一种分离制备矮牵牛素-3-O-(6-O-对香豆酰)葡萄糖苷的方法
文献发布时间:2023-06-19 09:47:53
技术领域
本发明涉及天然产物的分离纯化技术领域,具体涉及一种分离制备矮牵牛素-3-O-(6-O-对香豆酰)葡萄糖苷的方法。
背景技术
葡萄是葡萄科葡萄属植物的果实,在世界各地均有栽培。葡萄具有很高的营养价值。中医认为葡萄性平、味甘,能补气血,强心利尿。现代研究表明,葡萄中不仅有维生素B1、维生素B2、维生素C、维生素E等维生素,还含有白藜芦醇,原花青素,花色苷等丰富的多酚类化合物,具有抗氧化,保护心血管,抗肿瘤,抗菌等生物活性。
花色苷是葡萄中重要的多酚类物质之一,研究表明,葡萄中含有飞燕草素、锦葵素、芍药素和矮牵牛素等花青素苷元与葡萄糖结合形成的花色苷。近年来的研究证实了天然果蔬来源的花色苷具有抗氧化、抗肿瘤、控制肥胖和预防心血管疾病等生物活性。但由于花色苷对光、温度和pH敏感,导致其化学性质相对不稳定,而且生物利用度较低,这些性质极大地限制了花色苷的实际应用。已有研究表明,相对于非酰化花色苷,酰化花色苷同样具有生物活性,并且有更稳定的化学性质以及更高的生物利用度。而葡萄中含有大量对香豆酰酰化的花色苷,这说明葡萄花色苷具有更好的市场前景。
近年来,固相萃取技术(SPE)、制备型高效液相色谱技术(preparative-HPLC)和高速逆流色谱技术(HSCCC)等新型纯化技术开始得到发展和应用。其中逆流色谱是一种液液色谱技术,其固定相和流动相都是液态的,原理是根据不同化合物分子在固定相和流动相的分配系数的差异而分离。逆流色谱法的分离效果,主要取决于使用的两相溶剂体系是否合适。逆流色谱技术具有样品前处理简单、适用范围广、样品损失少、处理量大等优点。而固相萃取技术是使用固体吸附剂吸附样品中的目标化合物,与样品的基体和其他干扰化合物分离,然后用洗脱液洗脱,从而能富集和分离出目标化合物。固相萃取法具有节省时间、溶剂用量少、易放大生产等优点。目前在葡萄花色苷的制备中,主要采用萃取、大孔树脂和单一的柱层析或色谱技术进行分离纯化。
如公开号CN104177460A的中国专利文献中公开了一种3,5-二糖类花色苷的制备方法,该方法使用超声辅助提取、萃取、纯化等步骤,其中使用了优化的超声辅助提取方法减少了提取剂的使用,采用凝胶柱和大孔树脂结合的方法,简化了操作。但获得的产物为花色苷混合物,含有三种不同的二糖类花色苷,并且产物不涉及酰化花色苷。
又如公开号CN102229633A的中国专利文献中公开了一种从葡萄皮中分离制备五种高纯度花色苷单体的方法。该方法使用浸提,大孔树脂纯化,制备液相的方法纯化得到了五种花色苷,然而由于使用了两步制备液相纯化,导致样品损耗,降低了纯化得率,且其中两种酰化花色苷(锦葵素乙酰化葡萄糖苷,锦葵素反式香豆酰化葡萄糖苷)的纯度相对较低,分别仅为91.7%和95.5%。
公开号CN108976268A的中国专利文献中公开了一种制备刺葡萄两个主要花色苷标准品的方法,该方法采用大孔树脂对刺葡萄浊汁进行吸附富集,随后经过洗脱,冷冻干燥得到花色苷粗品,再使用高速逆流色谱,以水-正丁醇-甲基叔丁基醚-乙腈-三氟乙酸(体积比5:4:1:2:0.001或5:3:1:1:0.001)为两相溶剂体系分离,得到两种花色苷,纯度分别为95.8%和92.2%。该技术方案虽然公开其分离得到两种花色苷,但是从图1的刺葡萄浊汁HPLC图中,可以看到其花色苷组成简单,由高速逆流色谱的局限性可推测出,当分离的样品花色苷组分复杂时,使用该方法可能难以得到目标花色苷。
由于酰化花色苷的分离纯化困难,导致目前市面上没有商业化的酰化花色苷标准品。因此,研究开发一种从复杂花色苷原料如葡萄中纯化酰化花色苷单体的工艺,对推动花色苷标准品市场及开发葡萄深加工产品具有重要意义。因此,本发明公开了一种基于高速逆流色谱与固相萃取联用的方法,可以实现从花色苷组分复杂的葡萄中,大量制备高纯度的矮牵牛素-3-O-(6-O-对香豆酰)葡萄糖苷单体。
发明内容
针对本领域存在的不足之处,本发明提供了一种分离制备矮牵牛素-3-O-(6-O-对香豆酰)葡萄糖苷的方法,为我国葡萄资源的开发利用提供新的思路。
一种分离制备矮牵牛素-3-O-(6-O-对香豆酰)葡萄糖苷的方法,包括步骤:
(1)醇提浓缩:以葡萄为原料,经醇提浓缩得到葡萄皮花色苷粗提液;
(2)大孔树脂纯化:将所述葡萄皮花色苷粗提液注入大孔树脂,经洗脱,浓缩得到花色苷洗脱液;
(3)萃取:使用有机溶剂萃取所述花色苷洗脱液,再经减压浓缩,冻干后得到花色苷冻干粉;
(4)高速逆流色谱分离:将甲基叔丁基醚、甲醇、水和三氟乙酸混合作为两相溶剂体系,且以上相为固定相,下相为流动相,依次将固定相和流动相泵入高速逆流色谱仪器中,两相在管路中达到平衡后,将花色苷冻干粉用流动相溶解,进样并在紫外检测器下检测,检测波长为280nm,收集保留时间为100-120min的组分并减压浓缩后,得到花色苷单体粗品溶液;
(5)固相萃取柱纯化:将花色苷单体粗品溶液注入固相萃取柱中,使用乙腈体积百分浓度为0-40%的酸性乙腈溶液进行梯度洗脱,收集乙腈体积百分浓度为28%-32%的酸性乙腈洗脱液,再经减压浓缩、冻干,得到目标化合物矮牵牛素-3-O-(6-O-对香豆酰)葡萄糖苷;
所述花色苷单体粗品溶液的浓度为5-20mg/mL,进样量为2-10mL;
所述酸性乙腈溶液中的酸选自盐酸、甲酸、乙酸、草酸中的至少一种。
如无特殊说明,本发明中出现的原料百分比均指体积百分浓度,本发明中出现的各种溶液,如无特别说明,均以水作为溶剂。
步骤(1)中,所述醇提浓缩,具体为:将葡萄洗净取皮,与酸性乙醇溶液混合,混匀后打浆,50℃以下(优选室温)超声提取,过滤,滤液在40-50℃减压浓缩除去乙醇,得到葡萄皮花色苷粗提液;
葡萄皮与酸性乙醇溶液的料液比为1g:4-8mL;
所述酸性乙醇溶液中,乙醇体积浓度为50%-80%,优选60%-70%,酸体积浓度为0.1%-1%;
所述超声提取时间为40-120min。
步骤(1)中,所述酸性乙醇溶液中,酸选自盐酸、甲酸、乙酸、草酸中的至少一种。
步骤(2)中,所述大孔树脂纯化方法具体为:
将葡萄皮花色苷粗提液注入大孔树脂中,之后分别用乙醇体积浓度为0,5%,20%,40%的酸性乙醇溶液各4倍柱体积(4BV)洗脱,收集乙醇体积浓度为40%的酸性乙醇洗脱液,40-50℃减压蒸发除去乙醇,得到花色苷洗脱液;
所述大孔树脂选自AB-8,D101,XAD-7,HPD-100或DM-130,其比表面积为450-550m
所述酸性乙醇溶液选自酸的体积百分浓度为0.1%~1.5%的乙醇溶液,其中酸选自盐酸、甲酸、乙酸中的至少一种。
步骤(3)中,所述有机溶剂为乙酸乙酯。
步骤(3)中,优选以所述有机溶剂与花色苷洗脱液体积比1:1的比例萃取,并萃取2次以上。
步骤(4)中,所述两相溶剂体系中,甲基叔丁基醚、甲醇、水和三氟乙酸的体积比为2:2:3:0.001。
步骤(4)中,高速逆流色谱仪器温度稳定在20-30℃,正接正转,泵入固定相,之后调节转速至800-950r/min,以3mL/min的流速通入流动相并使之平衡,每次进样量以花色苷冻干粉计为10-50mg。
步骤(5)中,采用C18固相萃取柱,分别使用乙腈体积百分浓度为0、4%、8%、12%、16%、20%、24%、28%、32%、36%、40%的酸性乙腈溶液各2倍柱进行梯度洗脱;
所述酸性乙腈溶液中,酸体积百分浓度为0.1%~2%。
本发明与现有技术相比,主要优点包括:
1、首次建立了从葡萄皮中分离矮牵牛素-3-O-(6-O-对香豆酰)葡萄糖苷(分子结构如图1所示)的方法,产率可达到不低于8mg/kg葡萄皮,纯度可达到不低于98%。
2、通过将高速逆流色谱和固相萃取柱联用,能够在多酚组分复杂的葡萄原料中,大批量地制备得到矮牵牛素-3-O-(6-O-对香豆酰)葡萄糖苷,有处理量大、重复性好等优点,便于实现工业化生产。
附图说明
图1为矮牵牛素-3-O-(6-O-对香豆酰)葡萄糖苷的分子结构图;
图2为实施例1中,葡萄皮花色苷粗提液的高效液相色谱图;
图3为实施例1中,经大孔树脂分离纯化后,含有矮牵牛素-3-O-(6-O-对香豆酰)葡萄糖苷部分的高效液相色谱图;
图4为实施例1中的高速逆流色谱图;
图5为实施例1中,经逆流色谱分离后,含有矮牵牛素-3-O-(6-O-对香豆酰)葡萄糖苷部分的高效液相色谱图;
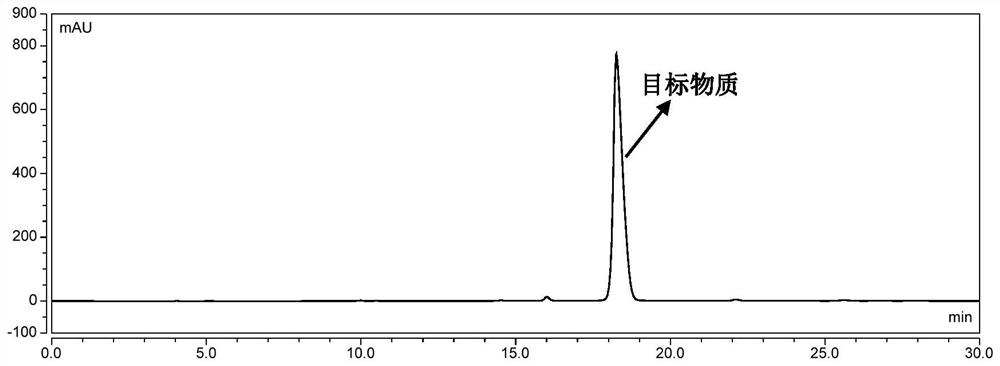
图6为实施例1中,最终产物矮牵牛素-3-O-(6-O-对香豆酰)葡萄糖苷的高效液相色谱图;
图7为实施例1中,矮牵牛素-3-O-(6-O-对香豆酰)葡萄糖苷的一级质谱图和二级质谱图;
图8为对比例1中最终产物的液相色谱图;
图9为对比例3中最终产物的液相色谱图。
具体实施方式
下面结合附图及具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的操作方法,通常按照常规条件,或按照制造厂商所建议的条件。
实施例1
将葡萄洗净剥皮,得到1kg葡萄皮,按照料液比1g:6mL的比例加入含0.5%(v/v)盐酸的70%的乙醇溶液充分混合,超声提取60min,(控制温度在50℃以下,避光),纱布过滤,滤液在4000rpm下离心10min,取上清液。滤渣按相同的方法再提取1次。将滤液合并,使用布氏漏斗再过滤一次。滤液于45℃下减压蒸发除去乙醇并浓缩,得到葡萄皮花色苷粗提液。葡萄皮花色苷粗提液的高效液相色谱图如图2所示。
将AB-8大孔树脂装入层析柱,并依次使用乙醇,0.5mol/L的盐酸溶液,0.5mol/L的氢氧化钠溶液,水清洗后,将花色苷粗提液以0.2BV/h的流速注入层析柱中。上样后,依次使用酸水(含0.5%盐酸),5%、20%、40%的酸性乙醇(含0.5%盐酸)各4倍柱体积洗脱,收集40%酸性乙醇洗脱液,减压蒸发除去乙醇。然后以1:1的比例,使用乙酸乙酯萃取2遍,取水相,适当减压浓缩,冻干后,得到花色苷粗提物冻干粉。经大孔树脂分离纯化后,含有矮牵牛素-3-O-(6-O-对香豆酰)葡萄糖苷部分的高效液相色谱图如图3所示。
将甲基叔丁基醚:甲醇:水:三氟乙酸按2:2:3:0.001的体积比置于分液漏斗中,充分摇匀,静置30min后,将上下相分离,分别超声脱气30min。使高速逆流色谱系统的仪器温度稳定在20℃,泵入固定相,之后调节转速至850r/min,正接正转,以3mL/min的流速通入流动相直到平衡,以每3mg冻干粉溶于1mL流动相的比例溶解花色苷冻干粉,使用微孔滤膜过滤后进样,单次进样10mL,并在紫外检测器下检测,检测波长为280nm。收集100-120min的组分(如图4)并减压浓缩,得到矮牵牛素-3-O-(6-O-对香豆酰)葡萄糖苷单体粗品溶液。经逆流色谱分离后,含有矮牵牛素-3-O-(6-O-对香豆酰)葡萄糖苷部分的高效液相色谱图如图5所示。
将固相萃取柱用甲醇活化,然后注入纯水置换柱子内的甲醇,然后每次将4mL花色苷单体粗品溶液缓慢注入固相萃取柱中,再分别使用含0.5%(v/v)盐酸的0、4%、8%、12%、16%、20%、24%、28%、32%、36%、40%的乙腈溶液各2倍柱体积进行梯度洗脱,收集乙腈体积百分浓度为28%-32%的洗脱液。之后经减压浓缩,冻干,即得到9.2mg目标化合物矮牵牛素-3-O-(6-O-对香豆酰)葡萄糖苷,高效液相色谱图如图6所示,HPLC纯度为99.1%。
将制得花色苷样品在质谱仪中进样,根据质谱图对样品进行分析(图7),确认分离得到的花色苷质量数正常。
实施例2
将葡萄洗净剥皮,得到4kg葡萄皮,按照料液比1g:6mL的比例加入含0.5%(v/v)盐酸的70%的乙醇溶液充分混合,超声提取90min,(控制温度在50℃以下,避光),纱布过滤,滤液在4000rpm下离心10min,取上清液。滤渣按相同的方法再提取1次。将滤液合并,使用布氏漏斗再过滤一次。滤液于45℃下减压蒸发除去乙醇并浓缩,得到葡萄皮花色苷粗提液。
将AB-8大孔树脂装入层析柱,并依次使用乙醇,0.5mol/L的盐酸溶液,0.5mol/L的氢氧化钠溶液,水清洗后,将花色苷粗提液以0.2BV/h的流速注入层析柱中。上样后,依次使用酸水(含0.5%盐酸),5%、20%、40%的酸性乙醇(含0.5%盐酸)各4倍柱体积洗脱,收集40%酸性乙醇洗脱液,减压蒸发除去乙醇。然后以1:1的比例,使用乙酸乙酯萃取1遍,取水相,适当减压浓缩,冻干后,得到花色苷粗提物冻干粉。
将甲基叔丁基醚:甲醇:水:三氟乙酸按2:2:3:0.001的体积比置于分液漏斗中,充分摇匀,静置30min后,将上下相分离,分别超声脱气30min。使高速逆流色谱系统的仪器温度稳定在20℃,泵入固定相,之后调节转速至850r/min,正接正转,以3mL/min的流速通入流动相直到平衡,以每5mg冻干粉溶于1mL流动相的比例溶解花色苷冻干粉,使用微孔滤膜过滤后进样,单次进样10mL,并在紫外检测器下检测,检测波长为280nm。收集100-120min的组分并减压浓缩,得到矮牵牛素-3-O-(6-O-对香豆酰)葡萄糖苷单体粗品溶液。
将固相萃取柱用甲醇活化,然后注入纯水置换柱子内的甲醇,然后每次将4mL花色苷单体粗品溶液缓慢注入固相萃取柱中,再分别使用含0.5%(v/v)盐酸的0、4%、8%、12%、16%、20%、24%、28%、32%、36%、40%的乙腈溶液各2倍柱体积进行梯度洗脱,收集乙腈体积百分浓度为28%-32%的洗脱液。之后经减压浓缩,冻干,即得到40mg目标化合物矮牵牛素-3-O-(6-O-对香豆酰)葡萄糖苷,HPLC纯度为98.1%。
实施例3
将葡萄洗净剥皮,得到10kg葡萄皮,按照料液比1g:6mL的比例加入含0.5%(v/v)盐酸的70%的乙醇溶液充分混合,超声提取60min,(控制温度在50℃以下,避光),纱布过滤,滤液在4000rpm下离心10min,取上清液。滤渣按相同的方法再提取1次。将滤液合并,使用布氏漏斗再过滤一次。滤液于45℃下减压蒸发除去乙醇并浓缩,得到葡萄皮花色苷粗提液。
将AB-8大孔树脂装入层析柱,并依次使用乙醇,0.5mol/L的盐酸溶液,0.5mol/L的氢氧化钠溶液,水清洗后,将花色苷粗提液以0.2BV/h的流速注入层析柱中。上样后,依次使用酸水(含0.5%盐酸),5%、20%、40%的酸性乙醇(含0.5%盐酸)各4倍柱体积洗脱,收集40%酸性乙醇洗脱液,减压蒸发除去乙醇。然后以1:1的比例,使用乙酸乙酯萃取2遍,取水相,适当减压浓缩,冻干后,得到花色苷粗提物冻干粉。
将甲基叔丁基醚:甲醇:水:三氟乙酸按2:2:3:0.001的体积比置于分液漏斗中,充分摇匀,静置30min后,将上下相分离,分别超声脱气30min。使高速逆流色谱系统的仪器温度稳定在20℃,泵入固定相,之后调节转速至850r/min,正接正转,以3mL/min的流速通入流动相直到平衡,以每5mg冻干粉溶于1mL流动相的比例溶解花色苷冻干粉,使用微孔滤膜过滤后进样,单次进样10mL,并在紫外检测器下检测,检测波长为280nm。收集100-120min组分并减压浓缩,得到矮牵牛素-3-O-(6-O-对香豆酰)葡萄糖苷单体粗品溶液。
将固相萃取柱用甲醇活化,然后注入纯水置换柱子内的甲醇,然后每次将6mL花色苷单体粗品溶液缓慢注入固相萃取柱中,再分别使用含0.5%(v/v)盐酸的0、4%、8%、12%、16%、20%、24%、28%、32%、36%、40%的乙腈溶液各2倍柱体积进行梯度洗脱,收集乙腈体积百分浓度为28%-32%的洗脱液。之后经减压浓缩,冻干,即得到95mg目标化合物矮牵牛素-3-O-(6-O-对香豆酰)葡萄糖苷,HPLC纯度为98.6%。
对比例1
与实施例1的区别仅在于使用10kg葡萄皮,且固相萃取纯化过程每次使用15mL花色苷单体粗品溶液,其他步骤不变,最终得到87mg目标化合物矮牵牛素-3-O-(6-O-对香豆酰)葡萄糖苷,由于固相萃取纯化过程进样量过大,HPLC纯度仅为92.2%(图8)。
对比例2
对比于实施例1,去掉固相萃取柱的步骤,其他步骤不变,最终产物的高效液相色谱图如图5所示。可知,该对比例只能得到含矮牵牛素-3-O-(6-O-对香豆酰)葡萄糖苷的混合物,无法得到矮牵牛素-3-O-(6-O-对香豆酰)葡萄糖苷单体。
对比例3
制备工艺与实施例1的相比,区别仅在于将高速逆流色谱分离的溶剂体系替换为正丁醇:甲基叔丁基醚:甲醇:水:三氟乙酸按2:2:1:5:0.001的体系。经测试,目标化合物矮牵牛素-3-O-(6-O-对香豆酰)葡萄糖苷无法在100-120min时间段内收集到,并且包含目标化合物的部分含有大量杂质(如图9)。
对比例4
制备工艺与实施例1的相比,区别仅在于再固相萃取柱的收集范围,若不收集乙腈体积百分浓度28%-32%的洗脱液,则无法得到矮牵牛素-3-O-(6-O-对香豆酰)葡萄糖苷单体;若收集范围大于28-32%的范围,则分离得到的矮牵牛素-3-O-(6-O-对香豆酰)葡萄糖苷单体的纯度低于90%。
此外应理解,在阅读了本发明的上述描述内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本申请所附权利要求书所限定的范围。
- 一种分离制备矮牵牛素-3-O-(6-O-对香豆酰)葡萄糖苷的方法
- 一种芍药素-3-O-(6-O-对香豆酰)葡萄糖苷-5-葡萄糖苷的分离制备方法