,Kim,Y.B.,Packer,M.S.,Zuris,J.A.&Liu,D.R.Programmableediting of a target base in genomic DNA without double-stranded DNAcleavage.Nature 533,420-424(2016).
14.Gaudelli,N.M.,Komor,A.C.,Rees,H.A.,Packer,M.S.,Badran,A.H.,Bryson,D.I.&Liu,D.R.Programmable base editing of A·T to G·C in genomic DNA withoutDNA cleavage.Nature 551,464-471(2017).
15.Billon,P.,Bryant,E.E.,Joseph,S.A.,Nambiar,T.S.,Hayward,S.B.,Rothstein,R.&Ciccia,A.CRISPR-Mediated Base Editing Enables EfficientDisruption of Eukaryotic Genes through Induction of STOP Codons.Mol.Cell 67,1068-1079.e4(2017).
16.Kuscu,C.,Parlak,M.,Tufan,T.,Yang,J.,Szlachta,K.,Wei,X.,Mammadov,R.&Adli,M.CRISPR-STOP:gene silencing through base-editing-induced nonsensemutations.Nat.Methods 14,710-712(2017).
17.Zafra,M.P.,Schatoff,E.M.,Katti,A.,Foronda,M.,Breinig,M.,Schweitzer,A.Y.,Simon,A.,Han,T.,Goswami,S.,Montgomery,E.,Thibado,J.,Kastenhuber,E.R.,
18.Liu,Z.,Lu,Z.,Yang,G.,Huang,S.,Li,G.,Feng,S.,Liu,Y.,Li,J.,Yu,W.,Zhang,Y.,Chen,J.,Sun,Q.&Huang,X.Efficient generation of mouse models of humandiseases via ABE-and BE-mediated base editing.Nat.Commun.9,2338(2018).
19.Loughran,G.,Chou,M.-Y.,Ivanov,I.P.,Jungreis,I.,Kellis,M.,Kiran,A.M.,Baranov,P.V.&Atkins,J.F.Evidence of efficient stop codon readthrough infour mammalian genes.Nucleic Acids Res.42,8928-8938(2014).
20.Andreev,D.E,O’Connor,P.B.F.,Zhdanov,A.V.,Dmitriev,R.I.,Shatsky,I.N.,Papkovsky,D.B.&Baranov,P.V.Oxygen and glucose deprivation induceswidespread alterations in mRNA translation within 20 minutes.Genome Biol.16,90(2015).
21.Scotti,M.M.&Swanson,M.S.RNA mis-splicing indisease.Nat.Rev.Genet.17,19-32(2016).
22.Komor,A.C.,Zhao,K.T.,Packer,M.S.,Gaudelli,N.M.,Waterbury,A.L.,Koblan,L.W.,Kim,Y.B.,Badran,A.H.&Liu,D.R.Improved base excision repairinhibition and bacteriophage Mu Gam protein yields C:G-to-T:A base editorswith higher efficiency and product purity.1-10(2017).
23.Hendel,A.,Bak,R.O.,Clark,J.T.,Kennedy,A.B.,Ryan,D.E.,Roy,S.,Steinfeld,I.,Lunstad,B.D.,Kaiser,R.J.,Wilkens,A.B.,Bacchetta,R.,Tsalenko,A.,Dellinger,D.,Bruhn,L.&Porteus,M.H.Chemically modified guide RNAs enhanceCRISPR-Cas genome editing in human primary cells.Nat.Biotechnol.33,985-989(2015).
24.Kluesner,M.G.,Nedveck,D.A.,Lahr,W.S.,Garbe,J.R.,Abrahante,J.E.,Webber,B.R.&Moriarity,B.S.EditR:A Method to Quantify Base Editing from SangerSequencing.The CRISPR Journal 1,239-250(2018).
25.Koblan,L.W.,Doman,J.L.,Wilson,C.,Levy,J.M.,Tay,T.,Newby,G.A.,Maianti,J.P.,Raguram,A.&Liu,D.R.Improving cytidine and adenine base editorsby expression optimization and ancestral reconstruction.Nat.Biotechnol.(2018).doi:10.1038/nbt.4172
26.Tsai,S.Q.,Zheng,Z.,Nguyen,N.T.,Liebers,M.,Topkar,V.V.,Thapar,V.,Wyvekens,N.,Khayter,C.,Iafrate,A.J.,Le,L.P.,Aryee,M.J.&Joung,J.K.GUIDE-seqenables genome-wide profiling of off-target cleavage by CRISPR-Casnucleases.Nat.Biotechnol.33,187-197(2015).
27.Mahnke,Y.D.,Brodie,T.M.,Sallusto,F.,Roederer,M.&Lugli,E.The who’swho of T-cell differentiation:Human memory T-cell subsets.Eur.J.Immunol.43,2797-2809(2013).
28.Wang,L.,Xue,W.,Yan,L.,Li,X.,Wei,J.,Chen,M.,Wu,J.,Yang,B.,Yang,L.&Chen,J.Enhanced base editing by co-expression of free uracil DNA glycosylaseinhibitor.Cell Res.27,1289-1292(2017).
29.von Kalle,C.,Deichmann,A.&Schmidt,M.Vector integration andtumorigenesis.Hum.Gene Ther.25,475-481(2014).
30.Eyquem,J.,Mansilla-Soto,J.,Giavridis,T.,van der Stegen,S.J.C.,Hamieh,M.,Cunanan,K.M.,Odak,A.,
31.Ellis,J.Silencing and variegation of gammaretrovirus andlentivirus vectors.Hum.Gene Ther.16,1241-1246(2005).
32.Kim,Y.B.,Komor,A.C.,Levy,J.M.,Packer,M.S.,Zhao,K.T.&Liu,D.R.Increasing the genome-targeting scope and precision of base editing withengineered Cas9-cytidine deaminase fusions.Nat.Biotechnol.35,371-376(2017).
33.Hirano,H.,Gootenberg,J.S.,Horii,T.,Abudayyeh,O.O.,Kimura,M.,Hsu,P.D.,Nakane,T.,Ishitani,R.,Hatada,I.,Zhang,F.,Nishimasu,H.&Nureki,O.Structureand Engineering of Francisella novicida Cas9.Cell 164,950-961(2016).
34.Ran,F.A.,Hsu,P.D.,Lin,C.-Y.,Gootenberg,J.S.,Konermann,S.,Trevino,A.E.,Scott,D.A.,Inoue,A.,Matoba,S.,Zhang,Y.&Zhang,F.Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity.Cell 154,1380-1389(2013).
35.Hoopes,J.I.,Cortez,L.M.,Mertz,T.M.,Malc,E.P.,Mieczkowski,P.A.&Roberts,S.A.APOBEC3A and APOBEC3B Preferentially Deaminate the Lagging StrandTemplate during DNA Replication.Cell Rep.14,1273-1282(2016).
36.Lewis,C.A.,Jr,Crayle,J.,Zhou,S.,Swanstrom,R.&Wolfenden,R.Cytosinedeamination and the precipitous decline of spontaneous mutation during Earth’s history.Proc.Natl.Acad.Sci.U.S.A.113,8194-8199(2016).
37.Gibson,D.G.Enzymatic assembly of overlapping DNA fragments.MethodsEnzymol.498,349-361(2011).
38.Wang,X.,Tilford,C.,Neuhaus,I.,Mintier,G.,Guo,Q.,Feder,J.N.&Kirov,S.CRISPR-DAV:CRISPR NGS data analysis and visualizationpipeline.Bioinformatics 33,3811-3812(2017).
39.Philip,B.,Kokalaki,E.,Mekkaoui,L.,Thomas,S.,Straathof,K.,Flutter,B.,Marin,V.,Marafioti,T.,Chakraverty,R.,Linch,D.,Quezada,S.A.,Peggs,K.S.&Pule,M.A highly compact epitope-based marker/suicide gene for easier andsafer T-cell therapy.Blood 124,1277-1287(2014).
本领域技术人员认识到或仅使用常规实验就能够确定本文所述的本发明的特定实施方案的许多等同方案。这样的等同方案旨在由所附权利要求书涵盖。
本文公开的所有参考文献(包括专利文件)通过引用整体并入。
- 使用Cas9碱基编辑器的淋巴造血改造
- 腺苷脱氨酶碱基编辑器及使用其修饰靶标序列中的核碱基的方法