一种催化氢胺化反应制备2-甲基吡咯烷化合物的合成方法
文献发布时间:2023-06-19 10:13:22
技术领域
本发明涉及有机合成化学技术领域,具体为一种催化氢胺化反应制备2-甲基吡咯烷化合物的合成方法。
背景技术
氮杂环化合物是非常重要的有机化合物,而且大多数具有较强的生物活性,其大量存在于天然产物中,尤其是在抗肿瘤药物的合成中具有极其重要的意义((a)Pinder,R.A.Nat.Prod.Rep.1992,9,17-23.(b)Ricci,A.,Amino Group Chemistry:FromSynthesis to the Life Sciences,1st ed.,Wiley-VCH,Weinheim,2007)。因此,发展新颖高效、环境友好的含氮合物合成方法在有机化学、生物化学和药物化学中的均具有重要意义和应用价值。
在各类制备方法中,氢胺化反应是制备这类化合物最直接最有效的手段之一((a)Müller,T.E.;Beller,M.Chem.Rev.1998,98,675-704.(b)Pohlki,F.;Doye,S.“Chem.Soc.Rev.2003,32,104-114.(c)Hultzsch,K.C.Adv.Synth.Catal.2005,347,367-391.(d)Hong,S.;Marks,T.J.Acc.Chem.Res.2004,37,673-686.(e)Müller,T.E.;Hultzsch,K.C.;Yus,M.;Foubelo,F.;Tada,M.Chem.Rev.2008,108,3795-3892.)。这类反应可以从易得的起始原料出发,经一步反应直接得到目标化合物。与传统的合成方法相比,氢胺化反应有很多优点,例如原子经济性为100%,符合绿色化学理念;过对催化剂和配体的调控,可以实现区域选择性和立体选择性,合成手性胺类化合物。
但由于氮原子的孤对电子和烯烃上富电子的π体系存在电子排斥,反应的熵变为负值,以及N-H键对碳碳不饱和键的[2+2]环加成是轨道禁止的,这些内在因素使得氢胺化反应在无催化剂存在下无法进行。在早期研究过程中,这类反应所使用的催化剂包括碱金属和碱土金属化合物。这些强碱与胺作用产生强亲核性的活性中间体,对碳碳双键发生加成得到相应的氢胺化反应产物。近年来镧系金属化合物和第IVB族过渡金属化合在氢胺化反应中得到了较大的发展,同时后过渡金属化合物在碳碳不饱和键的氢胺化反应中也给出了较好的结果。一般来说,镧系及第IVB族金属催化剂对空气和水高度敏感,同时,反应物中存在的其它极性官能团对反应结果的影响很大,这些局限性在一定程度上限制了它们在有机合成中的使用。后过渡金属催化剂比镧系金属化合物稳定,但大多数研究所使用的都是金、钯和铂等贵金属,同时这类催化剂只对某些特定的底物有效,也在某种程度上限制了它们在有机合成中的使用。
有鉴于此,开发简单易行有效的催化氢胺化反应制备2-甲基吡咯烷化合物具有重要意义,可以为制备一些高附加值的氮杂环化合物提供一条简洁高效、清洁绿色的途径。这也是本发明得以完成的基础和动力所在。
发明内容
本发明的目的在于提供一种催化氢胺化反应制备2-甲基吡咯烷化合物的合成方法,以解决上述背景技术中提出的问题。
为实现上述目的,本发明提供如下技术方案:一种催化氢胺化反应制备2-甲基吡咯烷化合物的合成方法,包括以下步骤:
A、在反应器中依次加入化合物1、KI和KHSO
B、在敞口条件及温度条件下将上述化合物1、KI和KHSO
C、反应结束后减压蒸除溶剂得粗产物;
D、经柱层析提纯得到2-甲基吡咯烷化合物2。
优选的,所述步骤(A)中,反应器为圆底烧瓶,通过化学反应制备得到了2-甲基吡咯烷化合物,其反应方程式如下:
式中化合物1为各类4-戊烯磺酰胺。
优选的,所述步骤(A)中,化合物1、KI和KHSO
优选的,所述步骤(B)中,在敞口条件及室温条件下将化合物1、KI和KHSO
优选的,所述步骤(C)中,溶剂为丙酮与水的混合溶剂、或乙腈与水的混合溶剂中的一种。
优选的,所述步骤(D)中,柱层析提纯所用的洗脱液为石油醚与乙酸乙酯的混合溶剂,所述石油醚:乙酸乙酯的体积比为(2~20):1。
与现有技术相比,本发明的有益效果是:本发明制备方法以4-戊烯磺酰胺原料,以KI和KHSO
附图说明
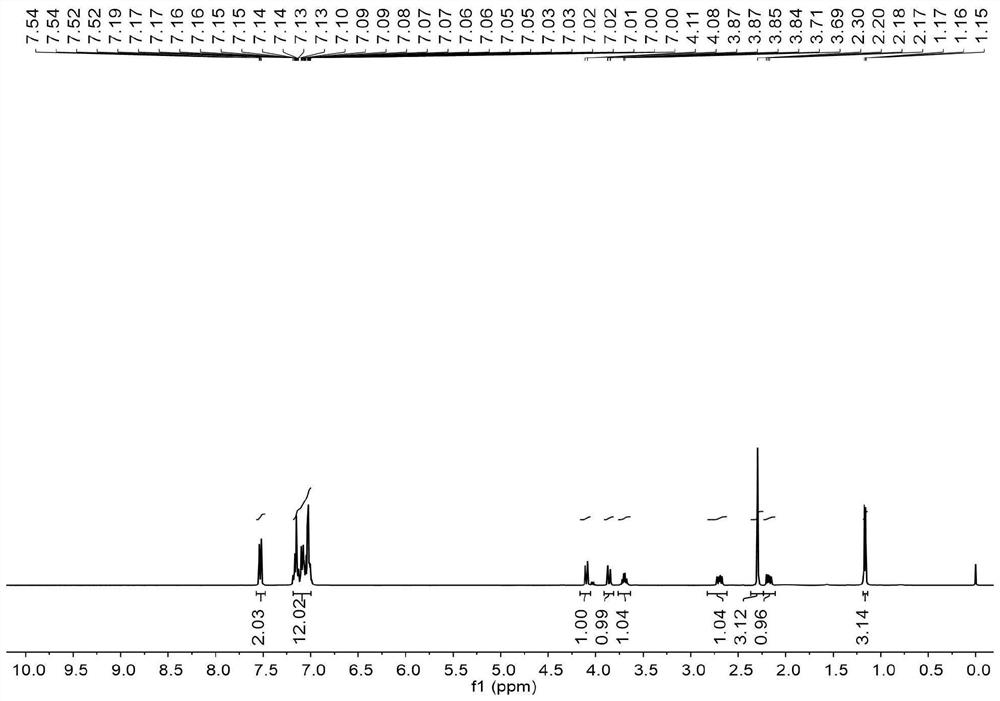
图1为本发明实施例1所得产物的氢谱图;
图2为本发明实施例1所得产物的碳谱图;
图3为本发明实施例2所得产物的氢谱图;
图4为本发明实施例2所得产物的碳谱图;
图5为本发明实施例3所得产物的氢谱图;
图6为本发明实施例3所得产物的碳谱图;
图7为本发明实施例4所得产物的氢谱图;
图8为本发明实施例4所得产物的碳谱图;
图9为本发明实施例5所得产物的氢谱图;
图10为本发明实施例5所得产物的碳谱图;
图11为本发明实施例6所得产物的氢谱图;
图12为本发明实施例6所得产物的碳谱图;
图13为本发明实施例7所得产物的氢谱图;
图14为本发明实施例7所得产物的碳谱图;
图15为本发明实施例8所得产物的氢谱图;
图16为本发明实施例8所得产物的碳谱图;
图17为本发明实施例9所得产物的氢谱图;
图18为本发明实施例9所得产物的碳谱图;
图19为本发明实施例10所得产物的氢谱图;
图20为本发明实施例10所得产物的碳谱图。
具体实施方式
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
请参阅图1-20,本发明提供一种技术方案:一种催化氢胺化反应制备2-甲基吡咯烷化合物的合成方法,包括以下步骤:
A、在反应器中依次加入化合物1、KI和KHSO
B、敞口条件下,在一定温度条件下进行将上述化合物1、KI和KHSO
C、反应结束后减压蒸除溶剂得粗产物;
D、经柱层析提纯得到2-甲基吡咯烷化合物2。
一种以4-戊烯磺酰胺1为原料,KI和KHSO
实施例1
室温下,向装有磁力搅拌子的10毫升圆底烧瓶中装入N-(2,2-二苯基-4-戊烯基)-4-甲基苯磺酰胺(0.2毫摩尔),碘化钾(0.01毫摩尔),硫酸氢钾(0.01毫摩尔),水/丙酮混合溶剂(1/1,2毫升),加毕,敞口条件下室温反应10小时,反应完成后,将有机相通过旋转蒸发器除去溶剂,残余物用硅胶柱进行纯化(硅胶规格为200目~300目,洗脱剂为石油醚/乙酸乙酯=10:1),得到白色固体74毫克,产率为95%,
所得产物核磁图谱数据为:
实施例2
室温下,向装有磁力搅拌子的10毫升圆底烧瓶中装入N-(2,2-二苯基-4-戊烯基)甲磺酰胺(0.2毫摩尔),碘化钾(0.01毫摩尔),硫酸氢钾(0.01毫摩尔),水/丙酮混合溶剂(1/1,2毫升),加毕,敞口条件下室温反应10小时,反应完成后,将有机相通过旋转蒸发器除去溶剂,残余物用硅胶柱进行纯化(硅胶规格为200目~300目,洗脱剂为石油醚/乙酸乙酯=12:1),得到白色固体59毫克,产率为94%,
所得产物核磁图谱数据为:
实施例3
室温下,向装有磁力搅拌子的10毫升圆底烧瓶中装入N-(2,2-二甲基-4-戊烯基)-苯磺酰胺(0.2毫摩尔),碘化钾(0.01毫摩尔),硫酸氢钾(0.01毫摩尔),水/丙酮混合溶剂(1/1,2毫升),加毕,敞口条件下室温反应10小时,反应完成后,将有机相通过旋转蒸发器除去溶剂,残余物用硅胶柱进行纯化(硅胶规格为200目~300目,洗脱剂为石油醚/乙酸乙酯=10:1),得到黄色液体86毫克,产率为94%,
所得产物核磁图谱数据为:
实施例4
室温下,向装有磁力搅拌子的10毫升圆底烧瓶中装入N-(2,2-二甲基-4-戊烯基)-4-硝基苯磺酰胺(0.2毫摩尔),碘化钾(0.01毫摩尔),硫酸氢钾(0.01毫摩尔),水/丙酮混合溶剂(1/1,2毫升),加毕,敞口条件下室温反应10小时,反应完成后,将有机相通过旋转蒸发器除去溶剂,残余物用硅胶柱进行纯化(硅胶规格为200目~300目,洗脱剂为石油醚/乙酸乙酯=8:1),得到黄色固体73毫克,产率为86%,
所得产物核磁图谱数据为:
实施例5
室温下,向装有磁力搅拌子的10毫升圆底烧瓶中装入N-(2,2-二甲基-4-戊烯基)-4-甲基苯磺酰胺(0.2毫摩尔),碘化钾(0.01毫摩尔),硫酸氢钾(0.01毫摩尔),水/丙酮混合溶剂(1/1,2毫升),加毕,敞口条件下室温反应10小时,反应完成后,将有机相通过旋转蒸发器除去溶剂,残余物用硅胶柱进行纯化(硅胶规格为200目~300目,洗脱剂为石油醚/乙酸乙酯=5:1),得到白色固体52毫克,产率为97%,
所得产物核磁图谱数据为:
实施例6
室温下,向装有磁力搅拌子的10毫升圆底烧瓶中装入N-4-戊烯基-4-甲基苯磺酰胺(0.2毫摩尔),碘化钾(0.01毫摩尔),硫酸氢钾(0.01毫摩尔),水/丙酮混合溶剂(1/1,2毫升),加毕,敞口条件下室温反应10小时,反应完成后,将有机相通过旋转蒸发器除去溶剂,残余物用硅胶柱进行纯化(硅胶规格为200目~300目,洗脱剂为石油醚/乙酸乙酯=6:1),得到白色固体43毫克,产率为90%,
所得产物核磁图谱数据为:
实施例7
室温下,向装有磁力搅拌子的10毫升圆底烧瓶中装入N-(2-烯丙基苯基)-4-甲基苯磺酰胺(0.2毫摩尔),碘化钾(0.01毫摩尔),硫酸氢钾(0.01毫摩尔),水/丙酮混合溶剂(1/1,2毫升),加毕,敞口条件下室温反应10小时,反应完成后,将有机相通过旋转蒸发器除去溶剂,残余物用硅胶柱进行纯化(硅胶规格为200目~300目,洗脱剂为石油醚/乙酸乙酯=10:1),得到白色固体53毫克,产率为92%,
所得产物核磁图谱数据为:
实施例8
室温下,向装有磁力搅拌子的10毫升圆底烧瓶中装入N-((1-烯丙基环己基)甲基)-4-甲基苯磺酰胺(0.2毫摩尔),碘化钾(0.01毫摩尔),硫酸氢钾(0.01毫摩尔),水/丙酮混合溶剂(1/1,2毫升),加毕,敞口条件下室温反应10小时,反应完成后,将有机相通过旋转蒸发器除去溶剂,残余物用硅胶柱进行纯化(硅胶规格为200目~300目,洗脱剂为石油醚/乙酸乙酯=10:1),得到无色液体58毫克,产率为95%,
所得产物核磁图谱数据为:
实施例9
室温下,向装有磁力搅拌子的10毫升圆底烧瓶中装入N-(2,2-二烯丙基-4-戊烯基)-4-甲基苯磺酰胺(0.2毫摩尔),碘化钾(0.01毫摩尔),硫酸氢钾(0.01毫摩尔),水/丙酮混合溶剂(1/1,2毫升),加毕,敞口条件下室温反应10小时,反应完成后,将有机相通过旋转蒸发器除去溶剂,残余物用硅胶柱进行纯化(硅胶规格为200目~300目,洗脱剂为石油醚/乙酸乙酯=10:1),得到无色液体61毫克,产率为96%,
所得产物核磁图谱数据为:
实施例10
室温下,向装有磁力搅拌子的10毫升圆底烧瓶中装入N-(2,2-二苯基-4-己烯基)-4-甲基苯磺酰胺(0.2毫摩尔),碘化钾(0.01毫摩尔),硫酸氢钾(0.01毫摩尔),水/丙酮混合溶剂(1/1,2毫升),加毕,敞口条件下室温反应10小时,反应完成后,将有机相通过旋转蒸发器除去溶剂,残余物用硅胶柱进行纯化(硅胶规格为200目~300目,洗脱剂为石油醚/乙酸乙酯=12:1),得到黄色液体73毫克,产率为90%,
所得产物核磁图谱数据为:
综上所述,本发明制备方法以4-戊烯磺酰胺原料,以KI和KHSO
本发明未详述之处,均为本领域技术人员的公知技术。
对于本领域技术人员而言,显然本发明不限于上述示范性实施例的细节,而且在不背离本发明的精神或基本特征的情况下,能够以其他的具体形式实现本发明。因此,无论从哪一点来看,均应将实施例看作是示范性的,而且是非限制性的,本发明的范围由所附权利要求而不是上述说明限定,因此旨在将落在权利要求的等同要件的含义和范围内的所有变化囊括在本发明内。不应将权利要求中的任何附图标记视为限制所涉及的权利要求。
- 一种催化氢胺化反应制备2-甲基吡咯烷化合物的合成方法
- 一种催化氢胺化反应制备2-甲基吡咯烷化合物的合成方法