吡咯烷酮类化合物在制备治疗炎症性疾病的药物中的用途
文献发布时间:2023-06-19 12:08:44
技术领域
本发明涉及生物医药领域,具体涉及吡咯烷酮类化合物在制备治疗炎症性疾病的药物中的用途。
背景技术
一点红药材系菊科植物(Compositae)一点红Emilia sonchifolia(Linnaeus)deCandolle的新鲜或干燥全草。本品长10~50cm。根细而弯曲,有须根。茎细圆柱形,表面暗绿色,下部被茸毛。叶多皱缩,展平后基生叶呈琴状分裂,长5~10cm,宽2.5~5cm,灰绿色或暗绿色,先端裂片大,近三角形,基部抱茎,边缘具疏钝齿;茎生叶渐狭。头状花序2~3个排成聚伞状,总苞圆柱形,苞片1层,呈条状披针形或近条状,长约1cm;管状花棕黄色,冠毛白色。瘦果狭矩圆形,长约3mm,有棱。
中国华南、华中和西南等地均有野生。一点红性平,味苦微辛,凉血解毒,其以食嫩梢嫩叶为主,可炒食、作汤或作火锅料,质地爽脆,类似茼蒿的品味,具有清热解毒、活血散淤等功效,还可以治泌尿系统感染、咽喉炎、毒疮等。
神经保护剂是一种保护易受损神经元,减轻或阻止疾病进展的神经调节剂。可减少神经元的死亡,常用的神经保护剂包括钙通道阻滞剂、自由基清除剂、谷氨酸拮抗剂、细胞膜稳定剂等。目前,神经保护剂是急性脑卒中的治疗手段之一。神经保护药物可以抑制细胞死亡和阻止缺血区组织再灌注损伤,借此可以延长脑卒中发作后的使用纤维蛋白溶解疗法的治疗时间。
发明内容
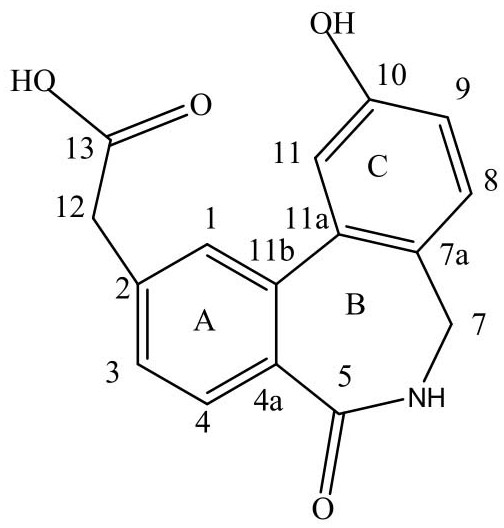
本发明公开了式I的化合物及其药学上可接受的盐;
所述的式I的化合物的制备方法包括以下步骤:
A、取一点红药材经体积分数80-95%的乙醇回流提取1-3次,合并提取液,浓缩干燥后得浸膏;
B、将浸膏与硅藻土以1:1-3的重量比例拌样后,依次用石油醚、二氯甲烷、乙酸乙酯回流洗脱,取乙酸乙酯回流提取物,回收溶剂,得乙酸乙酯提取物;
C、将乙酸乙酯提取物用硅胶柱色谱分离,采用氯仿-甲醇溶液体系以体积比20:1-0:1进行梯度洗脱,取其中11:1-9:1段的洗脱留分;
D、将步骤C获得的洗脱留分经硅胶柱色谱分离,采用氯仿-甲醇溶液体系以体积比50:1-0:1进行梯度洗脱,取其中以氯仿-甲醇29:1-20:1洗脱收集段的洗脱留分;
E、将步骤D获得的洗脱留分继续经硅胶柱色谱分离,采用氯仿-甲醇溶液体系以体积比100:0-0:100进行梯度洗脱,取其中以氯仿-甲醇40-21:60-79洗脱收集段的洗脱留分;
F、将步骤E获得的洗脱留分经MCIgel柱色谱分离,采用甲醇-水溶液体系以体积比10:90-100:0进行梯度洗脱,取其中甲醇-水30:70段的洗脱留分回收溶剂后,即得。
本发明还公开了式I的化合物在制备神经细胞保护药物中的用途。
本发明还公开了一种神经细胞保护药物组合物,含有式I的化合物和/或其药学上可接受的盐。
所述的药物组合物,还含有一点红、白花蛇舌草、鸡血藤、桃金娘根、白背叶根、地桃花、菥蓂中的一种或几种。
所述的药物组合物,还含有一点红、白花蛇舌草、鸡血藤、桃金娘根、白背叶根、地桃花、菥蓂中的一种或几种的提取物。
所述的药物组合物的剂型包括片剂、胶囊剂、颗粒剂、散剂、丸剂、溶液剂、混悬剂、糖浆剂、注射剂、软膏剂、栓剂或喷雾剂。
本发明所述的式Ⅰ的化合物为一点红中新发现的化合物,经过本实验例2的细胞水平实验表明其对皮质酮损伤的PC12细胞具有明显的保护作用,体现出了较好的神经细胞保护作用,是一种新型的神经细胞保护药物。
附图说明
本发明附图如下:
图1是不同浓度的式Ⅰ的化合物对皮质酮损伤的PC12细胞存活力的影响结果图;
图2是本发明化合物的式Ⅰ。
具体实施方式
实施例1
所述化合物的制备方法包括以下步骤:
A、取一点红药材30kg,干燥、切碎,用6倍量体积分数为80%的乙醇回流提取2次,每次2小时,合并提取液,浓缩干燥得浸膏备用;将浸膏与硅藻土以1:1的重量比例拌样
B、将步骤A中浸膏与硅藻土混合物用石油醚进行回流洗脱,取石油醚回流洗脱后的残渣,用二氯甲烷进行回流洗脱,取二氯甲烷回流洗脱后的残渣,用乙酸乙酯进行回流洗脱,收集洗脱液,命名为ZF-3;
C、将洗脱液ZF-3用60-180目的柱层析硅胶进行色谱分离,洗脱溶剂为氯仿-甲醇,氯仿和甲醇体积比逐步变化,20:1to 0:1;其按顺序收集洗脱留分,其中以氯仿-甲醇(20:1、19:1、18:1)洗脱收集为Fr1;以氯仿-甲醇(17:1、16:1、15:1)洗脱收集为Fr2;以氯仿-甲醇(14:1、13:1、12:1)洗脱收集为Fr3;以氯仿-甲醇(11:1、10:1、9:1)洗脱收集为Fr4;以氯仿-甲醇(8:1、7:1、6:1)洗脱收集为Fr5;以氯仿-甲醇(5:1、4:1、3:1)洗脱收集为Fr6;以氯仿-甲醇(2:1、1:1、0:1)洗脱收集为Fr7;
D、将步骤C获得的洗脱留分经硅胶柱色谱分离,采用氯仿-甲醇溶液体系以体积比50:1-0:1进行梯度洗脱,其中以氯仿-甲醇(50:1-45:1)洗脱收集为Fr4.1;以氯仿-甲醇(44:1-40:1)洗脱收集为Fr4.2;以氯仿-甲醇(39:1-35:1)洗脱收集为Fr4.3;以氯仿-甲醇(34:1-30:1)洗脱收集为Fr4.4;以氯仿-甲醇(29:1-20:1)洗脱收集为Fr4.5;以氯仿-甲醇(19:1-10:1)洗脱收集为Fr4.6;以氯仿-甲醇(9:1-0:1)洗脱收集为Fr4.7。取其中以氯仿-甲醇(29:1-20:1)洗脱收集段的洗脱留分Fr4.5;
E、将步骤D获得的洗脱留分继续经硅胶柱色谱分离,采用氯仿-甲醇溶液体系以体积比100:0-0:100进行梯度洗脱,其中以氯仿-甲醇(100-81:0-19)洗脱收集为Fr4.5.1;以氯仿-甲醇(80-61:20-39)洗脱收集为Fr4.5.2;以氯仿-甲醇(60-41:40-59)洗脱收集为Fr4.5.3;以氯仿-甲醇(40-21:60-79)洗脱收集为Fr4.5.4;以氯仿-甲醇(20-100:80-0)洗脱收集为Fr4.5.5。取其中以氯仿-甲醇(40-21:60-79)洗脱收集段的洗脱留分Fr4.5.4;
F、将步骤E获得的洗脱留分经MCIgel柱色谱分离,采用甲醇-水溶液体系以体积比10:90-100:0进行梯度洗脱,取其中甲醇-水30:70段的洗脱留分回收溶剂后,即得式1的化合物。
将制备得到的化合物进行质谱、核磁共振氢谱、核磁共振碳谱的检测,结果证明所得化合物为式1的化合物。
实施例2
所述化合物的制备方法包括以下步骤:
A、取一点红药材30kg,干燥、切碎,用9倍量体积分数为95%的乙醇回流提取2次,每次2小时,合并提取液,浓缩干燥得浸膏备用;将浸膏与硅藻土以1:3的重量比例拌样
B、将步骤A中浸膏与硅藻土混合物用石油醚进行回流洗脱,取石油醚回流洗脱后的残渣,用二氯甲烷进行回流洗脱,取二氯甲烷回流洗脱后的残渣,用乙酸乙酯进行回流洗脱,收集洗脱液,命名为ZF-3;
C、将洗脱液ZF-3用60-180目的柱层析硅胶进行色谱分离,洗脱溶剂为氯仿-甲醇,氯仿和甲醇体积比逐步变化,20:1to 0:1;其按顺序收集洗脱留分,其中以氯仿-甲醇(20:1、19:1、18:1)洗脱收集为Fr1;以氯仿-甲醇(17:1、16:1、15:1)洗脱收集为Fr2;以氯仿-甲醇(14:1、13:1、12:1)洗脱收集为Fr3;以氯仿-甲醇(11:1、10:1、9:1)洗脱收集为Fr4;以氯仿-甲醇(8:1、7:1、6:1)洗脱收集为Fr5;以氯仿-甲醇(5:1、4:1、3:1)洗脱收集为Fr6;以氯仿-甲醇(2:1、1:1、0:1)洗脱收集为Fr7;
D、将步骤C获得的洗脱留分经硅胶柱色谱分离,采用氯仿-甲醇溶液体系以体积比50:1-0:1进行梯度洗脱,其中以氯仿-甲醇(50:1-45:1)洗脱收集为Fr4.1;以氯仿-甲醇(44:1-40:1)洗脱收集为Fr4.2;以氯仿-甲醇(39:1-35:1)洗脱收集为Fr4.3;以氯仿-甲醇(34:1-30:1)洗脱收集为Fr4.4;以氯仿-甲醇(29:1-20:1)洗脱收集为Fr4.5;以氯仿-甲醇(19:1-10:1)洗脱收集为Fr4.6;以氯仿-甲醇(9:1-0:1)洗脱收集为Fr4.7。取其中以氯仿-甲醇(29:1-20:1)洗脱收集段的洗脱留分Fr4.5;
E、将步骤D获得的洗脱留分继续经硅胶柱色谱分离,采用氯仿-甲醇溶液体系以体积比100:0-0:100进行梯度洗脱,其中以氯仿-甲醇(100-81:0-19)洗脱收集为Fr4.5.1;以氯仿-甲醇(80-61:20-39)洗脱收集为Fr4.5.2;以氯仿-甲醇(60-41:40-59)洗脱收集为Fr4.5.3;以氯仿-甲醇(40-21:60-79)洗脱收集为Fr4.5.4;以氯仿-甲醇(20-100:80-0)洗脱收集为Fr4.5.5。取其中以氯仿-甲醇(40-21:60-79)洗脱收集段的洗脱留分Fr4.5.4;
F、将步骤E获得的洗脱留分经MCIgel柱色谱分离,采用甲醇-水溶液体系以体积比10:90-100:0进行梯度洗脱,取其中甲醇-水30:70段的洗脱留分回收溶剂后,即得式1的化合物。
将制备得到的化合物进行质谱、核磁共振氢谱、核磁共振碳谱的检测,结果证明所得化合物为式1的化合物。
实施例3
所述化合物的制备方法包括以下步骤:
A、取一点红药材30kg,干燥、切碎,用7倍量体积分数为90%的乙醇回流提取2次,每次2小时,合并提取液,浓缩干燥得浸膏备用;将浸膏与硅藻土以1:2的重量比例拌样
B、将步骤A中浸膏与硅藻土混合物用石油醚进行回流洗脱,取石油醚回流洗脱后的残渣,用二氯甲烷进行回流洗脱,取二氯甲烷回流洗脱后的残渣,用乙酸乙酯进行回流洗脱,收集洗脱液,命名为ZF-3;
C、将洗脱液ZF-3用60-180目的柱层析硅胶进行色谱分离,洗脱溶剂为氯仿-甲醇,氯仿和甲醇体积比逐步变化,20:1to 0:1;其按顺序收集洗脱留分,其中以氯仿-甲醇(20:1、19:1、18:1)洗脱收集为Fr1;以氯仿-甲醇(17:1、16:1、15:1)洗脱收集为Fr2;以氯仿-甲醇(14:1、13:1、12:1)洗脱收集为Fr3;以氯仿-甲醇(11:1、10:1、9:1)洗脱收集为Fr4;以氯仿-甲醇(8:1、7:1、6:1)洗脱收集为Fr5;以氯仿-甲醇(5:1、4:1、3:1)洗脱收集为Fr6;以氯仿-甲醇(2:1、1:1、0:1)洗脱收集为Fr7;
D、将步骤C获得的洗脱留分经硅胶柱色谱分离,采用氯仿-甲醇溶液体系以体积比50:1-0:1进行梯度洗脱,其中以氯仿-甲醇(50:1-45:1)洗脱收集为Fr4.1;以氯仿-甲醇(44:1-40:1)洗脱收集为Fr4.2;以氯仿-甲醇(39:1-35:1)洗脱收集为Fr4.3;以氯仿-甲醇(34:1-30:1)洗脱收集为Fr4.4;以氯仿-甲醇(29:1-20:1)洗脱收集为Fr4.5;以氯仿-甲醇(19:1-10:1)洗脱收集为Fr4.6;以氯仿-甲醇(9:1-0:1)洗脱收集为Fr4.7。取其中以氯仿-甲醇(29:1-20:1)洗脱收集段的洗脱留分Fr4.5;
E、将步骤D获得的洗脱留分继续经硅胶柱色谱分离,采用氯仿-甲醇溶液体系以体积比100:0-0:100进行梯度洗脱,其中以氯仿-甲醇(100-81:0-19)洗脱收集为Fr4.5.1;以氯仿-甲醇(80-61:20-39)洗脱收集为Fr4.5.2;以氯仿-甲醇(60-41:40-59)洗脱收集为Fr4.5.3;以氯仿-甲醇(40-21:60-79)洗脱收集为Fr4.5.4;以氯仿-甲醇(20-100:80-0)洗脱收集为Fr4.5.5。取其中以氯仿-甲醇(40-21:60-79)洗脱收集段的洗脱留分Fr4.5.4;
F、将步骤E获得的洗脱留分经MCIgel柱色谱分离,采用甲醇-水溶液体系以体积比10:90-100:0进行梯度洗脱,取其中甲醇-水30:70段的洗脱留分回收溶剂后,即得式1的化合物。
将制备得到的化合物进行质谱、核磁共振氢谱、核磁共振碳谱的检测,结果证明结果证明所得化合物为式1的化合物。
实验例1
结构鉴定:
式I化合物
白色粉末;UV(MeOH)λ
式I化合物的碳谱和氢谱
实验例3
通式I的生物碱类化合物神经保护作用的研究:
一点红生物碱对皮质酮诱导的PC12细胞损伤的保护作用
以式I化合物为研究对象,考察其对神经细胞的保护作用。PC12细胞株为大鼠肾上腺嗜铬细胞克隆化细胞株,分化的PC12细胞有典型的神经细胞特征,其胞膜上糖皮质激素受体表达丰富。用高浓度的皮质酮诱导PC12细胞损伤,已成为研究抑郁症的一种常用体外模型。本研究用高浓度的皮质酮与PC12细胞共同孵育,模拟抑郁症病人的大脑神经元损伤状态,以此观察一点红生物碱碱对神经细胞的保护作用。
1材料与试剂
1.1药物式I的化合物。
1.2试剂大鼠嗜铬细胞瘤PC12细胞(上海细胞库);精致胎牛血清、马血清、青霉素钠、链霉素(Gibco公司);皮质酮,DMEM,MTT,二甲基亚砜(Sigma公司)。
1.3仪器纯水仪(美国Millipore公司);二氧化碳培养箱(NAPCO 6500TC法国);连续波长酶标仪(美国BIO-TEK)。
2方法
2.1PC12细胞培养
PC12细胞液置于15mL的离心管中,1000rpm,离心5min,弃上清液,将细胞用含有10%胎牛血清、5%马血清的高糖DMEM培养基悬浮,分装于25mL的培养瓶中,置于37℃的二氧化碳培养箱中培养,每2-3d换液1次。
2.2供试品对皮质酮损伤的PC12细胞存活力的影响
PC12细胞培养基养至对数生长期,用含10%胎牛血清、5%马血清的DMEM培养基(含青霉素钠100U/ml、链霉素100μg/ml)重悬细胞,调细胞浓度为每毫升1×10
实验结果见图1,从图1中可见式I的化合物在5μg/mL)时显示出较强的保护作用(P<0.05),细胞存活率为87%.在更高的浓度时(20μg/mL)会导致细胞生存率下降。
- 吡咯烷酮类化合物在制备治疗炎症性疾病的药物中的用途
- 二色波罗蜜中的二苯乙烯类化合物及其在制备治疗炎症性疾病药物中的用途