EGFR基因突变检测系统
文献发布时间:2023-06-19 10:19:37
技术领域
本发明属于医药技术领域,涉及基因突变检测系统,具体涉及EGFR基因突变检测系统。
背景技术
非小细胞肺癌(non-small cell lung cancer,NSCLC)包括鳞状细胞癌、腺癌、大细胞癌,与小细胞癌相比其癌细胞生长分裂较慢,扩散转移相对较晚,非小细胞肺癌约占所有肺癌的80%。表皮生长因子受体(epidermal growth factor receptor,EGFR)是一种跨膜酪氨酸激酶受体,该受体激酶域的激活对癌细胞增殖、生长的相关信号传递具有重要意义。EGFR基因是非小细胞肺癌中最常见的驱动基因之一。突变的EGFR是使用酪氨酸激酶抑制剂(TKIs)作为药物治疗非小细胞肺癌(NSCLC)的治疗靶点。吉非替尼和厄洛替比等TKIs是此类靶向治疗的推荐药物,可以显著延长患者的中位生存时间。大量的临床研究已证实EGFR靶向治疗能显著降低疾病进展或死亡风险、改善患者生活质量。而在EGFR基因未发生突变的肺癌患者中,研究数据提示不宜使用TKIs靶向药物,EGFR基因突变检测是晚期肺癌患者使用EGFR-TKIs治疗的先决条件。EGFR基因外显子21(L858R和L861Q)点突变的患者对TKI敏感,然而,外显子20(T790M)点突变的患者具有TKI抗性。
现有技术中对L858R、L861Q和T790M的点突变的检测通常是采用基因测序的方法,但是这种方法成本高,样品制备、测序反应和数据解释等所需时间长,因此目前的方法不适合常规的临床应用。鉴于上述原因,临床实践中EGFR基因突变的受检率并不高,导致基于驱动基因变异靶点的个体化治疗策略难以实施。因此,开发一种能够快速将非小细胞肺癌患者归类为TKIs药物敏感或耐药的检测设备和方法具有非常高的临床意义。
发明内容
本发明的目的在于提供EGFR基因突变检测系统,该检测系统可以实现对EGFR基因突变的快速且准确的检查,克服了EGFR基因突变检测的检测成本高和检测周期长的缺陷。
为解决上述技术问题,本发明技术方案如下:
EGFR基因突变检测系统,包括用于扩增待测DNA样品的扩增单元、微流控检测设备和用于识别目的DNA片段的杂交单元;所述杂交单元包括探针和洗脱缓冲液,所述探针用于与目的DNA片段杂交,所述探针与目的DNA片段杂交的温度为47℃;所述洗脱缓冲液中含有纳米金颗粒和稳定寡核苷酸。
采用上述技术方案,技术原理以及有益效果如下:
使用扩增单元扩大待测DNA样品中目的基因的拷贝数,便于后续有足够多的的待测DNA用于分子杂交。探针和目的DNA片段发生杂交,识别上述目的DNA片段,从而实现对EGFR基因是否突变以及发生何种突变进行判断。EGFR基因的突变为点突变(SNP),用于检测突变型EGFR基因的探针和用于检测野生型EGFR基因的探针差异较小,又加之目的DNA片段存在较为复杂的二级结构,导致用分子杂交的方法检测EGFR基因突变的假阳性率高,检测准确率低,从而也导致了现在主流的检测EGFR的手段为基因测序,基因测序成本高,且样品制备、测序反应和数据解释等所需时间长,并不适合常规的临床应用。发明人经过长期研究发现,将探针与目的DNA片段杂交的温度调整为47℃,且使用含有纳米金颗粒和稳定寡核苷酸的洗脱缓冲液,来洗脱探针与目的DNA片段杂交后的非目的DNA片段,可极大程度的减少非特异性结合,降低假阳性率以及提高检测准确率。通常来说,DNA分子杂交的温度并不适合采用较高温度,这样会导致碱基之间的氢键不稳定,两条单链DNA不能稳定的结合,这样探针和目的DNA片段也不能稳定结合,从而无法有效识别目的DNA。但是,发明人将杂交温度提升,温度的升高虽然一定程度上会影响探针和目的DNA片段的结合的稳定性,但是使用纳米金颗粒溶液并不能破坏探针和目的DNA片段的结合,目的DNA片段仍然与探针结合以等待后续可视化检测。高温对探针和非目的DNA片段的结合也产生了一定影响,降低了探针和非目的DNA片段的结合的稳定性,通过纳米金颗粒溶液的洗脱可以将大量的非稳定结合的探针和非目的DNA片段分离,减少非特异性结合。而采用普通的PBS是不能很好的将上述非稳定结合的探针和非目的DNA片段分离的。在47℃的杂交温度下,特异性结合的目的DNA与探针结合稳定,不会被AuNP洗脱缓冲液洗脱;非特异性结合的非目的DNA与探针结合不稳定,会被AuNP洗脱缓冲液洗脱,从而提高了DR值,使得检测结果更为准确。因此,本技术方案将高温杂交和纳米金颗粒溶液洗脱相结合,降低了探针和非目的DNA片段的非特异性结合,增加了对EGFR基因的突变检测的准确率。
进一步,所述探针包括用于特异性结合EGFR基因的野生型外显子20的E20_WT探针、用于特异性结合EGFR基因的野生型外显子21的E21_WT探针、用于特异性结合EGFR基因的T790M突变型外显子20的T790M探针、用于特异性结合EGFR基因的L858R突变型外显子21的L858R探针和用于特异性结合EGFR基因的L861Q突变型外显子21的L858R探针。
采用上述技术方案,可实现对野生型和突变型的EGFR基因的检测,从而将非小细胞肺癌患者归类为TKIs药物敏感型或耐药型。EGFR基因外显子21(L858R和L861Q)点突变的患者对TKI敏感,外显子20(T790M)点突变的患者具有TKI抗性,对于非EGFR基因突变的非小细胞肺癌不宜采用TKI药物。如果待测DNA样品中,有单链DNA被L858R探针或L858R探针杂交,则说明待测DNA样品中有EGFR基因突变型外显子21,则该患者为TKIs药物敏感型,可采用TKIs治疗;如果待测DNA样品中,有单链DNA被T790M探针杂交,则说明待测DNA样品中有EGFR基因突变型外显子20,则该患者为TKIs药物耐药型,不宜采用TKIs治疗;如果待测DNA样品中,有单链DNA被E20_WT探针以及E21_WT探针杂交,则该患者为非EGFR基因突变型癌症,不宜采用TKIs治疗。
进一步,所述纳米金颗粒的粒径为5nm,浓度为5nM。
采用上述技术方案,纳米金颗粒的使用可以减少洗脱过程中的链的错配,减少非特异性结合。
进一步,所述稳定寡核苷酸的序列为5’-CGCCAGAGAATACCAAAACTC-3’,浓度为8nM。
采用上述技术方案,稳定寡核苷酸可以避免纳米金颗粒的自聚集。
进一步,所述微流控检测设备包括用于固定所述探针的功能化基板和微流控芯片,所述微流控芯片设有第一表面和与第一表面相对的第二表面,所述第一表面上设置有至少两条不交叉的凹槽,在凹槽的两端设有贯穿第一表面和第二表面的通孔,所述第一表面用于与功能化基板可拆卸地密封接合;所述微流控芯片包括第一微流控芯片和第二微流控芯片,第一微流控芯片先与功能化基板密封接合形成第一流道,第二微流控芯片再与功能化基板密封接合形成第二流道;第一流道和第二流道位于功能化基板的同一表面,第一流道和第二流道的交叉点为检测位点;所述第一流道用于孵育含有探针的溶液,所述第二流道用于孵育含有单链化的待测DNA样品的溶液。
采用上述技术方案,微流控检测设备具有结构简单、便于制备、检测速度快以及可根据实际需求灵活调整上样模式等优点。
进一步,所述探针的5’端连接有氨基,所述功能化基板为表面具有醛基化修饰的载玻片。
采用上述技术方案,探针通过5’端的氨基可结合在醛基化修饰的载玻片,实现探针的固定。
进一步,所述E20_WT探针的序列为NH
所述E21_WT探针的序列为NH
所述T790M探针的序列为NH
所述L858R探针的序列为NH
所述L861Q探针探针的序列为NH
其中,NH
采用上述技术方案,上述探针在特定的杂交温度和洗脱条件下,可以较为特异性地与目的DNA片段结合,实现对基因突变的检测。
进一步,所述扩增单元包括用于扩增EGFR基因的外显子20的第一引物对和用于扩增EGFR基因的外显子21的第二引物对。
采用上述技术方案,原始DNA样品中的DNA拷贝数通常较低,探针不能结合上足够的目的DNA片段,造成假阴性,需要针对外显子20和外显子21进行扩增,增加DNA拷贝数,避免假阴性的产生。
进一步,所述第一引物对包括第一上游引物和第一下游引物,所述第二引物对包括第二上游引物和第二下游引物,所述第一上游引物和第二上游引物的5’端均标记有生物素。
采用上述技术方案,生物素标记可实现定量检测。
进一步,所述第一上游引物的序列为:Bio-5’-AAGCCTACGTGATGGCCAG-3’;
所述第一下游引物的序列为:5’-CTTTGCGATCTGCACACACCAG-3’;
所述第二上游引物的序列为:Bio-5’-GGGCATGAACTACTTGGAGGAC-3’;
所述第二下游引物的序列为:5’-TTTGCCTCCTTCTGCATGGTAT-3’;
其中,Bio表示生物素。
采用上述技术方案,待测DNA样品中的DNA通常不含有标记,后续无法对结合上探针的目的DNA片段进行可视化检测。在上游引物的5’端标记上生物素,可实现对外显子20和外显子21扩增产物的标记,通过生物素-亲和素结合的方式,在目的DNA片段上进行可视化的标记。
进一步,所述杂交单元还包括Cy-5标记的链霉亲和素溶液。
采用上述技术方案,Cy-5是常用的荧光染料,将Cy-5标记在链霉亲和素上的技术成熟,且Cy-5在激发光的作用下,可以产生稳定的荧光信号以供检测。
综上,采用本技术方案的EGFR基因突变检测系统进行基因突变检测的过程如下:
(1)使用标记有生物素的第一引物对和第二引物对分别对原始DNA样品进行基因扩增,获得待测DNA样品,待测DNA样品包括含有大量外显子20拷贝的E20待测DNA样品和含有大量外显子21拷贝的E21待测DNA样品。
(2)将第一微流控芯片密封在醛基化修饰的载玻片上,在形成的第一流道中分别加入探针溶液(E20_WT探针、E21_WT探针、T790M探针、L858R探针和L858R探针分别加入不同的第一流道),经孵育后,移除探针溶液,并用固定缓冲液冲洗第一流道,将第一流道中的液体吸干之后,将第一微流控芯片从载玻片上拆卸。载玻片上形成若干探针条带(非可视化条带),即已经将探针固定在了载玻片上。
(3)将第二微流控芯片密封在载玻片上,第一微流控芯片和第二微流控芯片均位于载玻片的同一面。在形成的第二流道中分别加入经过变性处理的待测DNA样品(第一流道与探针条带形成交叉,交叉点为检测位点)。待测DNA样品中的E20待测DNA样品和E21待测DNA样品经变性处理后,分别加入不同的第二流道。经孵育后(即高温杂交,每个检测位点上形成探针与DNA片段的杂合体,即杂合DNA双链),使用含有纳米金颗粒和稳定寡核苷酸的洗脱缓冲液冲洗第二流道。然后将第二流道中的液体吸干之后,在第二流道中加入Cy-5标记的链霉亲和素溶液,链霉亲和素与生物素结合,将Cy-5标记在探针与DNA片段的杂合体上。将第二流道中的液体吸干之后,将第二微流控芯片从载玻片上拆卸,获得待检测的载玻片。
(4)对待检测的载玻片进行荧光信号的检测,获得每个位点的荧光信号值。检测位点被分为5种:检测位点A(固定有E20_WT探针,并使用E20待测DNA样品)、检测位点B(固定有E21_WT探针,并使用E21待测DNA样品)、检测位点C(固定有T790M探针,并使用E20待测DNA样品)、检测位点D(固定有L858R探针,并使用E21待测DNA样品),检测位点E(固定有L858R探针,并使用E21待测DNA样品)。
结果判定原则:
如果检测位点D或检测位点E的荧光信号超过阈值,或者检测位点D和检测位点E的荧光信号同时超过阈值,且检测位点A荧光信号超过阈值,其他位点的荧光信号未超过阈值,则说明原始DNA样品中有EGFR基因突变型外显子21,则该患者为TKIs药物敏感型,可采用TKIs治疗。
如果检测位点C的荧光信号超过阈值,且检测位点B荧光信号超过阈值,其他位点的荧光信号未超过阈值,则说明原始DNA样品中有EGFR基因突变型外显子20,则该患者为TKIs药物耐药型,不宜采用TKIs治疗。
如果检测位点A的荧光信号超过阈值,且检测位点B荧光信号超过阈值,其他位点的荧光信号未超过阈值,则说明原始DNA样品中无EGFR基因突变(指在外显子20和外显子21上无突变),则该患者为非EGFR基因突变型癌症,不宜采用TKIs治疗。
出现非以上三种情况的检测结果,说明检测误差,均为检测无效,需要重新检测。
附图说明
图1为实施例1的微流控检测设备示意图(展示第一微流控芯片和功能化基板)。
图2为实施例1的微流控检测设备示意图(展示第二微流控芯片和功能化基板)。
图3为实施例1的外显子21的荧光检测结果图(处理条件1E21)。
图4为实施例1的外显子21的荧光检测结果图(处理条件2E21)。
图5为实施例1的外显子21的荧光检测结果图(处理条件3E21)。
图6为实施例1的外显子21的荧光检测结果图(处理条件4E21)。
图7为实施例1的外显子21的荧光检测结果图(处理条件5E21)。
图8为实施例2的外显子20的荧光检测结果图(处理条件1E20)。
图9为实施例2的外显子20的荧光检测结果图(处理条件2E20)。
图10为实施例2的外显子20的荧光检测结果图(处理条件3E20)。
图11为实施例2的外显子20的荧光检测结果图(处理条件4E20)。
具体实施方式
附图标记如下:功能化基板1、第一微流控芯片2、第一流道3、第一上样孔4、第一吸样孔5、第二微流控芯6、探针条带7、第二流道8、检测位点9、第二上样孔10、第二吸样孔11。
实施例1
(1)微流控检测设备的制备
微流控芯片使用聚二甲基硅氧烷(PDMS)作为原材料进行制备,制备的方法参见论文“L.Wang et al,Flexible microarray construction and fast DNA hybridizationconducted on a microfluidic chip for greenhouse plant fungal pathogendetection J.Agric.Food Chem,2007,55,10509-16”。微流控芯片的尺寸为2英寸×2英寸,厚度约为2mm。微流控芯片上平行分布有通道(16个通道),通道的深度为35μm,宽度为150μm。每个通道的两端设有通孔(一端叫做上样孔,一端叫做吸样孔)。
取载玻片(基板的具体形式),首先用100mL(由70mL 98%H
微流控设备包括微流控芯片(包括两块完全一样的第一微流控芯片和第二微流控芯片)和功能化基板,使用时需要将微流控芯片密封在功能化基板的表面上,微流控芯片上的通道和功能化基板形成样品的流道,从上样孔打入样品,在吸样孔施加负压吸引样品,加速样品在流道中的流动。
(2)DNA序列、探针、引物等的设计
根据EGFR基因序列以及其突变序列(NCBI编号:NM_001346941.1,NM005228.4,NG_007726.3)设计探针(E20_WT探针、T790M探针、E21_WT探针、L858R探针和L861Q探针)、引物(包括第一引物对和第二引物对,第一引物对包括第一上游引物(E20_F-Bio)和第一下游引物(E20_R),第二引物对包括第二上游引物(E21_F-Bio)和第二下游引物(E21_R))和60-mer聚寡核苷酸(均为单链,包括E20W60、E20M60、E21W60和E21M60),并委托国际DNA技术公司(Coralville,IA,USA)进行合成和修饰。序列情况请详见表1。5种探针的5'端通过间隔物(本实施例中具体为(CH2)
表1:DNA序列、探针、引物等序列信息
(3)探针固定和目标分子杂交
用含1M NaCl和0.15M NaHCO
探针固定和目标分子杂交的过程如图1和图2所示(原理参见L.Wang et al,Flexible microarray construction and fast DNA hybridization conducted on amicrofluidic chip for greenhouse plant fungal pathogen detection J.Agric.FoodChem,2007,55,10509-16)。将第一微流控芯片2密封在功能化基板1的表面上,形成第一流道3,在第一流道3的一端的第一上样孔4中分别加入探针溶液。在第一流道3的另一端的第一吸样孔5中吸取探针溶液,使得探针溶液浸润整个第一流道3。经孵育1h后,胺化的探针被固定在功能化基板1上。将第一流道3中的探针溶液吸出,并使用固定缓冲液冲洗第一流道3。然后将第一微流控芯片2从功能化基板1上拆卸下来,此时,探针再功能化基板1上已经形成探针条带7。然后将第二微流控芯6片密封在功能化基板1的表面上,第二微流控芯6片上的通道和功能化基板1之间形成第二流道8,第二流道8与第一流道3(探针条带7)垂直,第二流道8与第一流道3(探针条带7)之间的交叉点即为检测位点9。其中,第一微流控芯片2和第二微流控芯6片均密封在功能化基板1的同一面上。在第二流道8的一端的第二上样孔10中加入60-mer聚核苷酸溶液,在第二流道8的另一端的第二吸样孔11中吸取60-mer聚核苷酸溶液,使得60-mer聚核苷酸溶液浸润整个第二流道8。使得60-mer聚核苷酸与探针(已经固定在功能化基板1上)杂交1h,杂交完成后,使用指定洗脱缓冲液冲洗第二流道8。然后,将1μL的50ng/mL链霉-Cy5溶液(Cy-5标记的链霉亲和素溶液)引入所有第二流道8中,并使其与生物素标记物(60-mer聚核苷酸)结合15min,然后用含吐温的1×PBS溶液洗涤第二流道8。之后将第二微流控芯6片拆卸,拆卸第二微流控芯6片后的载玻片(功能化基板1),获得待检测基板,将用于荧光扫描。
在本实施例中,60-mer聚核苷酸与探针杂交以及洗涤第二流道的洗脱缓冲液参数选择如表2所示。本实施例是针对EGFR基因的外显子21的突变情况进行检测,使用的探针溶液为50μM E21_WT探针溶液、75μM T790M探针溶液、50μM L858R探针溶液、75μM L858R探针溶液、50μM L861Q探针溶液和75μM L861Q探针溶液(分别在不同的第一流道中加入)。使用的60-mer聚核苷酸溶液为25μM E21W60溶液和25μM E21M60溶液(分别在不同的第二流道中加入)。AuNP洗脱缓冲液为含有5nM AuNP和8nM稳定寡核苷酸的溶液(核苷酸序列:5’-CGCCAGAGAATACCAAAACTC-3’,SEQ ID NO.14),1×PBS洗脱缓冲液为现有技术中常用的缓冲液,再此不做赘述。
表2:检测外显子21使用的杂交以及洗脱时间选择
(4)荧光检测以及评价
用phosphoimager(Typhoon 9410,多功能激光扫描成像系统)检测到待检测基板上的荧光信号,用ImageQuant 5.2成像软件对图像做进一步处理。
为了评估探针的结合特异性,本方案设置了反应结合特异性的参数分化比率(DR,differentiation ratio),计算方法为该探针正确结合位点(完全匹配,PM)的荧光强度和该探针错误结合位点(错误匹配,MM)的荧光强度的比值。例如,针对E20_WT探针,该探针结合在含有野生型外显子20的序列上为正确结合,但是该探针也可能错误结合在含有突变型外显子20的序列上。为了量化评价这个探针结合目的序列的特异性程度,我们将E20_WT探针与野生型外显子20结合的位点的荧光强度除以E20_WT探针与突变型外显子20结合的位点的荧光强度,获得DR值。计算公式为:DR=FI(PM)/FI(MM),FI(PM)代表该探针正确结合位点(完全匹配,PM)的荧光强度,FI(MM)代表该探针错误结合位点(错误匹配,MM)的荧光强度。
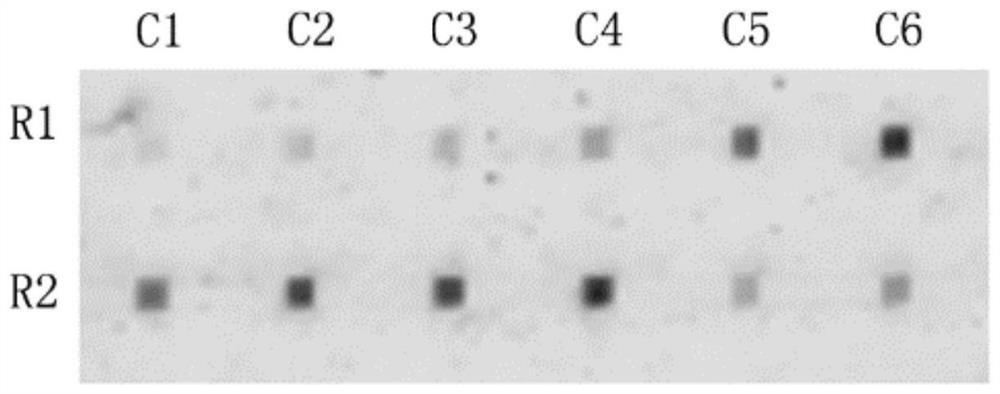
本实施例的检测结果如图3-图7所示。在图3中,C1列使用的探针溶液为50μML858R探针溶液,C2列使用的探针溶液为75μM L858R探针溶液,C3列使用的探针溶液为50μML861Q探针溶液,C4列使用的探针溶液为75μM L861Q探针,C5列使用的探针溶液为50μME21_WT探针溶液,C6列使用的探针溶液为75μM E21_WT探针溶液;R1排使用的60-mer聚核苷酸溶液为25μM E21W60溶液,R2排使用的60-mer聚核苷酸溶液为25μM E21M60溶液。所以在图3中,位于检测位点C1R1上双链DNA为L858R探针和E21W60形成的杂合体(错误匹配,MM),位于C1R2上双链DNA为L858R探针和E21M60形成的杂合体(完全匹配,PM),位于检测位点C2R1上双链DNA为L858R探针和E21W60形成的杂合体(MM),位于C2R2上双链DNA为L858R探针和E21M60形成的杂合体(PM);位于检测位点C3R1上双链DNA为L861Q探针和E21W60形成的杂合体(MM),位于C3R2上双链DNA为L861Q探针和E21M60形成的杂合体(PM);位于检测位点C4R1上双链DNA为L861Q探针和E21W60形成的杂合体(MM),位于C4R2上双链DNA为L861Q探针和E21M60形成的杂合体(PM);位于检测位点C5R1上双链DNA为E21_WT探针和E21W60形成的杂合体(PM),位于C5R2上双链DNA为E21_WT探针和E21M60形成的杂合体(MM);位于检测位点C6R1上双链DNA为E21_WT探针和E21W60形成的杂合体(PM),位于C6R2上双链DNA为E21_WT探针和E21M60形成的杂合体(MM)。图4-图7中,C1-C6以及R1和R2的设置情况同图3一致,不同点在于杂交和洗脱条件的选择(表2),图3-图7分别采用了处理条件1E21-5E21中的杂交和洗脱条件。根据图3-图7的荧光检测结果计算了DR值,如表3所示,结合表2中的参数条件,针对L858R探针和L861Q探针,4E21和5E21组的DR值最高(DR值越大,检测准确程度越高),说明结合使用47℃和AuNP洗脱缓冲液(含有5nM AuNP和8nM稳定寡核苷酸的溶液)
表3:外显子21检测的DR值
(5)L858R探针和L861Q探针的使用条件研究
发明人在研发过程中发现,外显子21上存在两个突变,在使用L858R探针、L861Q探针进行检测的时候,这两个探针的DR值很难到达比较理想的状态。发明人经过长时间探索,发现结合使用47℃和AuNP洗脱缓冲液(含有5nM AuNP和8nM稳定寡核苷酸的溶液)这两个条件,可以使得这两个L858R探针、L861Q探针获得较高的DR值。以下为发明人为寻找最适合的L858R探针和L861Q探针的使用条件所进行的实验。实验过程基本同本实施例(3),不同点在于探针浓度只设置为50μM(未采用两个浓度梯度),在杂交温度上采用梯度设置,杂交时间采用1h,洗脱时间采用3sec,洗脱缓冲液采用8nM稳定寡核苷酸的溶液或者1×PBS,每个条件重复10次,具体参数和DR测试结果如表4所示。由表4中的数据可知,L858R探针平均DR在组13中最高,经统计分析,组13的L858R探针平均DR显著高于其他组(组13与其他组两两进行t-test,p<0.05)。值得注意的是组13的L858R探针平均DR比组5的L858R探针平均DR显著提高,说明AuNP洗脱缓冲液可协同加强在47℃杂交温度下的DR值。组13的L861Q探针平均DR和组5的L861Q探针平均DR均产生了与L858R探针相似的现象。并且两两对比组1和组9、组2和组10、组3和组11、组4和组12、组6和组14、组7和组15、组8和组16,发现均无显著差异(P>0.1)。即,组1的L858R探针平均DR和组9的L858R探针平均DR相对比(t-test比较显著性),且组1的L861Q探针平均DR和组9的L861Q探针平均DR相对比(t-test比较显著性),其他组的两两对比依次类推。说明在非47℃的温度下,是否使用AuNP洗脱缓冲液并不能对DR造成显著影响。发明采用47℃的温度和AuNP洗脱缓冲液的结合,对L858R和L861Q两个突变位点的检测产生了预料不到的技术效果,大大提高了这两个突变位点的检测准确率。在47℃的杂交温度下,特异性结合的目的DNA与探针结合稳定,不会被AuNP洗脱缓冲液洗脱;非特异性结合的非目的DNA与探针结合不稳定,会被AuNP洗脱缓冲液洗脱,从而提高了DR值,使得检测结果更为准确。
表4:L858R探针和L861Q探针的使用条件研究结果
实施例2
本实施例基本同实施例1,不同点在于,本实施例是针对于EGFR基因的外显子20的突变情况进行检测,使用的探针溶液为50μM E20_WT探针、75μM E20_WT探针、50μM T790M探针、75μM T790M探针(分别在不同的第一流道中加入)。使用的60-mer聚核苷酸溶液为25μME20W60溶液和25μM E20M60溶液(分别在不同的第二流道中加入)。在本实施例中,使得60-mer聚核苷酸与探针杂交以及洗涤第二流道的洗脱缓冲液参数选择如表5所示。
表5:检测外显子20的杂交以及洗脱时间选择
本实施例的检测结果如图8-图11所示,在图8中,C1列使用的探针溶液为50μMT790M探针溶液,C2列使用的探针溶液为75μM T790M探针溶液,C3列使用的探针溶液为50μME20_WT探针溶液,C4列使用的探针溶液为75μM E20_WT探针;R1排使用的60-mer聚核苷酸溶液为25μM E21W60溶液,R2排使用的60-mer聚核苷酸溶液为25μM E21M60溶液。所以在图8中,位于检测位点C1R1上双链DNA为T790M探针和E20W60形成的杂合体(错误匹配,MM),位于C1R2上双链DNA为T790M探针和E20M60形成的杂合体(完全匹配,PM),位于检测位点C2R1上双链DNA为T790M探针和E20W60形成的杂合体(MM),位于C2R2上双链DNA为T790M探针和E20M60形成的杂合体(PM);位于检测位点C3R1上双链DNA为T790M探针和E20W60形成的杂合体(MM),位于C3R2上双链DNA为T790M探针和E20M60形成的杂合体(PM);位于检测位点C4R1上双链DNA为T790M探针和E20W60形成的杂合体(MM),位于C4R2上双链DNA为T790M探针和E20M60形成的杂合体(PM)。图9-图11中,C1-C4以及R1和R2的设置情况同图8一致,不同点在于杂交和洗脱条件的选择(表5),图8-图11依次使用1E20-4E20中的处理条件。根据图8-图11的荧光检测结果计算了DR值,实验数据说明,采用47℃的杂交条件以及AuNP洗脱缓冲液,在调节适当的洗脱时间的条件下可以获得较为满意的检测DR值。例如,3E20组中,使用75μMT790M探针的DR值为4.7(洗脱时间3sec);4E20组中,使用50μM E20_WT探针的DR值为3.3(洗脱时间30min)。
以上所述的仅是本发明的实施例,方案中公知的具体技术方案和/或特性等常识在此未作过多描述。应当指出,对于本领域的技术人员来说,在不脱离本发明技术方案的前提下,还可以作出若干变形和改进,这些也应该视为本发明的保护范围,这些都不会影响本发明实施的效果和专利的实用性。本申请要求的保护范围应当以其权利要求的内容为准,说明书中的具体实施方式等记载可以用于解释权利要求的内容。
SEQUENCE LISTING
<110> 珠海澳加动力生物科技有限公司
<120> EGFR基因突变检测系统
<130> 2020-12-10
<160> 14
<170> PatentIn version 3.5
<210> 1
<211> 22
<212> DNA
<213> 人工序列
<400> 1
catgagctgc gtgatgagct gc 22
<210> 2
<211> 22
<212> DNA
<213> 人工序列
<400> 2
catgagctgc atgatgagct gc 22
<210> 3
<211> 21
<212> DNA
<213> 人工序列
<400> 3
agcagtttgg ccagcccaaa a 21
<210> 4
<211> 20
<212> DNA
<213> 人工序列
<400> 4
ttggcccgcc caaaatctgt 20
<210> 5
<211> 21
<212> DNA
<213> 人工序列
<400> 5
tcttccgcac ccagctgttt g 21
<210> 6
<211> 19
<212> DNA
<213> 人工序列
<400> 6
aagcctacgt gatggccag 19
<210> 7
<211> 22
<212> DNA
<213> 人工序列
<400> 7
ctttgcgatc tgcacacacc ag 22
<210> 8
<211> 22
<212> DNA
<213> 人工序列
<400> 8
gggcatgaac tacttggagg ac 22
<210> 9
<211> 22
<212> DNA
<213> 人工序列
<400> 9
tttgcctcct tctgcatggt at 22
<210> 10
<211> 60
<212> DNA
<213> 人工序列
<400> 10
cctcacctcc accgtgcagc tcatcacgca gctcatgccc ttcggctgcc tcctggacta 60
<210> 11
<211> 60
<212> DNA
<213> 人工序列
<400> 11
cctcacctcc accgtgcagc tcatcatgca gctcatgccc ttcggctgcc tcctggacta 60
<210> 12
<211> 60
<212> DNA
<213> 人工序列
<400> 12
caagatcaca gattttgggc tggccaaact gctgggtgcg gaagagaaag aataccatgc 60
<210> 13
<211> 60
<212> DNA
<213> 人工序列
<400> 13
caagatcaca gattttgggc gggccaaaca gctgggtgcg gaagagaaag aataccatgc 60
<210> 14
<211> 21
<212> DNA
<213> 人工序列
<400> 14
cgccagagaa taccaaaact c 21
- EGFR基因突变检测系统
- 用于检测EGFR基因突变的引物Blocker组、试剂盒及方法