甲基化标记和标靶甲基化探针板
文献发布时间:2023-06-19 12:16:29
本申请要求2018年9月27日提交的美国临时专利申请案第62/737,836号及2019年4月2日提交的国际专利申请案第PCT/US2019/025358号的权益,在此通过引用将其全部内容合并于本文。
本申请包含一序列表,其已以ASCII格式以电子方式提交,现通过引用将其整体并入本申请。该ASCII副本创建于2019年9月26日,名为50251-846_601_SL.txt,大小为52371626位元组(bytes)。
背景技术
脱氧核糖核酸(DNA)甲基化在调节基因表达中起重要作用。异常的DNA甲基化与许多疾病过程有关,包含癌症。使用甲基化测序(例如,全基因组亚硫酸氢盐测序(WGBS))的DNA甲基化分析越来越被认为是用于检测、诊断和/或监测癌症的有价值的诊断工具。例如,不同的甲基化区域的特定模式可用作各种疾病的分子标记。
然而,WGBS目前不适合于产品化验。原因是绝大多数的基因组在癌症中没有差异甲基化,或局部CpG密度太低而无法提供可靠的信号。仅百分之几的基因组可能对分类有用。使用WGBS,以目前的测序成本计算,由于成本限制,深度测序(高达~1000×)只能在一小部分的多个基因组区域内完成,可能占基因组的0.1%。
此外,在识别各种疾病中的多个差异甲基化区域方面存在各种挑战。首先,确定一疾病群组中的差异甲基化区域,只有与一群组的多个对照组对象比较才有分量,因此,如果对照组的人数较少,则所述确定将对较小对照组失去信心。另外,在一群组的多个对照组对象中,甲基化状态可以变化,这在当确定一疾病组中所述多个区域存在差异甲基化时很难解释。另一方面,在一CpG位点的胞嘧啶甲基化与在一随后的CpG位点的甲基化强烈相关。概括这种依赖性本身就是一个挑战。
因此,还没有能够通过从多个差异甲基化区域分析DNA来准确诊断疾病的经济有效的方法。
发明内容
在多个对象中及早检测到癌症是很重要的,因为它可以及早治疗,从而获得更大的生存机会。利用无细胞DNA(cell-free DNA,cfDNA)片段,标靶检测癌症特定的甲基化模式,可以通过提供一种成本效益高、非侵入性的方法用于获得相关于癌症的存在或不存在、癌症的起源组织、或癌症类型的信息,使癌症的早期检测成为可能。通过使用一标靶基因组区域化验板而非在一测试样本中的所有核酸进行测序(也称为“全基因组测序”),该方法可以增加标靶区域的测序深度,并降低成本。
为此,本说明书提供了用于检测标靶基因组区域中癌症特异性甲基化模式的癌症化验板(或者称为“诱饵集”),以及使用癌症化验板诊断癌症的方法。本文进一步提供通过识别具有癌症特异性甲基化模式的基因组位点以及可用于本文提供的各种方法的基因组位点或基因组区域的列表来设计和制造癌症化验板的方法。本文所描述的方法还包含设计探针以有效地扩增与所选基因组区域相对应或衍生自所选基因组区域的cfDNA而不下拉过量不需要的DNA的方法。
在一个方面,本文提供一种用于杂交捕获的诱饵组,所述诱饵组包含不同的多个含有寡核苷酸的探针,其中所述多个含有寡核苷酸的探针中的每个包含一序列,所述序列的长度为至少30个碱基,所述序列与以下互补:(1)一基因组区域的一序列;或(2)上述(1)的所述序列仅通过一个或多个转换而变异生成的一序列,其中所述一个或多个转换中的每个相应转换发生在所述基因组区域中的一胞嘧啶处,以及其中所述不同的多个含有寡核苷酸的探针的每个探针与对应于一CpG位点的一序列互补,所述CpG位点在多个癌症样本中相对于多个非癌症样本是差异甲基化的。
所述诱饵组可以包含至少500、1,000、2,000、2,500、5,000、6,000、7,500、10,000、15,000、20,000、25,000、50,000或100,000个不同的含有寡核苷酸的探针。在一个方面,所述CpG位点在多个癌症样本中相对于多个非癌症样本基于一标准被认为是差异甲基化的,所述标准包含多个癌症样本的一数量,所述多个癌症样本的所述数量包含与所述CpG位点重叠的一异常甲基化cfDNA片段。在一个方面,所述CpG位点被认为在多个癌症样本中相对于多个非癌症样本基于一标准是差异甲基化的,所述标准包含N
在一个方面,在所述不同的多个含有寡核苷酸的探针中的每个,长度为至少30个碱基的所述序列可以为:(1)与从列表1至列表8中的任一个所列的多个基因组区域中选择的一基因组区域内的一序列互补;或者(2)与上述(1)的所述序列仅通过一个或多个转换而变异生成的一序列互补,其中所述一个或多个转换中的每个相应转换发生在所述基因组区域中的一胞嘧啶处。在一个方面,所述不同的多个含有寡核苷酸的探针是每个与一亲和部分相结合。所述亲合部分是生物素。在一个方面,对于所述不同的多个含有寡核苷酸的探针中的至少一个,至少30个碱基的序列与上述(1)的所述序列仅通过一个或多个转换而变异生成的一序列互补,其中所述一个或多个转换的每个相应转换发生在所述基因组区域中的一胞嘧啶处。在另一个方面,对于至少500、1000、2000、2500、5000、6000、10000、15000、20000、25000或50000个的所述不同的多个含有寡核苷酸的探针中的每个,至少30个碱基的序列与上述(1)的所述序列仅通过一个或多个转换而变异生成的一序列互补,其中所述一个或多个转换的每个相应转换发生在所述基因组区域中的一胞嘧啶处。在一个方面,所述诱饵组中至少80%、90%或95%的所述多个含有寡核苷酸的探针不包含至少30、至少40或至少45个碱基的一序列,所述序列具有20个或以上的脱靶基因组区域。在另一个方面,所述诱饵组中所述多个含有寡核苷酸的探针不包含至少30、至少40或至少45个碱基的一序列,所述序列具有20个或以上的脱靶基因组区域。至少30个碱基的所述序列的长度为至少40个碱基、至少45个碱基、至少50个碱基、至少60个碱基、至少75个或至少100个碱基。所述多个含有寡核苷酸的探针中的每个具有长度为至少45、40、75、100或120个碱基的一核酸序列。所述多个含有寡核苷酸的探针中的每个具有长度不超过300、250、200或150个碱基的一核酸序列。
在一个方面,所述不同的多个含有寡核苷酸的探针可以包含至少500、至少1,000、至少2,000、至少2,500、至少5,000、至少6,000、至少7,500和至少10,000、至少15,000、至少20,000或至少25,000对的不同探针,其中每对探针包含一第一探针和一第二探针,其中所述第二探针不同于所述第一探针,并通过长度为至少30、至少40、至少50或至少60个核苷酸的一重叠序列与所述第一探针相重叠。在一个方面,所述诱饵组可以包含多个含有寡核苷酸的探针,配置为标靶列表1至8中任一个列表中所识别的至少20%、至少25%、至少30%、至少40%、至少50%、至少60%、至少70%、至少80%、至少90%、至少95%或100%的基因组区域。在另一个方面,所述诱饵组可以包含多个含有寡核苷酸的探针,被配置成标靶列表1中所识别的至少20%、至少25%、至少30%、至少40%、至少50%、至少60%、至少70%、至少80%、至少90%、至少95%或100%的基因组区域。所述诱饵组可以包含多个含有寡核苷酸的探针,被配置成标靶列表3中所识别的至少20%、至少25%、至少30%、至少40%、至少50%、至少60%、至少70%、至少80%、至少90%、至少95%或100%的基因组区域。所述诱饵组中全部的含有寡核苷酸的探针可以被配置成与由选自列表1至8中任一个列表中对应于至少30%、40%、50%、60%、70%、80%、90%或95%的基因组区域的多个cfDNA分子所获得的多个片段杂交。所述诱饵组中全部的含有寡核苷酸的探针可以被配置成与由对应于列表1至8中任一个列表中的至少500、1,000、5,000、10,000、15,000、20,000、至少25,000或至少30,000个基因组区域的多个cfDNA分子所获得的多个片段杂交。所述诱饵组中全部含有寡核苷酸的探针可以被配置成与由对应于列表1至8中任一个列表中的至少50、60、70、80、90、100、120、150或200个基因组区域的多个cfDNA分子所获得的多个片段杂交。
所述多个含有寡核苷酸的探针可以包含至少500、1,000、5,000或10,000个不同的探针子集,其中每个探针子集包含多个探针,以2倍平铺的方式共同延伸跨过从列表1至8中任一个列表的多个基因组区域中选择的一基因组区域。以所述2倍平铺的方式共同延伸跨过所述基因组区域的所述多个探针可以包含至少一对探针,所述至少一对探针通过长度至少为30个碱基、至少40个碱基、至少50个碱基或至少60个碱基的一序列相重叠。所述多个探针可以共同延伸跨过所述基因组的多个部分,总体上的一总合大小为0.2MB(百万碱基)至15MB之间、0.5MB至15MB之间、1MB至15MB之间、3MB至12MB之间、3MB至7MB之间、5MB至9MB之间或7MB至12MB之间。所述诱饵组中所述不同的多个含有寡核苷酸的探针的至少一个子集可以被设计成与从列表4或列表6中的一个或多个基因组区域衍生的多个cfDNA片段杂交。所述诱饵组中所述不同的含有寡核苷酸的探针的所述子集可以被设计成标靶来自列表4或列表6中的至少2个、至少10个、至少50个、至少100个、至少1,000个或至少5,000个、至少8,000个、至少10,000个或至少20,000个基因组区域。所述不同的多个含有寡核苷酸的探针中可以每一个包含少于20、15、10、8或6个CpG检测位点。在一个方面,所述诱饵组中至少80%、85%、90%、92%、95%或98%的所述多个含有寡核苷酸的探针在所有的CpG检测位点上可以仅具有CpG或仅具有CpA。所述诱饵组的所述多个含有寡核苷酸的探针可以对应于从列表1至列表8中的任一个列表的所述多个基因组区域中选择的多个基因组区域的一数量,其中与所述诱饵组中的多个探针对应的所述多个基因组区域中至少30%为外显子或内含子。所述诱饵组的所述多个含有寡核苷酸的探针可以对应于多个基因组区域的一数量,其中与所述诱饵组中的多个探针对应的至少15%或至少20%的基因组区域为外显子。所述诱饵组的所述多个含有寡核苷酸的探针可以对应于多个基因组区域的一数量,其中与所述诱饵组中的多个探针对应的少于10%的基因组区域是基因间区域。在一个方面,对于所述不同的多个含有寡核苷酸的探针中的每一个,所述至少30个核苷酸序列可以与另一序列互补,所述另一序列是所述基因组区域的所述序列通过在所述序列内的所有CpG位点处的一个或多个转换而变异生成的。在另一个方面,对于多个含有寡核苷酸的探针,所述多个含有寡核苷酸的探针相对于所述基因组区域内的所述序列通过一个或多个转换而变异,且可以在所述基因组区域内的每个CpG位点处均发生一转换。所述不同的多个含有寡核苷酸的探针可以与多个cfDNA片段互补,所述多个cfDNA片段已被转化以尿嘧啶取代胞嘧啶,其中在来自多个癌症对象的cfDNA中发现的所述多个cfDNA片段是在来自多个非癌症对象的cfDNA中发现的所述多个cfDNA片段的频率的至少2倍、10倍、20倍、50倍、100倍、或者1000倍。
在一个实施例中,本文提供一种混合物,包含:一诱饵组及转化的cfDNA,所述转化的cfDNA可以包含亚硫酸氢盐转化的cfDNA。所述转化的cfDNA可以包含已通过胞嘧啶脱氨酶转化的cfDNA。在一个方面,本文提供一种用于扩增一转化的cfDNA样本的方法,包含:将无细胞DNA样本与所述诱饵组相接触;以及通过杂交捕获来扩增所述样本并用于多个基因组区域的一第一集合。
在另一个方面,本文提供一种用于提供关于一癌症的存在或不存在、癌症的一阶段或癌症的一类型的序列信息的方法,所述方法包含:用一脱氨基剂处理来自一生物样本的无细胞DNA,以产生一无细胞DNA样本,所述无细胞DNA样本包含多个脱氨基核苷酸;扩增所述无细胞DNA样本,以用于获取多个无细胞DNA分子的信息,其中扩增所述无细胞DNA样本以获取所述多个无细胞DNA分子的信息包含:将所述无细胞DNA与多个探针相接触,所述多个探针被配置成杂交到对应于列表1至列表8的任一个列表中所识别的多个区域的多个无细胞DNA分子;以及对扩增后的所述多个无细胞DNA分子进行测序,从而获得多个序列读数的一集合,以提供一癌症存在或不存在、癌症的一阶段或癌症的一类型的信息。所述多个探针包含多个引物,及扩增所述无细胞DNA可以包含:使用所述多个引物通过PCR扩增所述多个无细胞DNA片段。在一个方面,扩增所述无细胞DNA不涉及杂交捕获。在一个方面,所述多个探针可以被配置为杂交到从所述多个cfDNA分子获得的多个转换片段,所述多个转换片段对应于或衍生自列表1至列表8中任何一个列表中的至少30%、40%、50%、60%、70%、80%、90%或95%的基因组区域。所述多个探针可以包含所述多个含有寡核苷酸的探针。
在另一方面,所述方法进一步包含以下步骤:通过评估多个序列读数的集合来确定一癌症分类,其中所述癌症分类为:(a)癌症存在或不存在;(b)癌症的一阶段;或(c)一种类型的癌症存在或不存在。所述癌症分类可以是癌症存在或不存在。所述癌症的分类可以是癌症的一阶段。癌症的所述阶段可以选自从阶段I、阶段II、阶段III和阶段IV。所述癌症分类可以是一种类型的癌症的存在或不存在。在一个方面,所述确定一癌症分类的步骤可以进一步包含:(a)基于多个序列读数的集合生成一测试特征向量;以及(b)将所述测试特征向量应用于一分类器。在一个方面,所述分类器还包含一模型,所述模型通过一训练程序训练,所述训练过程具有来自一个或多个患有癌症的训练对象的多个片段的一癌症集合和来自一个或多个没有癌症的训练对象的多个片段的一非癌症集合,其中所述多个片段的所述癌症集合和所述多个片段的所述非癌症集合均包含多个训练片段。所述分类器在接收者操作特性曲线下可以具有大于0.70、大于0.75、大于0.77、大于0.80、大于0.81、大于0.82或大于0.83的一面积。在99%的特异性下,所述分类器可以具有至少35%、至少40%、至少45%或至少50%的敏感度。在一个方面,确定一癌症分类的步骤进一步包含:(a)基于多个序列读数的集合生成一测试特征向量;以及(b)将所述测试特征向量应用于一分类器。在一个方面,所述分类器包含一模型,所述模型通过一训练程序训练,所述训练过程具有来自一个或多个患有癌症的训练对象的多个片段的一癌症集合和来自一个或多个没有癌症的训练对象的多个片段的一非癌症集合,其中所述多个片段的所述癌症集合和所述多个片段的所述非癌症集合均包含多个训练片段。
在一个方面,所述类型的癌症选自由以下组成的群组:头颈癌、肝癌/胆道癌、上消化道癌、胰腺癌/胆囊癌、结直肠癌、卵巢癌、肺癌、多发性骨髓瘤、淋巴肿瘤、黑色素瘤、肉瘤、乳腺癌和子宫癌。在一个方面,在分类为头颈癌,及在99.4%特异性下,所述分类器可以具有至少70%、至少80%、至少85%、或至少87%的敏感度。在分类是肝癌/胆道癌,及在99.4%特异性下,所述分类器可以具有至少60%、至少65%、至少70%、或至少73%的敏感度。在分类为上消化道癌,及在99.4%特异性下,所述分类器可以具有至少70%、至少75%、至少80%、或至少85%的敏感度。在分类为胰腺癌或胆囊癌,及在99.4%特异性下,所述分类器可以具有至少70%、至少80%、至少85%、或至少90%的敏感度。在分类为结直肠癌,及在99.4%特异性下,所述分类器可以具有至少70%、至少80%、至少90%、至少95%、或至少98%的敏感度。在分类为卵巢癌,及在99.4%特异性下,所述分类器可以具有至少60%、至少70%、至少80%、至少85%、或至少87%的敏感度。在分类为肺癌,及在99.4%特异性下,所述分类器可以具有至少70%、至少80%、至少90%、至少95%、或至少97%的敏感度。在分类为多发性骨髓瘤,及在99.4%特异性下,所述分类器可以具有至少70%、至少80%、至少85%、或至少90%、或至少93%的敏感度。在分类为淋巴肿瘤,及在99.4%特异性下,所述分类器可以具有至少70%、至少80%、至少90%、或至少95%、或至少98%的敏感度。在分类为黑色素瘤,及在99.4%特异性下,所述分类器可以具有至少70%、至少80%、至少90%、或至少95%、或98%的敏感度。在分类为肉瘤,及在99.4%特异性下,所述分类器可以具有至少35%、至少40%、至少45%、或至少50%的敏感度。在分类为乳腺癌,及在99.4%特异性下,所述分类器可以具有至少70%、至少80%、至少90%、或至少95%、或至少98%的敏感度。在分类为子宫癌,及在99.4%特异性下,所述分类器可以具有至少70%、至少80%、至少90%、或至少95%、或至少97%的敏感度。
在另一个方面,本文提供一种用于确定一癌症分类的步骤包含步骤:(a)基于多个序列读数的集合生成一测试特征向量;以及(b)将所述测试特征向量应用于通过一训练程序获得一模型,所述模型具有来自具有癌症的一个或多个训练对象的多个片段的一癌症集合和来自不具有癌症的一个或多个训练对象的多个片段的一非癌症集合,其中所述多个片段的所述癌症集合和所述多个片段的所述非癌症集合均包含多个训练片段。所述训练程序可以包含:(a)从多个训练对象中获取多个训练片段的序列信息;(b)对于每一个训练片段,确定所述训练片段是低甲基化或高甲基化,其中所述多个低甲基化和高甲基化的训练片段中的每一个包含至少一个阈值数的多个CpG位点,所述多个CpG位点分别具有至少一个阈值百分比为未甲基化或甲基化;(c)
对于每一个训练对象,基于所述多个低甲基化的训练片段及基于所述多个高甲基化的训练片段产生一训练特征向量;及(d)利用来自不具有癌症的一个或多个训练对象的所述多个训练特征向量和来自具有癌症的一个或多个训练对象的所述多个训练特征向量训练所述模型。在另一个方面,所述训练程序包含:(a)从多个训练对象中获取多个训练片段的序列信息;(b)对于每一个训练片段,确定所述训练片段是低甲基化或高甲基化,其中所述多个低甲基化和高甲基化的训练片段中的每一个包含至少一个阈值数的多个CpG位点,所述多个CpG位点分别具有至少一个阈值百分比为未甲基化或甲基化;(c)对于在一参考基因组中的多个CpG位点中的每一个:量化与所述CpG位点重叠的多个低甲基化的训练片段的一数量和与所述CpG位点重叠的多个高甲基化的训练片段的一数量;及基于多个低甲基化的训练片段和多个高甲基化的训练片段的所述数量,生成一低甲基化得分和一高甲基化得分;(d)对于每一个训练片段,基于所述训练片段中所述多个CpG位点的所述低甲基化得分生成一总合的低甲基化得分,以及基于所述训练片段中所述多个CpG位点的所述高甲基化得分生成一总合的高甲基化得分;(e)对于每一个训练对象:基于总合的低甲基化得分对所述多个训练片段进行排名,并基于总合的高甲基化得分对所述多个训练片段进行排名;及基于所述多个训练片段的所述排名生成一特征向量;(f)获取不具有癌症的一个或多个训练对象的多个训练特征向量,以及具有癌症的一个或多个训练对象的多个训练特征向量;以及(g)利用所述不具有癌症的一个或多个训练对象的所述多个特征向量及具有癌症的一个或多个训练对象的所述多个特征向量训练所述模型。所述模型可以包含一内核逻辑回归分类器、一随机森林分类器、一混合模型、一卷积神经网络和一自动编码器模型中的一种。在另一个方面,所述方法还包含以下步骤:基于所述模型获得测试样本的一癌症概率;以及将所述癌症概率与一阈值概率进行比较,以确定所述测试样本是否来自具有癌症的一患者或不具有癌症的一患者。所述方法还可以包含:对所述对象施用一抗癌剂。
在另一方面,本文提供一种治疗癌症患者的方法,所述方法包含:通过上面所述方法识别为一癌症对象,及向一对象施用一抗癌剂。所述抗癌剂可以是选自以下群组所组成的化学治疗剂:烷基化剂、抗代谢物、蒽环类、抗肿瘤抗生素、细胞骨架破坏剂(紫杉醇)、拓扑异构酶抑制剂、有丝分裂抑制剂、皮质类固醇、激酶抑制剂、核苷酸类似物和铂基药物。
本文提供一种评估一对象是否患有癌症的方法,所述方法可以包含:
获取所述对象cfDNA;通过杂交捕获从所述对象分离cfDNA的一部分;从捕获的cfDNA获得多个序列读数,以确定多个cfDNA片段的甲基化状态;将一分类器应用于所述多个序列读数;以及
基于所述分类器的应用判断所述对象是否具有癌症;其中所述分类器在接收者操作特性曲线下具有大于0.70、大于0.75、大于0.77、大于0.80、大于0.81、大于0.82、或大于0.83的一面积。在一个方面,所述方法还包含:在通过杂交捕获从所述对象分离cfDNA的所述部分之前,将cfDNA中的未甲基化的胞嘧啶转化为尿嘧啶。在另一个方面,所述方法还包含:在通过杂交捕获从所述对象分离cfDNA的所述部分后,将cfDNA中的未甲基化的胞嘧啶转化为尿嘧啶。所述分类器可以是二进制分类器。在一个方面,通过杂交捕获从所述对象分离cfDNA的一部分可以包含:将所述无细胞DNA与不同的多个含有寡核苷酸的探针的一诱饵组相接触。
在另一个方面,本文提供一种用于识别在多个癌症样本中相对于多个非癌症样本列表现出差异甲基化的多个基因组区域的方法,所述方法可以包含:(a)从多个癌症对象和多个非癌症对象获得转化的cfDNA的多个序列读数;(b)基于所述多个序列读数,确定多个cfDNA片段:(i)在多个非癌性样本中具有一p值稀有度低于一阈值;及(ii)具有至少X个CpG位点,其中至少Y%的CpG位点被甲基化,其中X为至少4、5、6、7、8、9或10,及Y为至少70;(c)对于一参考基因组中的多个CpG位点中的每一个,计算具有在步骤(b)中识别的一片段的:(1)多个癌症对象的一数量(N
(a)从多个癌症对象和多个非癌症对象获得转化的cfDNA的多个序列读数;(b)基于所述多个序列读数,确定多个cfDNA片段:(i)具有至少X个CpG位点,其中至少Y%的CpG位点未甲基化,其中X为4、5、6、7、8、9或10及Y为至少70;及(ii)在多个非癌性样本中具有一p值稀有度低于一阈值;
(c)对于一参考基因组中的多个CpG位点中的每一个,计算具有在步骤(b)中识别的一片段的:(1)多个癌症对象的一数量(N
在一个方面,本文提供一种用于选择cfDNA杂交捕获的多个探针的方法,所述方法可以包含:识别一第一集合的多个基因组区域,所述第一集合的多个基因组区域为多个癌症对象相对于多个非癌症对象在cfDNA是优先高甲基化的;识别一第二集合的多个基因组区域,所述第二集合的多个基因组区域为多个癌症对象相对于多个非癌症对象在cfDNA是优先低甲基的;以及选择对应所述第一集合的多个基因组区域及所述第二集合的多个基因组区域的cfDNA杂交捕获的多个探针,其中所述多个探针可以包含一第一集合的多个探针及一第二集合的多个探针,所述第一集合的多个探针对应所述第一集合的多个基因组区域的cfDNA杂交捕获,及所述第二集合的多个探针对应所述第二集合的多个基因组区域的cfDNA杂交捕获;其中所述多个探针可以包含至少500、至少1000、至少2500、至少5000、至少10000、至少20000个探针子集,其中每个探针子集可以包含以2倍平铺的方式延伸跨过一基因组区域的多个探针。用于杂交捕获的所述第二集合的多个探针可以包含以下步骤:选择仅通过一个或多个转换与所述基因组区域中的一序列不同的多个探针,其中每个转换发生在所述基因组区域中对应于一胞嘧啶的一核苷酸处。在一个方面,选择用于杂交捕获的多个探针的步骤可以包含:滤除具有超过多个脱标靶区域的一阈值数量的多个探针。每个探针子集可以包含至少三个探针。每个探针的长度为75到200个核苷酸之间、100到150个核苷酸之间、110到130个核苷酸之间、或120个核苷酸。
在另一个方面,本文提供一种用于扩增供癌症诊断的多个cfDNA分子的化验板,所述化验板可以包含:至少不同的500对的多核苷酸探针,其中所述至少500对的探针中的每一对:(i)包含两个不同的探针,配置为通过30个或更多个核苷酸的一重叠序列彼此相重叠,及(ii)被配置为与从所述多个cfDNA分子的处理中获得的一修饰的片段杂交,其中所述多个cfDNA分子中的每一个对应于或衍生自一个或多个基因组区域,其中所述一个或多个基因组区域中的每一个可以包含至少5个甲基化位点以及在多个癌性训练样本中相对于多个非癌性训练样本具有一异常甲基化模式。所述重叠序列可以包含至少40、50、75或100个核苷酸。所述化验板可以包含至少1000、2000、2500、5000、6000、7500、10000、15000、20000或25000对探针。
在另一个方面,本文提供一种用于扩增供癌症诊断的多个cfDNA分子的化验板,所述化验板可以包含:至少1000个多核苷酸探针,其中所述至少1000个探针中的每一个被配置成与一修饰的多核苷酸杂交,所述修饰的多核苷酸杂交从所述多个cfDNA分子的处理中获得,其中所述多个cfDNA分子中的每一个对应于或衍生自一个或多个基因组区域,其中所述一个或多个基因组区域中的每一个可以包含至少5个甲基化位点以及在多个癌性训练样本中相对于多个非癌性训练样本具有一异常甲基化模式。所述多个cfDNA分子的处理可以包含:将所述多个cfDNA分子中的未甲基化C(胞嘧啶)转化为U(尿嘧啶)。所述化验板上的所述多个多核苷酸探针中的每一个与一亲和部分接合。所述亲和部分为一生物素部分。所述一个或多个基因组区域中的每一个在所述多个癌性训练样本中相对于多个非癌性参考样本是高甲基化或低甲基化。在一个方面,所述化验板上至少80%、85%、90%、92%、95%或98%的探针在多个CpG检测位点上仅具有CpG或仅具有CpA。所述化验板上的所述多个探针中的每一个可以包含少于20、15、10、8或6个CpG检测位点。所述化验板上的所述多个探针中的每一个被设计为少于20、15、10或8个脱靶基因组区域。在一个方面,所述少于20个脱靶基因组区域使用k-mer接种策略来识别。在另一个方面,所述少于20个脱靶基因组区域使用k-mer接种策略结合在多个接种位置处的局部对位来识别。
所述化验板可以包含至少1000、2000、2500、5000、10000、12000、15000、20000或25000个探针。在一个方面,所述至少500对探针或所述至少1000个探针同时可以包含至少20万、40万、60万、80万、100万、200万、400万、或600万个核苷酸。所述化验板上的所述多个探针中的每一个可以包含至少50、75、100或120个核苷酸。所述化验板上的所述多个探针中的每一个可以包含少于300、250、200或150个核苷酸。所述化验板上的所述多个探针中的每一个可以包含100至150个核苷酸。在一个方面,所述多个基因组区域的至少30%是外显子或内含子。在另一个方面,所述多个基因组区域的至少15%是外显子。在另一个方面,所述多个基因组区域的至少20%是外显子。所述一个或多个基因组区域中的每一个可以选自列表1至列表8中的一个。所述化验板上的多个探针的一整体一起被配置为与多个修饰的片段杂交,所述多个修饰的片段可以从对应于或衍生自列表1至列表8的一个或多个中的至少30%、40%、50%、60%、70%、80%、90%或95%的所述多个基因组区域的所述多个cfDNA分子获得。所述化验板上的多个探针的一整体部一起被配置为与多个修饰的片段杂交,所述多个修饰的片段从对应于或衍生自列表1至列表8中的至少500、1000、5000、10000或15000个基因的所述多个cfDNA分子获得。所述cfDNA分子的处理可包含将所述cfDNA分子中未甲基化的C(胞嘧啶)转化为U(尿嘧啶)。所述化验板上的多个探针中的每一个都可以与一亲和部分相结合。所述亲和部分可以是生物素。所述化验板上至少80%、85%、90%、92%、95%或98%的探针可以在多个CpG检测点上仅具有CpG或仅具有CpA。
在另一方面,本文提供一种提供癌症存在或不存在的信息的序列信息的方法,所述方法可以包含以下步骤:(a)获取一测试样本,所述测试样本可以包含多个cfDNA测试分子;(b)处理所述多个cfDNA测试分子,从而获得多个转化的测试片段;(c)将所述多个转化的测试片段与一化验板相接触,从而通过杂交捕获来扩增所述多个转化的测试片段的一子集;及(d)对所述多个转化的测试片段的所述子集进行测序,从而获得多个序列读数的一集合。所述多个转化的测试片段可以是多个亚硫酸氢盐转化的测试片段。在另一个方面,所述方法还可以包含以下步骤:通过评估所述多个序列读数的所述集合确定一癌症分类,其中所述癌症分类为:(a)癌症存在或不存在;(b)癌症的一阶段;或(c)一种类型的癌症存在或不存在。在一个方面,所述确定一癌症分类的步骤可以包含以下步骤:(a)基于所述多个序列读数的所述集合生成一测试特征向量;以及(b)将所述测试特征向量应用于一分类器。所述分类器可以包含一模型,所述模型通过一训练程序训练,所述模型具有来自具有癌症的一个或多个训练对象的多个片段的一癌症集合和来自不具有癌症的一个或多个训练对象的多个片段的一非癌症集合,其中所述多个片段的所述癌症集合和所述多个片段的所述非癌症集合均可以包含多个训练片段。所述分类器在接收者操作特性曲线下可以具有大于0.70、大于0.75、大于0.77、大于0.80、大于0.81、大于0.82或大于0.83的一面积。所述分类器可以具有至少35%、至少40%、至少45%或至少50%的敏感度。在另一个方面,确定一癌症分类的步骤还包含:(a)基于多个序列读数的集合生成一测试特征向量;以及(b)将所述测试特征向量应用于一分类器。所述分类器可以包含一模型,所述模型通过一训练程序训练,所述训练过程具有来自一个或多个患有癌症的训练对象的多个片段的一癌症集合和来自一个或多个没有癌症的训练对象的多个片段的一非癌症集合,其中所述多个片段的所述癌症集合和所述多个片段的所述非癌症集合均包含多个训练片段。
所述类型的癌症选自由以下组成的群组:头颈癌、肝癌/胆道癌、上消化道癌、胰腺癌/胆囊癌、结直肠癌、卵巢癌、肺癌、多发性骨髓瘤、淋巴肿瘤、黑色素瘤、肉瘤、乳腺癌和子宫癌。在一个方面,对于分类为头颈癌,及在99.4%特异性下,所述分类器具有至少70%、至少80%、至少85%、或至少87%的敏感度。对于分类是肝癌/胆道癌,及在99.4%特异性下,所述分类器具有至少60%、至少65%、至少70%、或至少73%的敏感度。对于分类为上消化道癌,及在99.4%特异性下,所述分类器具有至少70%、至少75%、至少80%、或至少85%的敏感度。对于分类为胰腺癌/胆囊癌,及在99.4%特异性下,所述分类器具有至少70%、至少80%、至少85%、或至少90%的敏感度。对于分类为结直肠癌,及在99.4%特异性下,所述分类器具有至少70%、至少80%、至少90%、至少95%、或至少98%的敏感度。对于分类为卵巢癌,及在99.4%特异性下,所述分类器具有至少70%、至少80%、至少85%、或至少87%的敏感度。对于分类为肺癌,及在99.4%特异性下,所述分类器具有至少70%、至少80%、至少90%、至少95%、或至少97%的敏感度。对于分类为多发性骨髓瘤,及在99.4%特异性下,所述分类器具有至少70%、至少80%、至少85%、或至少90%、或至少93%的敏感度。对于分类为淋巴肿瘤,及在99.4%特异性下,所述分类器具有至少70%、至少80%、至少90%、或至少95%、或至少98%的敏感度。对于分类为黑色素瘤,及在99.4%特异性下,所述分类器具有至少70%、至少80%、至少90%、或至少95%、或98%的敏感度。对于分类为肉瘤,及在99.4%特异性下,所述分类器具有至少35%、至少40%、至少45%、或至少50%的敏感度。对于分类为乳腺癌,及在99.4%特异性下,所述分类器具有至少70%、至少80%、至少90%、或至少95%、或至少98%的敏感度。对于分类为子宫癌,及在99.4%特异性下,所述分类器具有至少70%、至少80%、至少90%、或至少95%、或至少97%的敏感度。
在一个方面,所述癌症分类的方法可以进一步包含步骤:(a)基于所述多个序列读数的所述集合生成一测试特征向量;以及(b)将所述测试特征向量应用于通过一训练程序获得一模型,所述模型具有来自具有癌症的一个或多个训练对象的多个片段的一癌症集合和来自不具有癌症的一个或多个训练对象的多个片段的一非癌症集合,其中所述多个片段的所述癌症集合和所述多个片段的所述非癌症集合均可以包含多个训练片段。所述训练程序可以包含步骤:(a)从多个训练对象中获取多个训练片段的序列信息;(b)对于每一个训练片段,确定所述训练片段是低甲基化或高甲基化,其中所述多个低甲基化和高甲基化的训练片段中的每一个可以包含至少一个阈值数的多个CpG位点,所述多个CpG位点分别具有至少一个阈值百分比为未甲基化或甲基化;(c)对于每一个训练对象,基于所述多个低甲基化的训练片段产生一训练特征向量及基于所述多个高甲基化的训练片段产生一训练特征向量;及(d)利用来自不具有癌症的一个或多个训练对象的所述多个训练特征向量和来自具有癌症的一个或多个训练对象的所述多个训练特征向量训练所述模型。所述训练程序可以进一步包含:(a)从多个训练对象中获取多个训练片段的序列信息;(b)对于每一个训练片段,确定所述训练片段是低甲基化或高甲基化,其中所述多个低甲基化和高甲基化的训练片段中的每一个可以包含至少一个阈值数的多个CpG位点,所述多个CpG位点分别具有至少一个阈值百分比为未甲基化或甲基化;(c)对于在一参考基因组中的多个CpG位点中的每一个:量化与所述CpG位点重叠的多个低甲基化的训练片段的一数量和与所述CpG位点重叠的多个高甲基化的训练片段的一数量;及基于多个低甲基化的训练片段和多个高甲基化的训练片段的所述数量,生成一低甲基化得分和一高甲基化得分;(d)对于每一个训练片段,基于所述训练片段中所述多个CpG位点的所述低甲基化得分生成一总合的低甲基化得分,以及基于所述训练片段中所述多个CpG位点的所述高甲基化得分生成一总合的高甲基化得分;(e)对于每一个训练对象:基于总合的低甲基化得分对所述多个训练片段进行排名,并基于总合的高甲基化得分对所述多个训练片段进行排名;及基于所述多个训练片段的所述排名生成一特征向量;(f)获取不具有癌症的一个或多个训练对象的多个训练特征向量,以及具有癌症的一个或多个训练对象的多个训练特征向量;以及(g)利用所述不具有癌症的一个或多个训练对象的所述多个特征向量及具有癌症的一个或多个训练对象的所述多个特征向量训练所述模型。所述模型可以包含一内核逻辑回归分类器、一随机森林分类器、一混合模型、一卷积神经网络和一自动编码器模型中的一种。在一个方面,所述癌症分类的方法进一步可以包含:(a)基于所述模型获得测试样本的一癌症概率;以及(b)将所述癌症概率与一阈值概率进行比较,以确定所述测试样本是否来自具有癌症的一患者或不具有癌症的一患者。在一个方面,所述识别一癌症对象的方法进一步可以包含:对所述对象施用一抗癌剂。所述抗癌剂是选自以下群组组成的化学治疗剂:烷基化剂、抗代谢物、蒽环类、抗肿瘤抗生素、细胞骨架破坏剂(紫杉醇)、拓扑异构酶抑制剂、有丝分裂抑制剂、皮质类固醇、激酶抑制剂、核苷酸类似物和铂基药物。
在另一个方面,本文提供一种方法,所述方法可以包含步骤:(a)获得多个修饰的测试片段的多个序列读数的一集合,其中所述多个修饰的测试片段是或已经通过处理来自一测试对象的一组的多个核酸片段而获得,其中所述多个核酸片段中的每一个对应于或衍生自选自列表1至列表8中的任何一个中的多个基因组区域;以及(b)将所述多个序列读数的所述集合或基于所述多个序列读数的所述集合获得的一测试特征向量应用于通过一训练程序获得的一模型,所述模型具有来自具有癌症的一个或多个训练对象的多个片段的一癌症集合和来自不具有癌症的一个或多个训练对象的多个片段的一非癌症集合,其中所述多个片段的所述癌症集合及所述多个片段的所述非癌症集合可以包含多个训练片段。所述方法进一步可以包含:获得所述测试特征向量的步骤,所述步骤可以包含:(a)对于所述多个核酸片段中的每一个,确定所述核酸片段是低甲基化或高甲基化,其中低甲基化和高甲基化的所述多个核酸片段中的每一个可以包含至少一个阈值数的多个CpG位点,所述多个CpG位点分别具有至少一个阈值百分比为未甲基化或甲基化;(b)对于一参考基因组中的多个CpG位点中的每一个:量化与所述CpG位点重叠的低甲基化的多个核酸片段的一数量和与所述CpG位点重叠的高甲基化的多个核酸片段的一数量;及
基于低甲基化的多个核酸片段和高甲基化的多个核酸片段的所述数量,生成一低甲基化得分和一高甲基化得分;(c)对于每一个核酸片段,基于所述核酸片段中所述多个CpG位点的所述低甲基化得分生成一总合的低甲基化得分和基于所述核酸片段中所述多个CpG位点的所述高甲基化得分生成一总合的高甲基化得分;(d)基于总合的低甲基化得分对所述多个核酸片段进行排名,及基于总合的高甲基化得分对所述多个核酸片段进行排名;以及(e)基于所述多个核酸片段的所述排名生成所述测试特征向量。所述训练程序可以包含步骤:(a)对于每一个训练片段,确定所述训练片段是低甲基化或高甲基化,其中所述多个低甲基化和高甲基化的训练片段中的每一个可以包含至少一个阈值数的多个CpG位点,所述多个CpG位点分别具有至少一个阈值百分比为未甲基化或甲基化;(b)对于每一个训练对象,基于所述多个低甲基化的训练片段产生一训练特征向量及基于所述多个高甲基化的训练片段产生一训练特征向量;及(c)利用来自不具有癌症的一个或多个训练对象的所述多个训练特征向量和来自具有癌症的一个或多个训练对象的所述多个特征向量训练所述模型。所述训练程序可以包含步骤:(a)对于每一个训练片段,确定所述训练片段是低甲基化或高甲基化,其中所述多个低甲基化和高甲基化的训练片段中的每一个可以包含至少一个阈值数的多个CpG位点,所述多个CpG位点分别具有至少一个阈值百分比为未甲基化或甲基化;(b)对于在一参考基因组中的多个CpG位点中的每一个:量化与所述CpG位点重叠的多个低甲基化的训练片段的一数量和与所述CpG位点重叠的多个高甲基化的训练片段的一数量;及基于多个低甲基化的训练片段和多个高甲基化的训练片段的所述数量,生成一低甲基化得分和一高甲基化得分;(c)对于每一个训练片段,基于所述训练片段中所述多个CpG位点的所述低甲基化得分生成一总合的低甲基化得分,以及基于所述训练片段中所述多个CpG位点的所述高甲基化得分生成一总合的高甲基化得分;(d)对于每一个训练对象:
基于总合的低甲基化得分对所述多个训练片段进行排名,并基于总合的高甲基化得分对所述多个训练片段进行排名;及
基于所述多个训练片段的所述排名生成一特征向量;(e)获取不具有癌症的一个或多个训练对象的多个训练特征向量,以及具有癌症的一个或多个训练对象的多个训练特征向量;以及(f)利用所述不具有癌症的一个或多个训练对象的所述多个特征向量及具有癌症的一个或多个训练对象的所述多个特征向量训练所述模型。
在一个方面,所述方法可以进一步包含:对于一参考基因组中的每一个CpG位点,量化与所述CpG位点重叠的多个低甲基化的训练片段的一数量以及与所述CpG位点重叠的多个高甲基化的训练片段的一数量进一步可以包含步骤:(a)量化来自与所述CpG位点重叠的具有癌症的一个或多个训练对象的多个低甲基化的训练片段的一癌症数量,以及量化来自与所述CpG位点重叠的不具有癌症的一个或多个训练对象的多个低甲基化的训练片段的一非癌症数量;以及(b)量化来自与所述CpG位点重叠的具有癌症的一个或多个训练对象的多个高甲基化的训练片段的一癌症数量,以及量化来自与所述CpG位点重叠的不具有癌症的一个或多个训练对象的多个高甲基化的训练片段的一非癌症数量。对于一参考基因组中的每一个CpG位点,基于多个低甲基化的训练片段和多个高甲基化的训练片段的所述数量来生成一低甲基化得分和一高甲基化得分还可以包含步骤:(a)对于生成所述低甲基化得分,计算多个低甲基化的训练片段的所述癌症数量与多个低甲基化的训练片段的所述癌症数量及多个低甲基化的训练片段的所述非癌症数量的一低甲基化总合的一低甲基化比率;以及(b)对于生成所述高甲基化得分,计算多个高甲基化的训练片段的所述癌症数量与多个高甲基化的训练片段的所述癌症数量及多个高甲基化的训练片段的所述非癌症数量的一高甲基化总合的一高甲基化比率。所述模型可以包含所述模型可以包含一内核逻辑回归分类器、一随机森林分类器、一混合模型、一卷积神经网络和一自动编码器模型中的一种。在一个方面,所述多个序列读数的所述集合可以是通过使用如本发明所述的化验板获得。
在一个方面,本文提供一种设计用于癌症诊断的化验板的方法,所述方法可以包含以下步骤:(a)识别多个基因组区域,其中所述多个基因组区域中的每一个:(i)可以包含至少30个核苷酸,及(ii)包含至少5个甲基化位点;(b)选择所述多个基因组区域的一子集,其中所述选择是当对应于或衍生自多个癌症训练样本中的所述多个基因组区域的每一个的多个cfDNA分子具有一异常甲基化模式时实行,其中所述异常甲基化模式包含至少5个甲基化位点已知为或被识别为的低甲基化或高甲基化;以及(c)设计所述化验板,所述化验板包含多个探针,其中所述多个探针中的每一个被配置成与从处理对应于或衍生自所述多个基因组区域的所述子集的一个或多个的多个cfDNA分子而获得的一修饰的片段杂交。所述多个cfDNA分子的处理可以包含将所述多个cfDNA分子中的未甲基化C(胞嘧啶)转化为U(尿嘧啶)。
在另一个方面,本文提供一种癌症化验板,所述癌症化验板可以包含:至少500对探针,其中所述至少500对的每一对包含两个探针,配置为通过一重叠序列彼此相重叠,其中所述重叠序列包含一30个核苷酸序列,及其中所述30个核苷酸序列被配置为与一个或多个基因组区域具有序列互补性,其中所述一个或多个基因组区域具有至少五个甲基化位点,及其中所述至少五个甲基化位点在多个非癌性样本或多个癌性样本中具有一异常甲基化模式。所述重叠序列可以包含至少40、50、75或100个核苷酸。所述癌症化验板包含至少1000、2000、2500、5000、6000、7500、10000、15000、20000或25000对探针。
在另一个方面,本文提供一种癌症化验板,所述癌症化验板可以包含:至少1000个探针,其中所述多个探针中的每一个被设计为一杂交探针与一个或多个基因组区域互补,其中所述多个基因组区域中的每一个可以包含:(i)至少30个核苷酸,以及(ii)至少五个甲基化位点,其中所述至少五个甲基化位点具有一异常甲基化模式,及在多个癌症样本或多个非癌症样本中为低甲基化或高甲基化。所述异常甲基化模式在所述多个非癌性样本中具有至少一阈值p值稀有度。在一个方面,所述多个探针中的每一个被设计为具有少于20个脱靶基因组区域。在一个方面,所述少于20个脱靶基因组区域使用k-mer接种策略来识别。在另一个方面,所述少于20个脱靶基因组区域使用k-mer接种策略结合在多个接种位置处的局部对位来识别。
在一个方面,基于标准可以被选择的所述多个基因组区域中的每一个,所述标准包含:(a)多个癌性样本包含具有所述异常甲基化模式的至少一个cfDNA片段的一数量(N
在一个方面,所述多个探针中的每一个被配置为具有少于20、15、10或8个脱靶基因组区域。在一个方面,所述多个基因组区域的至少30%是外显子或内含子。在另一个方面,所述多个基因组区域的至少15%是外显子。在另一个方面,所述多个基因组区域的至少20%是外显子。在另一个方面,少于10%的所述多个基因组区域是多个基因间区域。所述多个基因组区域选自列表1至列表8中的任一个。所述多个基因组区域选自列表3。所述多个基因组区域选自列表8。所述多个基因组区域包含列表1至列表8的任一个中至少30%、40%、50%、60%、70%、80%、90%或95%的基因组区域。所述多个基因组区域包含列表3中至少30%、40%、50%、60%、70%、80%、90%或95%的基因组区域。所述多个基因组区域包含列表5中至少20%、30%、40%、50%、60%、70%、80%、90%或95%的基因组区域。所述基因组区域包含列表8中至少20%、30%、40%、50%、60%、70%、80%、90%或95%的基因组区域。在一个方面,所述至少1000或至少2000个探针被配置为与列表1至列表8的任一个中的至少500、1000、5000、10000或15000个基因组区域互补。在另一个方面,所述至少1000或至少2000个探针被配置为与列表3中的至少500、1000、5000、10000或15000个基因组区域互补。在另一个方面,所述至少1000或至少2000个探针被配置为与列表5中的至少500、1000、5000、10000或15000个基因组区域互补。在另一个方面,所述至少1000或至少2000个探针被配置为与列表8中的至少1000、5000、10000或15000个基因组区域互补。所述30个核苷酸序列包含至少5个CpG检测位点,其中所述至少5个CpG检测位点中的至少80%包含CpG、UpG或CpA。
本文提供一种癌症化验的方法,所述方法包含步骤:接收一样本,所述样本包含多个核酸片段;处理所述多个核酸片段以将未甲基化的胞嘧啶转化为尿嘧啶,从而获得多个转化的核酸片段;将所述多个转化的核酸片段与上述权利要求任一项所述的癌症化验板上的所述多个探针相杂交;扩增所述多个转化的核酸片段的一子集;以及
对所述多个转化的核酸片段的扩增子集进行测序,从而提供多个序列读数的一集合。所述方法还包含以下步骤:通过评估所述多个序列读数的所述集合来确定一健康状况,其中所述健康状况是(i)癌症的存在或不存在,或(ii)癌症的一阶段。所述多个核酸片段的所述集合是从一人类对象获得。
在另一个方面,本文提供一种诊断癌症的方法,所述方法包含步骤:
(a)通过对来自一对象的多个核酸片段的一集合进行测序来获得多个序列读数的一集合;
(b)确定多个基因组区域的甲基化状态,所述多个基因组区域包含从列表1至列表8中的任一个的多个基因组区域中选择的多个基因组区域;以及(c)通过评估所述甲基化状态来确定所述对象的一健康状况,其中所述健康状况是(i)癌症的存在或不存在;或(ii)癌症的一阶段。所述多个基因组区域选自列表3。所述多个基因组区域选自列表5。所述多个基因组区域选自列表8。
在另一个方面,本文提供一种用于诊断癌症的方法,所述方法包含步骤:
(a)通过对来自一对象的多个核酸片段的一集合进行测序获得多个序列读数的一集合;
(b)确定多个基因组区域的甲基化状态,所述多个基因组区域包含列表1至列表8中任何一个中至少20%、30%、40%、50%、60%、70%、80%、90%或95%的基因组区域;以及(c)通过评估所述甲基化状态来确定所述对象的一健康状况,其中所述健康状况是(i)癌症的存在或不存在;或(ii)癌症的一阶段。所述多个基因组区域包含列表3中的至少20%、30%、40%、50%、60%、70%、80%、90%或95%的基因组区域。所述多个基因组区域包含列表5中的至少20%、30%、40%、50%、60%、70%、80%、90%或95%的基因组区域。所述多个基因组区域包含列表8中的至少20%、30%、40%、50%、60%、70%、80%、90%或95%的基因组区域。
在另一个方面,本文提供一种诊断癌症的方法,所述方法包含以下步骤:
(a)通过对来自一对象的多个核酸片段的一集合进行测序获得多个序列读数的一集合;
(b)确定列表1至列表8中的任一个的多个基因组区域中至少1000、2000、2500、5000、6000、7500、10000、15000、20000或25000个基因组区域中的甲基化状态;以及(c)通过评估所述甲基化状态来确定所述对象的一健康状况,其中所述健康状况是(i)癌症的存在或不存在;或(ii)癌症的一阶段。在一个方面,至少1000、2000个探针被配置为与列表3中的至少500、1000、5000、10000或15000个基因组区域互补。在一个方面,至少1000、2000个探针被配置为与列表5中的至少500、1000、5000、10000或15000个基因组区域互补。在一个方面,至少1000、2000个探针被配置为与列表8中的至少500、1000、5000、10000或15000个基因组区域互补。
在另一个方面,一种设计一癌症化验板的方法,所述方法包含步骤:识别多个基因组区域,其中所述多个基因组区域中的每一个(i)包含至少30个核苷酸,及(ii)包含至少五个甲基化位点,其中所述至少五个甲基化位点为低甲基化或高甲基化;比较多个癌性样本和多个非癌性样本之间的多个基因组区域中的每一个中的所述至少五个甲基化位点的甲基化状态;选择所述多个基因组区域的一子集,其中在多个癌性样本中所述多个基因组区域的所述子集的至少五个甲基化位点相对于多个非癌性样本具有一异常甲基化模式,以及设计一癌症化验板,所述癌症化验板包含多个探针集合,其中所述多个探针集合中的每一个包含至少一对探针,所述至少一对探针被配置成标靶所述多个基因组区域的子集中的一个。在一个方面,所述异常甲基化模式与一cfDNA片段的异常甲基化模式匹配,所述cfDNA片段来自所述多个癌性样本与所述至少五个甲基化位点中的至少一个相重叠,其中所述cfDNA相对于所述多个非癌性样本的一训练数据集具有至少一阈值p值稀有度。在一个方面,所述方法进一步包含所述选择步骤是基于一标准执行,所述标准包含:(a)所述多个癌性样本包含具有所述异常甲基化模式的多个cfDNA片段的所述多个癌性样本的一数量N
在另一个方面,本文提供一种癌症化验板,包含通过如上所述的方法制备的多个探针。所述癌症化验板中多个基因组区域的所述子集可以包含列表1至列表8中任一个的多个基因组区域。所述癌症化验板中多个基因组区域的所述子集可以包含列表3的多个基因组区域。所述癌症化验板中多个基因组区域的所述子集可以包含列表5的多个基因组区域。所述癌症化验板中多个基因组区域的所述子集可以包含列表8的多个基因组区域。所述癌症化验板中多个基因组区域的所述子集可以包含列表1至列表8中一个或多个中的至少20%、30%、40%、50%、60%、70%、80%或90%的基因组区域。所述癌症化验板中多个基因组区域的所述子集可以包含列表3中的至少20%、30%、40%、50%、60%、70%、80%或90%的基因组区域。所述癌症化验板中多个基因组区域的所述子集可以包含列表5中的至少20%、30%、40%、50%、60%、70%、80%或90%的基因组区域。所述癌症化验板中多个基因组区域的所述子集可以包含列表8中的至少20%、30%、40%、50%、60%、70%、80%或90%的基因组区域。所述癌症化验板中多个基因组区域的所述子集可以包含列表1至列表8中任何一个中的至少500、1000、5000、10000、15000、20000或25000个基因组区域。所述癌症化验板中多个基因组区域的所述子集可以包含列表3中的至少500、1000、5000、10000、15000、20000或25000个基因组区域。所述癌症化验板中多个基因组区域的所述子集可以包含列表5中的至少500、1000、5000、10000、15000、20000或25000个基因组区域。所述癌症化验板中多个基因组区域的所述子集可以包含列表8中的至少500、1000、5000、10000、15000、20000或25000个基因组区域。
在另一个方面,本文提供一种癌症化验板,所述癌症化验板包含多个探针,其中所述多个探针中的每一个被配置成与列表1至列表8中的任一个中的多个基因组区域中的一个相重叠,及所述多个探针与列表1至列表8中的任一个中的至少90%、95%或100%的基因组区域相重叠在一起。在另一个方面,本文提供一种癌症化验板,所述癌症化验板包含多个探针,其中所述多个探针中的每一个被配置成与列表1至列表8中的任一个中的多个基因组区域中的一个相重叠,及所述多个探针与列表1至列表8中任一个中的至少500、1000、5000、10000或15000个基因组区域重叠在一起。
本说明书提供一种癌症化验板,所述癌症化验板包含:至少500对探针,其中所述至少500对的每一对包含两个探针,配置为通过一重叠序列彼此相重叠,其中所述重叠序列包含一30个核苷酸片段,及其中所述30个核苷酸片段包含至少五个CpG位点,其中所述至少五个CpG位点中的至少80%包含CpG或所述至少五个CpG位点中的至少80%包含UpG,及其中所述30个核苷酸片段被配置以结合到多个癌性样本中的一个或多个基因组区域,其中所述一个或多个基因组区域具有至少五个甲基化位点,及其中所述至少五个甲基化位点在多个非癌性样本或多个癌性样本中具有一异常甲基化模式。
在一些实施例中,所述重叠序列包含至少40、50、75或100个核苷酸。在一些实施例中,所述癌症化验板包含至少1000、2000、2500、5000、6000、7500、10000、15000、20000或25000对探针。
本说明书的另一方面提供一种癌症化验板,所述癌症化验板包含:至少1000个探针,其中所述多个探针中的每一个被设计为一杂交探针与一个或多个基因组区域互补,其中所述多个基因组区域中的每一个包含:(i)至少30个核苷酸,以及(ii)至少五个甲基化位点,其中所述至少五个甲基化位点具有一异常甲基化模式,及在多个癌症样本或多个非癌症样本中为低甲基化或高甲基化。
在一些实施例中,所述异常甲基化模式在所述多个非癌性样本中具有至少一阈值p值稀有度。在一些实施例中,所述多个探针中的每一个被设计为与少于20个脱靶基因组区域互补。
在一些实施例中,基于标准选择所述多个基因组区域中的每一个,所述标准包含:多个癌性样本包含具有所述异常甲基化模式的多个cfDNA片段的一数量(N
在一些实施例中,所述多个基因组区域中的每一个是基于与N
在一些实施例中,所述癌症化验板包含至少1000、2000、2500、5000、10000、12000、15000、20000、30000、40000或50000、或100000个探针。
在一些实施例中,所述至少1000对探针或所述至少2000个探针共同包含至少20万、40万、60万、80万、100万、200万、300万、400万、500万、600万、700万、800万、900万、或1000万个核苷酸。在一些实施例中,所述多个探针中的每一个包含至少50、75、100或120个核苷酸。在一些实施例中,所述多个探针中的每一个包含少于300、250、200或150个核苷酸。在一些实施例中,所述多个探针中的每一个包含100至150个核苷酸。在一些实施例中,所述多个探针中的每一个包含少于20、15、10、8或6个甲基化位点。在一些实施例中,所述至少5个甲基化位点中的至少80%、85%、90%、92%、95%或98%在所述多个癌性样本中是甲基化或未甲基化。在一些实施例中,所述多个探针中的每一个被配置为具有少于20、15、10或8个脱靶基因组区域。
在一些实施例中,所述多个基因组区域的至少15%、20%、30%、或40%是外显子或内含子。在一些实施例中,所述多个基因组区域的至少5%、10%、15%、20%、30%、或40%是外显子。在一些实施例中,少于5%、10%、15%、20%、25%、或30%的所述多个基因组区域是多个基因间区域。在一些实施例中,20%至60%之间、30%至50%之间、或35%至55%之间的基因组区域是内含子或外显子。在一些实施例中,5%至30%之间、10%至25%之间、或12%至20%之间的基因组区域是外显子。在一些实施例中,5%至20%的基因组区域是基因间区域。
在一些实施例中,所述多个基因组区域选自列表1至列表8中的任一个。在一些实施例中,所述多个基因组区域包含列表1至列表8的任一个中至少30%、40%、50%、60%、70%、80%、90%或95%的基因组区域。在一些实施例中,所述至少1000个探针被配置为与列表1至列表8的任一个中的至少500、1000、5000、10000或15000个基因组区域互补。
在本说明书的另一方面,提供一种癌症化验的方法,所述方法包含步骤:接收一样本,所述样本包含多个核酸片段;处理所述多个核酸片段以将未甲基化的胞嘧啶转化为尿嘧啶,从而获得多个转化的核酸片段;将所述多个转化的核酸片段与上述权利要求任一项所述的癌症化验板上的所述多个探针相杂交;扩增所述多个转化的核酸片段的一子集;以及对所述多个转化的核酸片段的扩增子集进行测序,从而提供多个序列读数的一集合。
在一些实施例中,所述方法还包含以下步骤:通过评估所述多个序列读数的所述集合来确定一健康状况,其中所述健康状况是癌症的存在或不存在,或癌症的阶段。
在一些实施例中,所述多个核酸片段的所述集合是从一人类对象获得。
在其他方面,本说明书提供一种诊断癌症的方法,所述方法包含步骤:(a)通过对来自一对象的多个核酸片段的一集合进行测序来获得多个序列读数的一集合;(b)确定多个基因组区域的甲基化状态,所述多个基因组区域包含从列表1至列表8中的任一个中的多个基因组区域;以及(c)通过评估所述甲基化状态来确定所述对象的一健康状况,其中所述健康状况是(i)癌症的存在或不存在;或(ii)癌症的一阶段。在其他方面,本说明书提供一种用于诊断癌症的方法,所述方法包含步骤:(a)通过对来自一对象的多个核酸片段的一集合进行测序获得多个序列读数的一集合;(b)确定多个基因组区域的甲基化状态,所述多个基因组区域包含列表1至列表8中任何一个中至少20%、30%、40%、50%、60%、70%、80%、90%或95%的基因组区域;以及(c)通过评估所述甲基化状态来确定所述对象的一健康状况,其中所述健康状况是(i)癌症的存在或不存在;或(ii)癌症的一阶段。在其他方面,本说明书提供一种诊断癌症的方法,所述方法包含以下步骤:(a)通过对来自一对象的多个核酸片段的一集合进行测序获得多个序列读数的一集合;(b)确定列表1至列表8中的任一个的多个基因组区域中至少1000、2000、2500、5000、6000、7500、10000、15000、20000或25000个基因组区域中的甲基化状态;以及(c)通过评估所述甲基化状态来确定所述对象的一健康状况,其中所述健康状况是(i)癌症的存在或不存在;或(ii)癌症的一阶段。
另一方面提供了一种设计一癌症化验板的方法,所述方法包含步骤:识别多个基因组区域,其中所述多个基因组区域中的每一个(i)包含至少30个核苷酸,及(ii)包含至少五个甲基化位点;比较多个癌性样本和多个非癌性样本之间的多个基因组区域中的每一个中的所述至少五个甲基化位点的甲基化状态,其中所述至少五个甲基化位点为低甲基化或高甲基化;选择所述多个基因组区域的一子集,其中在多个癌性样本中所述多个基因组区域的所述子集的至少五个甲基化位点相对于多个非癌性样本具有一异常甲基化模式,以及设计一癌症化验板,所述癌症化验板包含多个探针集合,其中所述多个探针集合中的每一个包含至少一对探针,所述至少一对探针被配置成标靶(例如,与对应的多个转化的片段或多个未转化的片段互补的)所述多个基因组区域的子集中的一个。
在一些实施例中,所述异常甲基化模式与一cfDNA片段的异常甲基化模式匹配,所述cfDNA片段来自所述多个癌性样本与所述至少五个甲基化位点中的至少一个相重叠,其中所述cfDNA相对于所述多个非癌性样本的一训练数据集具有至少一阈值p值稀有度。
在一些实施例中,所述选择步骤是基于一标准执行,所述标准包含:所述多个癌性样本包含具有所述异常甲基化模式的多个cfDNA片段的所述多个癌性样本的一数量N
在一些实施例中,所述多个探针中的每一个具有少于20、15、10或8个脱靶基因组区域。在一些实施例中,所述方法还包含:制造所述癌症化验板的所述步骤,其中所述癌症化验板包含所述多个探针。
本说明书的另一个方面提供一种癌症化验板,所述癌症化验板是通过本文提供的所述方法制备。在一些实施例中,多个基因组区域的所述子集包含列表1至列表8中任一个的多个基因组区域。在一些实施例中,所述多个基因组区域的所述子集包含列表1至列表8中的至少20%、30%、40%、50%、60%、70%、80%或90%的基因组区域。在一些实施例中,所述多个基因组区域的所述子集包含列表1至列表8中任何一个中的至少500、1000、5000、10000、或15000个基因组区域。
本说明书的另一个方面提供一种癌症化验板,所述癌症化验板包含多个探针,其中所述多个探针中的每一个被配置成与列表1至列表8中的任一个中的多个基因组区域中的一个相重叠,及所述多个探针与列表1至列表8中的任一个中的至少90%、95%或100%的基因组区域相重叠在一起。在又一方面,本说明书提供了一种癌症化验板,所述癌症化验板包含多个探针,其中所述多个探针中的每一个被配置成与列表1至列表8中的任一个中的多个基因组区域中的一个相重叠,及所述多个探针与列表1至列表8中任一个中的至少500、1000、5000、10000或15000个基因组区域重叠在一起。
附图说明
图1A至图1B示出了2×平铺探针的设计,对于每一串DNA,标靶区域内的每个碱基(用点状矩形框起来)被恰恰两个探针覆盖(例如,互补)。
图2是根据一个实施例,描述为一对照组创建一数据结构的一程序的流程图。
图3是根据一个实施例,描述验证图2的所述对照组的所述数据结构的一附加步骤的流程图。
图4是根据一个实施例,描述用于选择多个基因组区域以用于设计多个探针从而用于一癌症化验板的一程序的流程图。
图5是根据一个实施例,示例p值得分计算的图示。
图6是根据一个实施例,描述基于甲基化状态的多个片段的训练一分类器的一程序的流程图。
图7A是根据一个实施例,描述对无细胞DNA(cfDNA)的一片段进行测序的一程序的流程图。
图7B是根据一个实施例,图7A对无细胞DNA(cfDNA)的一片段进行测序以获得一甲基化状态向量的一程序的图示。
图8A是根据一个实施例,用于多个核酸样本测序的多个装置的流程图。
图8B是根据一个实施例,提供分析cfDNA的甲基化状态的一分析系统。
图9A至图9B示出了验证一对照组测序一致性的三个数据图表。
图10是根据取决于多个DNA片段和多个探针之间重叠的大小,所述多个DNA片段接合到所述多个探针上的数量的一图表。
图11比较了标靶高甲基化的多个片段(高(Hyper))或低甲基化的多个片段(低(Hypo))的探针中高质量、中间质量和劣质的探针的数量。
图12A示出了对于各种化验板的数量,CpG的数量作为密度的一函数。
图12B示出了对于各种化验板,G/C分率作为密度的一函数。
图13A至图13C总结了在指定的化验板中多个标靶基因组区域(黑色)和多个随机选择区域(白色)的多个基因组注释(genomic annotation)的频率。
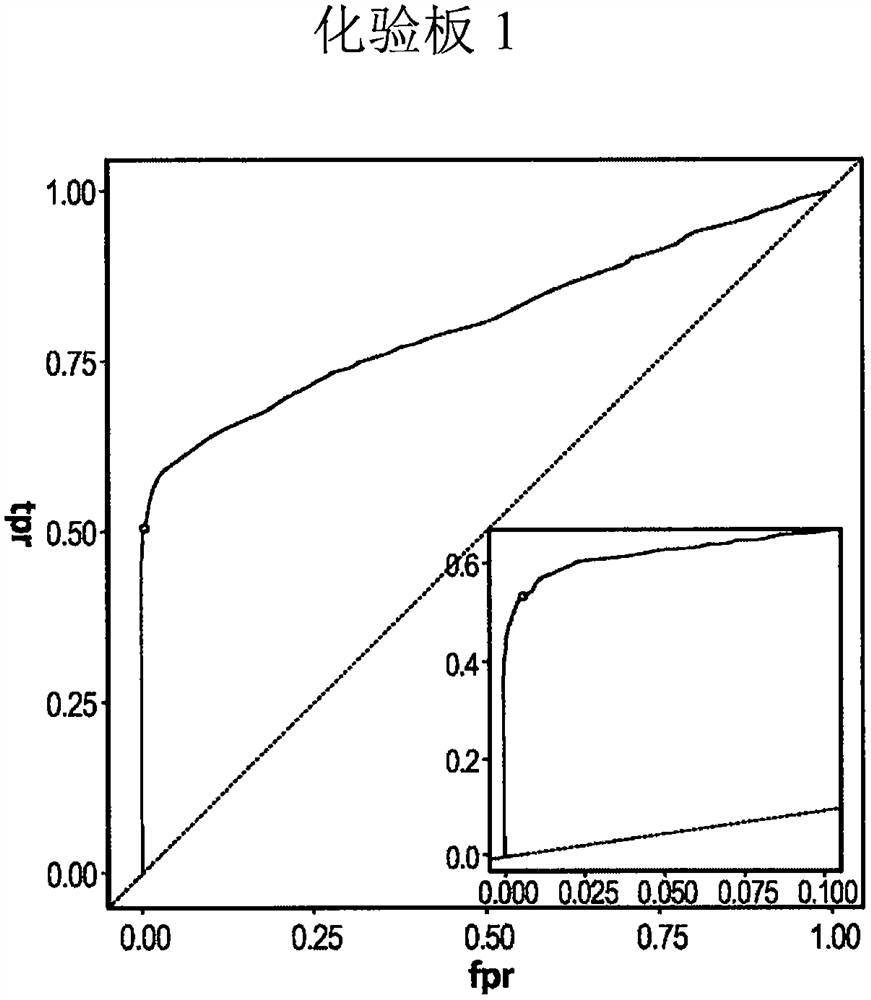
图14A描绘了一接收者操作特性曲线(ROC),示出了使用化验板1检测癌症的敏感度和特异性。
图14B是一混淆矩阵,描绘了使用化验板1对起源组织(TOO)分类的准确度。
图15A描绘了一接收者操作特性曲线(ROC),示出了使用化验板2检测癌症的敏感度和特异性。
图15B是一混淆矩阵,描绘了使用化验板2对起源组织(TOO)分类的准确度。
图16A描绘了一接收者操作特性曲线(ROC),示出了使用化验板3检测癌症的敏感度和特异性。
图16B是一混淆矩阵,描绘了使用化验板3对起源组织(TOO)分类的准确度。
图17A描绘了一接收者操作特性曲线(ROC),示出了使用化验板4检测癌症的敏感度和特异性。
图17B是一混淆矩阵,描绘了使用化验板4对起源组织(TOO)分类的准确度。
图18A描绘了一接收者操作特性曲线(ROC),示出了使用化验板5检测癌症的敏感度和特异性。
图18B是一混淆矩阵,描绘了使用化验板5对起源组织(TOO)分类的准确度。
图19A描绘了一接收者操作特性曲线(ROC),示出了使用化验板6检测癌症的敏感度和特异性。
图19B是一混淆矩阵,描绘了使用化验板6对起源组织(TOO)分类的准确度。
图20A描绘了一接收者操作特性曲线(ROC),示出了使用化验板3A检测癌症的敏感度和特异性。
图20B是一混淆矩阵,描绘了使用化验板3A对起源组织(TOO)分类的准确度。
图21A描绘了一接收者操作特性曲线(ROC),示出了使用化验板4A检测癌症的敏感度和特异性。
图21B是一混淆矩阵,描绘了使用化验板4A对起源组织(TOO)分类的准确度。
图22A描绘了一接收者操作特性曲线(ROC),示出了在化验板3中使用40%的标靶基因组区域的一随机选择(子集a)的癌症检测的敏感度和特异性。
图22B是一混淆矩阵,描绘了在化验板3中使用40%的标靶基因组区域的一随机选择(子集a)的起源组织(TOO)分类的准确度。
图23A描绘了一接收者操作特性曲线(ROC),示出了在化验板3中使用40%的标靶基因组区域的一随机选择(子集b)的癌症检测的敏感度和特异性。
图23B是一混淆矩阵,描绘了在化验板3中使用40%的标靶基因组区域的一随机选择(子集b)的起源组织(TOO)分类的准确度。
图24A描绘了一接收者操作特性曲线(ROC),示出了在化验板3中使用40%的标靶基因组区域的一随机选择(子集c)的癌症检测的敏感度和特异性。
图24B是一混淆矩阵,描绘了在化验板3中使用40%的标靶基因组区域的一随机选择(子集c)的起源组织(TOO)分类的准确度。
图25A描绘了一接收者操作特性曲线(ROC),示出了在化验板3中使用60%的标靶基因组区域的一随机选择的癌症检测的敏感度和特异性。
图25B是一混淆矩阵,描绘了在化验板3中使用60%的标靶基因组区域的一随机选择的起源组织(TOO)分类的准确度。
图26A描绘了一接收者操作特性曲线(ROC),示出了在化验板4中使用50%的标靶基因组区域的一随机选择的癌症检测的敏感度和特异性。
图26B是一混淆矩阵,描绘了在化验板4中使用50%的标靶基因组区域的一随机选择的起源组织(TOO)分类的准确度。
图27A描绘了一接收者操作特性曲线(ROC),示出了在化验板5中使用50%的标靶基因组区域的一随机选择的癌症检测的敏感度和特异性。
图27B是一混淆矩阵,描绘了在化验板5中使用50%的标靶基因组区域的一随机选择的起源组织(TOO)分类的准确度。
图中描述了本说明书的各种实施例仅用于说明。本领域技术人员将容易地从下面的讨论中认识到,在不脱离本文所描述的原理的情况下,可以使用本文所示的结构和方法的替代实施例。
具体实施方式
6.1.定义
除非另有定义,否则本文使用的所有技术和科学术语具有本说明书所属领域的技术人员通常理解的含义。如本文所用,以下术语具有如下所述含义。
本文中使用的术语“甲基化(methylation)”是指将甲基添加到DNA分子中的程序。例如,胞嘧啶碱基的嘧啶环上的一氢原子可以转化为一甲基,以形成5-甲基胞嘧啶。该术语还指将羟甲基添加到DNA分子的程序,例如通过胞嘧啶碱基的嘧啶环上的甲基氧化。甲基化和羟甲基化倾向于发生在胞嘧啶和鸟嘌呤的二核苷酸上,本文称为“CpG位点”。
在这些实施例中,用于检测甲基化的湿实验室(wet laboratory)化验可不同于如本文所述本领域公知的那些。
本文中使用的术语“甲基化位点(methylation site)”是指DNA分子上可以添加甲基的区域。“CpG”位点是最常见的甲基化位点,但甲基化位点并不局限于CpG位点。例如,DNA甲基化可能发生在CHG和CHH的胞嘧啶中,其中H是腺嘌呤、胞嘧啶或胸腺嘧啶。还可以使用本文公开的方法和程序来评估5-羟甲基胞嘧啶形式的胞嘧啶甲基化(参见例如通过引用并入本文的WO 2010/037001和WO 2011/127136)及其特征。
本文中使用的术语“CpG位点(CpG site)”是指DNA分子中的一个区域,其中胞嘧啶核苷酸在碱基的线性序列沿其5’至3’方向排列后接鸟嘌呤核苷酸。“CpG”是5’-C-磷酸盐-G-3’(5’-C-phosphate-G-3’)的简写,它是由仅有一个磷酸基分开的胞嘧啶和鸟嘌呤。CpG二核苷酸中的多个胞嘧啶可以被甲基化形成5-甲基胞嘧啶。
本文中使用的术语“CpG检测位点(CpG detection site)”是指一个探针中被配置成与一个标靶DNA分子的一个CpG位点杂交的一个区域。在所述标靶DNA分子上的所述CpG位点可以包含由一个磷酸基分离的胞嘧啶和鸟嘌呤,其中胞嘧啶被甲基化(methylated)或未甲基化(unmethylated)。在标靶DNA分子上的CpG位点可包含通过一个磷酸基分离的尿嘧啶和鸟嘌呤,其中尿嘧啶是通过未甲基化的胞嘧啶的转化生成。
术语“UpG”是5’-U-磷酸盐-G-3’(5’-U-phosphate-G-3’)的简写,即尿嘧啶和鸟嘌呤仅由一个磷酸基分开。UpG可由一个DNA通过一亚硫酸氢盐处理产生,将未甲基化的胞嘧啶转化为尿嘧啶。胞嘧啶可通过本领域已知的其他方法转化为尿嘧啶,例如化学修饰、合成或酶转化等方法。
本文中使用的术语“低甲基化(hypomethylated)”或“高甲基化(hypermethylated)”是指含有多个CpG位点(例如超过3、4、5、6、7、8、9、10个等)的一个DNA分子的甲基化状态,其中所述多个CpG位点的高百分比(例如,超过80%、85%、90%或95%,或在50%至100%范围内的任何其他百分比)分别为未甲基化或甲基化。
本文中使用的术语“甲基化状态向量(methylation state vector或methylationstatus vector)”是指包含多个元素的一向量,其中每个元素表示由包含多个甲基化位点的一个DNA分子中一个甲基化位点的甲基化状态,其顺序是在DNA分子中出现从5’到3’。例如, 本文中使用的术语“异常甲基化模式(abnormal methylation pattern或anomalous methylation pattern)”是指一DNA分子的甲基化模式或一甲基化状态向量,其期望在一样本中发现的频率低于一阈值。在本文提供的特定实施例中,在包含多个健康个体(例如未诊断为癌症的个人)的一健康对照组中找到一特定甲基化状态向量的期望值由p值(p-value)表示。低p值得分通常对应于与来自多个健康个体的多个样本中的其他甲基化状态向量相比相对意外的甲基化状态向量。高p值得分通常对应于与健康对照组中的多个健康个体的多个样本中发现的其他甲基化状态向量相比相对更期望的甲基化状态向量。一甲基化状态向量具有一p值低于一阈值(例如,0.1、0.01、0.001、0.0001等),则可以将其定义为一异常(abnormal/anomalous)甲基化模式。本领域已知的各种方法可用于计算甲基化模式或甲基化状态向量的p值或期望值。本文提供的示例性方法涉及使用马尔可夫链概率(Markov chain probability),其假设多个CpG位点的多个甲基化状态依赖于相邻的多个CpG位点的多个甲基化状态。本文提供的替代方法通过使用包含多个混合组分的一混合模型来计算观察在多个健康个体中一特定甲基化状态向量的期望值,其中每个混合组分都是一个独立位点模型,其中假设每个CpG位点处的甲基化与其他CpG位点处的甲基化状态相独立。 本文提供的方法将多个DNA片段表征为异常,当它们具有与多个参考样本中多个DNA片段的多个甲基化模式相比不寻常的一甲基化模式时,例如来自未被诊断为癌症的多个个体的多个样本。在多个参考样本中观察到一特定甲基化模式的可能性可以表示为一p值得分。本文提供的用于建模一特定甲基化模式的可能性的多个示例性方法涉及使用一马尔可夫链概率(Markov chain probability)和一滑动窗口。如果p值得分低于一阈值(例如,0.1、0.01、0.001、0.0001等),则具有该甲基化模式的所述DNA片段被分类为异常。在与所述阈值进行比较之前,与高甲基化或低甲基化的DNA片段相对应的多个p值得分可被进行加总或求平均。可以采用本领域已知的各种方法来比较对应于所述基因组区域和所述阈值的多个p值得分,包含但不限于算术平均值、几何平均值、调和平均值、中位数、众数等。 本文中使用的术语“癌性样本(cancerous sample)”是指包含来自被诊断为具有癌症的一个体的多个基因组DNA的样本。所述多个基因组DNA可以是但不限于多个cfDNA片段或来自具有癌症的一对象的多个染色体DNA。所述多个基因组DNA可被测序并且其甲基化状态可通过本领域已知的方法(例如亚硫酸氢盐测序)来评估。当多个基因组序列是从公共数据库(例如,癌症基因组图谱(The Cancer Genome Atlas,TCGA))中获得的,或通过对被诊断为具有癌症的一个体的一基因组进行测序实验而获得,癌性样本可以指具有所述多个基因组序列的多个基因组DNA或多个cfDNA片段。术语“多个癌性样本”作为复数是指包含来自多个个体的多个基因组DNA的多个样本,每个个体被诊断为具有癌症。在各种实施例中,使用来自100、300、500、1000、10000、20000、40000、50000个或更多个诊断为具有癌症的个体的多个癌性样本。 本文中使用的术语“非癌性样本(non-cancerous sample)”是指包含来自没被诊断为具有癌症的一个体的多个基因组DNA的一样本。所述多个基因组DNA可以是,但不限于来自不具有癌症的一对象的多个cfDNA片段或多个染色体DNA。所述多个基因组DNA可被测序并且其甲基化状态可通过本领域已知的方法(例如亚硫酸氢盐测序)来评估。当多个基因组序列是从公共数据库(例如,癌症基因组图谱(TCGA))中获得的,或通过对不具有癌症的一个体的一基因组进行测序实验而获得,非癌性样本可以指具有所述多个基因组序列的多个基因组DNA或多个cfDNA片段。术语“多个非癌性样本”作为复数是指包含来自多个个体的多个基因组DNA的多个样本,每个个体没被诊断为具有癌症。在各种实施例中,使用来自100、300、500、1000、10000、20000、40000、50000个或更多个不具有癌症的个体的癌性样本。 本文中使用的术语“训练样本(training sample)”是指用于训练本文所述的一分类器和/或以选择用于癌症诊断的一个或多个基因组区域的样本。所述多个训练样本可包含来自一个或多个健康对象以及具有用于诊断(例如,癌症、癌症的特定类型、癌症的特定阶段等)的一疾病状况的一个或多个对象。所述多个基因组DNA可以是但不限于多个cfDNA片段或多个染色体DNA。所述多个基因组DNA可被测序并且其甲基化状态可通过本领域已知的方法(例如亚硫酸氢盐测序)来评估。当多个基因组序列是从公共数据库(例如,癌症基因组图谱(TCGA))获得,或通过对一个体的一基因组进行测序实验而获得,一训练样本可以指具有所述多个基因组序列的多个基因组DNA或多个cfDNA片段。 本文中所述的“测试样本(test sample)”是指来自一对象的一样本,其健康状况已经、已被或将要使用本文所述的一分类器和/或一化验板进行测试。所述测试样本可以包含多个基因组DNA或其修饰。所述多个基因组DNA可以是但不限于多个cfDNA片段或多个染色体DNA。 本文中使用的术语“标靶基因组区域(target genomic region)”是指在一基因组中选择用于在多个测试样本中进行分析的一区域。用设计成与衍生自标靶基因组区域或其的一片段的多个核酸片段杂交(并可选地下拉(pull down))的多个探针生成一化验板。衍生自所述标靶基因组区域的一核酸片段是指通过降解、裂解、亚硫酸氢盐转化或来自标靶基因组区域的DNA的其他处理产生的一核酸片段。 本文所用的术语“脱靶基因组区域(off-target genomic region)”是指在一基因组中未曾被选择以用于在多个测试样本中的分析,但与一标靶基因组区域具有足够的同源性,而可通过设计用于标靶所述标靶基因组区域的一探针可能被绑定和下拉。在一个实施例中,一脱靶基因组区域是一基因组区域,所述基因组区域与一探针沿着至少45碱基对(bp)具有至少匹配率为90%对齐。 术语“转化DNA分子(converted DNA molecules)”、“转换cfDNA分子(convertedcfDNA molecules)”,及“从多个cfDNA分子的处理中获得的修饰片段”是指通过处理一样本中的多个DNA或cfDNA分子获得多个DNA分子,以区分所述多个DNA或cfDNA分子中的一甲基化核苷酸和一未甲基化核苷酸。例如,在一个实施例中,可以使用本领域已知的亚硫酸氢钠离子(例如,使用亚硫酸氢钠)处理样本,将未甲基化胞嘧啶(“C”)转化为尿嘧啶(“U”)。在另一实施例中,使用酶转化反应,例如使用胞苷脱氨酶(例如APOBEC)来完成未甲基化胞嘧啶到尿嘧啶的转换。处理后,转化的DNA分子或cfDNA分子包含原始cfDNA样本中不存在的额外尿嘧啶。通过DNA聚合酶复制包含一尿嘧啶的一DNA链导致在新生互补链中添加一腺嘌呤,而不是通常作为胞嘧啶或甲基胞嘧啶的补体添加的鸟嘌呤。 术语“无细胞核酸(cell free nucleic acid)”、“无细胞DNA(cell free DNA)”或“cfDNA”指的是在个体身体(例如血流)中循环并源自于一个或多个健康细胞和/或一个或多个癌细胞的多个核酸片段。此外,cfDNA可能来自其他来源,如病毒、胎儿等。 术语“循环肿瘤DNA(circulating tumor DNA)”或“ctDNA”是指源于肿瘤细胞的核酸片段,这些片段可能由于生物学过程(例如死亡细胞凋亡或坏死或活的肿瘤细胞主动释放)而释放到个体血液中。 本文中使用的术语“片段”可以指一核酸分子的一片段。例如,在一个实施例中,一片段可指一血液或血浆样本中的一cfDNA分子,或已从一血液或血浆样本中提取的一cfDNA分子。一cfDNA分子的一扩增产物也可称为一“片段”。在另一实施例中,术语“片段”是指已被处理以供后续分析(例如,用于基于机器学习的分类)的一序列读数或多个序列读数的一集合,如本文所述。例如,如本领域公知的,原始序列读数可以与一参考基因组对齐,并且匹配成对末端序列读数组装成一更长的片段以用于后续分析。 术语“个体(individual)”是指人的个体。术语“健康个体”是指没有癌症或疾病的个体。 术语“对象(subject)”是指正在分析DNA的个体。一对象可以是一个测试对象,其DNA可以使用本文所述的标靶化验板(panel)进行评估,以评估那个人是否患有癌症或其他疾病。一对象也可能是已知没有癌症或其他疾病的一对照组的一部分。一对象也可能是已知具有癌症或另一种疾病的一癌症或其他疾病组的一部分。对照组和癌症/疾病组可用于协助设计或验证标靶化验板。 本文所用的术语“多个序列读数(sequence reads)”是指来自一样本中的多个核苷酸序列读数。可以通过本文提供的各种方法或本领域已知的方法来获得多个序列读数。 本文所使用的术语“测序深度(sequencing depth)”是指在一样本中已被测序的一给定标靶核酸的计数的次数(例如,在一给定标靶区域读取的序列计数)。增加测序深度可以减少评估一疾病状态(例如癌症或起源组织)所需的核酸的数量。 本文所使用的术语“起源组织(tissue of origin)”或“TOO”是指癌症产生或来源的器官、器官群组、身体区域或细胞类型。起源组织或癌细胞类型的识别通常允许识别癌症连续护理中最合适的下一步步骤,以进一步诊断、分期和决定治疗。 术语“转换(transition)”通常是指从一种嘌呤到另一种嘌呤,或从一种嘧啶到另一种嘧啶的碱基组成的变化。例如,以下变化是转换:C→U、U→C、G→A、A→G、C→T、及T→C。 一化验板或诱饵组的“多个探针的一整体”或一化验板或诱饵组的“多个含有多核苷酸的探针的一整体”通常是指与特定化验板或诱饵组一起提供的所有探针。例如,在一些实施例中,一化验板或诱饵集可以同时包含:(1)多个探针,具有本文特定的多个特征(例如,用于与对应于或源自于本文阐述的一个或多个列表中多个基因组区域的多个无细胞DNA片段结合的多个探针);及(2)不包含这样(多个)特征的另外的多个探针。一化验板的多个探针的整体通常是指与所述化验板或诱饵组一起提供的所有探针,包含不含有特定的(多个)特征的探针。 6.2.其他解释惯例 本文所叙述的范围应理解为该范围内所有数值的简写形式,包含所叙述的端点。例如,范围1到50应该理解为包含从1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23、24、25、26、27、28、29、30、30、31、32、33、34、35、36、37、38、39、40、41、43、44、45、46、47、48、49和50组成的群组中的任何数字、多个数字的组合或其子范围。 6.3.癌症化验板 在第一方面,本说明书提供了一种癌症化验板(例如,一诱饵组),包含多个探针。所述多个探针可以是多个含有多核苷酸的探针,所述多个含有多核苷酸的探针特定地设计用于标靶一个或多个核酸分子,所述一个或多个核酸分子对应于或源自于在癌症与非癌性样本比较的差异甲基化的多个基因组区域,如本文提供的方法所识别。所述多个探针被用作诱饵来收集来源于癌症中差异甲基化的多个标靶基因组区域的cfDNA。在诊断化验中,扩增可提高灵敏度和准确度,同时减少测序费用。 为了设计所述癌症化验板,一分析系统可以收集与正在考虑的各种结果相对应的多个样本,例如已知患有癌症的多个样本,被认为是健康的多个样本,来自已知起源组织的多个样本等。用于选择多个标靶基因组区域的cfDNA和/或ctDNA的来源可以根据化验的目的而变化。例如,对于通常用于诊断癌症、特定类型的癌症、癌症阶段或起源组织的化验,可能需要不同的来源。这些样本可以用全基因组亚硫酸氢盐测序(WGBS)处理或从公共数据库(如TCGA)中获得。所述分析系统可以是具有一计算机处理器和一计算机可读存储介质的任何通用计算系统,具有用于执行所述计算机处理器以执行本公开所述的任何或所有操作的多个指令。 然后,所述分析系统可以基于多个核酸片段的多个甲基化模式选择多个标靶基因组区域。一种方法考虑多个区域或更具体地一个或多个CpG位点的成对结果之间的成对分辨率。另一种方法在考虑每个结果相对于其余结果时,考虑多个区域或更具体地一个或多个CpG位点的分辨率。从所选择的具有高分辨率能力的多个标靶基因组区域,所述分析系统可以设计多个探针以标靶包含所选的多个基因组区域的多个核酸片段。所述分析系统可以生成不同尺寸的癌症化验板,例如,一小型尺寸癌症化验板包含标靶信息最丰富的基因组区域的多个探针,一中型尺寸癌症化验板包含来自所述小型尺寸癌症化验板的多个探针和标靶第二级信息的基因组区域的多个附加探针,以及一大型癌症化验板包含来自所述小型尺寸和所述中型尺寸癌症化验板的多个探针,以及更多标靶第三级信息的基因组区域的多个探针。利用这种癌症化验板,所述分析系统可以训练具有各种分类技术的多个分类器,以预测一样本具有一特定结果(例如癌症、特定癌症类型、其他疾病等)的可能性。 多个标靶基因组区域可被选择,以最大化分类的准确度,但须受尺寸限制(由测序预算(sequencing budget)和所需测序深度决定)。潜在的多个标靶基因组区域可以根据本文所述的它们的暂定分类潜力进行排名,然后贪婪地(greedily)添加到一化验板中,直到达到尺寸限制。 一癌症化验板通常可用于检测癌症的存在或不存在、癌症的阶段(如I、II、III或IV)和/或癌症的起源组织。根据不同的目的,一化验板可以包含标靶多个一般癌症(泛癌)样本和多个非癌症样本之间差异甲基化的多个基因组区域的多个探针。在一些实施例中,一癌症化验板基于从多个癌症和非癌症个体的一集合的cfDNA和/或全基因组DNA生成的亚硫酸氢盐测序数据来设计。 每个探针、探针对或探针组可设计为通过选择性杂交来标靶一个或多个标靶基因组区域。所述多个标靶基因组区域被选择基于几个标准,所述几个标准旨在增强相关的多个cfDNA片段的选择性扩增,同时减少噪声和非特异性结合。例如,一化验板可以包含多个探针,所述多个探针可以选择性地杂交(即,结合)和扩增多个癌性样本中差异甲基化的多个cfDNA片段。此外,所述多个探针可进一步设计成标靶被确定为具有包含高甲基化或低甲基化的异常甲基化模式的多个基因组区域。在一些实施例中,一化验板(panel)包含标靶多个高甲基化片段的第一集合的多个探针和标靶多个低甲基化片段的第二集合的多个探针。在一些实施例中,标靶多个高甲基化片段的所述第一集合的多个探针和标靶多个低甲基化片段的所述第二集合的多个探针之间的比率(高:低比率)范围在0.4和2之间、0.5和1.8之间、0.5和1.6之间、1.4和1.6之间、1.2和1.4之间、1和1.2之间、0.8和1之间、0.6和0.8之间、或0.4和0.6之间。 扩增的多个片段的测序提供了与癌症诊断相关的信息。第6.4.2节(“异常甲基化片段”)详细提供了识别具有多个异常甲基化模式的多个片段的多种方法,第6.4.3节(“高甲基化或低甲基化片段的分析”)详细介绍了识别具有低甲基化或高甲基化模式的多个片段的方法。 例如,当与一基因组区域对齐的多个cfDNA和/或ctDNA片段具有一低p值的一甲基化模式时,可以根据在多个非癌性样本的一集合上训练的一马尔可夫模型选择多个基因组区域。用于选择多个基因组区域的多个cfDNA和/或ctDNA片段可能还需要包含至少一个阈值数(例如,5个)CpG,及70%、75%、80%、85%、90%、95%、98%或100%的CpG位点可能需要甲基化或未甲基化。所述多个探针中的每一个可标靶一基因组区域的至少25bp、30bp、35bp、40bp、45bp、50bp、60bp、70bp、80bp、90bp、100bp、110bp或120bp。在一些实施例中,所述多个基因组区域需要具有少于20、15、10、8或6个甲基化位点。 在一些实施例中,标靶基因组区域的选择涉及针对每个CpG位点执行一计算。具体地说,第一计数n 在一些实施例中,具有与一CpG位点重叠的一异常高甲基化和/或低甲基化cfDNA和/或ctDNA片段的多个非癌性样本(n 可以执行进一步的过滤,以选择具有高特异性的多个探针以用于从标靶的多个基因组区域衍生的多个核酸的扩增(即高结合效率)。多个探针可以被过滤以减少从非标靶的多个基因组区域衍生的多个核酸的非特异性结合(或脱靶(off-target)结合)。在一些实施例中,执行进一步过滤以选择用于一癌症化验板的多个探针,所述癌症化验板不会从下拉多个标靶基因组区域。在一些实施例中,可从下拉多个标靶基因组区域的多个探针被排除(eliminated)。在一些实施例中,设置一阈值以定义具有一不可接受的脱靶效应的高风险的多个探针。在一个实施例中,从一化验板中排除具有与多个脱靶基因组区域的阈值数(例如,1、2、3、4、5、10、15、20、25或更多)超过80%、85%、90%、95%或98%一致性的多个探针。在一些实施例中,如果多个探针包含至少20、25、30、35、40、45、50、55或60个核苷酸(nt)的一序列,并且与多个脱靶基因组区域的一阈值数(例如,1、2、3、4、5、10、15、20、25或更多)具有80%、85%、90%、95%或98%以上的一致性,则从一癌症化验板中排除所述多个探针。过滤去除了重复的探针,其可以下拉多个脱靶片段,这是不需要的,可以影响化验效率。 在一些实施例中,一癌症化验板的所述多个探针被设计为与衍生自多个标靶基因组区域的转化的cfDNA相杂交。杂交后,可回收和/或分离标靶的多核苷酸,选择性放大,并通过任何合适的方法进行测序。所述多个序列读数提供与癌症检测、癌症诊断和癌症起源组织或癌症类型的评估相关的信息。为此,一化验板可被设计成包含多个探针,所述多个探针可捕获多个片段,所述多个片段可一起提供与癌症检测和癌症诊断相关的信息。在一些实施例中,一化验板包含至少50、60、70、80、90、100、120、150或200对不同的探针。在其它实施例中,一化验板包含至少500、1000、2000、2500、5000、10000、12000、15000、20000、30000、40000、50000或100000个探针。所述多个探针共同可以包含至少20000、30000、40000、50000、75000、10万、20万、40万、60万、80万、100万、200万、300万、400万、500万、600万、700万、800万、900万或1000万个核苷酸。可选地,一探针的一端或两端的序列与标靶相同基因组区域或一相邻的基因组区域的其他探针的序列相重叠。 具体而言,在一些实施例中,所述癌症化验板包含至少50对探针,其中所述至少50对探针中的每一对包含两个探针,被配置为通过一重叠(例如,相同的)序列彼此相互重叠,其中所述重叠序列包含30个核苷酸的一序列,被配置成与从对应于一个或多个基因组区域的处理的多个cfDNA分子获得的一修饰片段杂交,其中所述多个基因组区域中的每一个包含至少5个甲基化位点,其中所述至少5个甲基化位点在多个训练样本中具有一异常甲基化模式。换句话说,当分析与所述基因组区域相对应的多个训练样本中的多个cfDNA分子时,它们具有多个甲基化状态向量出现的频率低于多个参考样本中的一阈值。 在其他实施例中,所述癌症化验板包含至少500对探针,其中所述至少500对探针中的每一对包含两个探针,被配置为通过一重叠序列彼此相互重叠,其中所述重叠序列包含30个核苷酸的一序列,被配置成与从对应于一个或多个基因组区域的处理的多个cfDNA分子获得的一修饰片段杂交,其中所述多个基因组区域中的每一个包含至少5个甲基化位点,其中所述至少5个甲基化位点在多个训练样本中具有一异常甲基化模式。同样,当分析与所述基因组区域相对应的多个训练样本中的多个cfDNA分子时,它们具有多个甲基化状态向量出现的频率低于多个参考样本中的一阈值。 在优选实施例中,所述至少5个甲基化位点在癌性和非癌性样本之间或在来自不同癌症类型的一对或多对样本之间为差异甲基化。在一些实施例中,转化的多个cfDNA分子包含多个cfDNA分子被处理(例如,通过亚硫酸氢盐处理或酶转化)以将未甲基化的C(胞嘧啶)转化为U(尿嘧啶)。在某些情况下,尿嘧啶进一步转化为胸腺嘧啶(例如,通过PCR扩增)。 所选的多个标靶基因组区域可选地位于一基因组中的各种位置,包含但不限于启动子、增强子、外显子、内含子和基因间区域。在某些实施例中,所述标靶基因组区域包含在癌症与非癌症、癌症的不同阶段和/或癌症中起源于不同起源组织的差异表达的基因和/或转录控制区域。 通过本文提供的方法设计的一种癌症化验板可包含至少1000对探针,每对探针包含两个探针,被配置为通过一重叠序列彼此相重叠,所述重叠序列包含30个核苷酸片段。所述30个核苷酸片段包含至少三个、至少四个、至少五个以上的CpG位点,其中所述至少三个、至少四个、至少五个或更多CpG位点中的至少70%、至少80%或至少90%为CpG或UpG。所述30个核苷酸片段被配置为结合衍生自多个癌性样本中的多个基因组区域的一个或多个cfDNA片段,其中所述一个或多个基因组区域与来自一非癌症样本的多个DNA分子相比具有一异常甲基化模式的至少三个、至少四个、至少五个或更多个甲基化位点。 另一个癌症化验板包含至少2000个探针,至少2000个探针中的每一个被设计为一杂交探针,所述杂交探针与一个或多个基因组区域互补。所述多个基因组区域中的每一个基于标准来选择,所述标准包含(i)至少30个核苷酸;及(ii)至少三个、至少四个、至少五个或更多个甲基化位点,其中所述至少五个甲基化位点具有一异常甲基化模式并且是低甲基化或高甲基化的。 在某些示例中,多个引物可用于特异性地放大感兴趣的多个标靶/多个生物标记物(例如,通过PCR),从而扩增样本所需的多个标靶/多个生物标记物(可选择不进行杂交捕获)。例如,可以为每个感兴趣的基因组区域制备正向和反向引物,并用于放大对应于或衍生自所需的基因组区域的多个片段。因此,虽然本发明特别关注多种癌症化验板和多种诱饵组,但本发明的范围足够广泛以包含用于扩增无细胞DNA的其他方法。因此,本领域技术人员在本发明的益处下将认识到,类似于本文所述的与杂交捕获相关的方法可以通过用一些其他扩增策略代替杂交捕获来实现,例如对应感兴趣的多个基因组区域的无细胞DNA的多个片段的PCR放大。在一些实施例中,亚硫酸氢盐锁式探针捕获用于扩增多个感兴趣区域,如Zhang等人(美国专利申请案公开第US2016/0340740号)中所述。在一些实施例中,使用附加或替代方法来扩增(例如,非标靶扩增),例如还原亚硫酸氢盐测序、甲基化限制酶测序、甲基化DNA免疫沉淀测序、甲基CpG结合域蛋白质测序、甲基DNA捕获测序,或微滴PCR。 6.3.1.探针 本文提供的癌症化验板可包含具有多个杂交探针(在本文中也称为“多个探针”)的一集合的化验板,其设计用于通过下拉感兴趣的多个核酸片段来扩增选定的多个标靶基因组区域。所述多个探针被设计用于询问(interrogate)被怀疑一般与癌症的存在或不存在、癌症阶段和/或起源组织相关的多个标靶基因组区域(例如,人类或其他生物体)的甲基化状态。 所述多个探针可以被设计与DNA或RNA的一标靶(互补)链(strand)进行退火(anneal)(或杂交)。所述标靶链可以是“正(positive)”链(例如,转录成mRNA的链,随后转化为蛋白质)或互补的“负(negative)”链。在一个特定实施例中,一癌症化验板包含多个探针的两个集合,一个标靶正链,另一个标靶一标靶基因组区域的负链。在某些实施例中,标靶一DNA序列的不同链的多个探针可以具有彼此序列互补的多个区域。 多个探针可选择地设计为与天然DNA或转化的DNA杂交。cfDNA、ctDNA和/或染色体DNA可通过本领域已知的各种方法转化以保存表观遗传标记,例如甲基化。可选地,通过亚硫酸氢盐或酶处理将DNA转化,其将未甲基化的胞嘧啶转化为尿嘧啶。因此,甲基化模式决定了一CpG序列中哪些胞嘧啶转化为尿嘧啶。一标靶区域中未甲基化的多个CpG位点通过亚硫酸氢盐(或酶促方式)转化为多个UpG位点,如果通过应用DNA聚合酶的一程序转化后放大标靶DNA,则多个UpG序列进一步转化为多个TpG序列,并且一互补探针将具有一CpA序列。标靶区域的多个甲基化CpG位点保留了它们的CpG序列,因此一互补探针也会具有一CpG序列。不在一CpG序列的胞嘧啶通常不被甲基化。这些胞嘧啶通常转化为尿嘧啶,尿嘧啶在放大后进一步转化为胸腺嘧啶,因此与亚硫酸氢盐转化DNA互补的多个探针对于不在一CpG位点的每个转化的C包含A。 多个探针可选地是在假设所有CpG位点在某些标靶基因组区域是甲基化(完全高甲基化)的情况下被设计的,而在其他标靶基因组区域没有CpG位点被甲基化(完全低甲基化)。换句话说,设计用于标靶一低甲基化区域的多个探针可设计为与其中所有CpG位点已被转换为UpG的一区域互补,而设计用于标靶一高甲基化区域的多个探针可设计为与其中没有CpG位点已被转换的一区域互补。 由于所述多个探针可被配置为与衍生自一个或多个基因组区域的一转化的DNA或转化的cfDNA分子杂交,因此所述多个探针可具有不同于所述标靶基因组区域的序列。例如,含有未甲基化的CpG位点的一DNA分子将转换后为包含UpG,因为未甲基化的多个胞嘧啶通过一转化反应(例如亚硫酸氢盐处理)转化为多个尿嘧啶。结果,一探针被配置成杂交到包含UpG的一序列,而不是自然存在的未甲基化的CpG。因此,所述探针中与所述未甲基化位点的一互补位点可以包含CpA而不是CpG,及标靶所有甲基化位点都未甲基化的一低甲基化位点的一些探针可以没有鸟嘌呤(G)碱基。在一些实施例中,所述多个探针的至少3%、5%、10%、15%、20%、30%或40%缺少G(鸟嘌呤)。在一些实施例中,所述化验板上所述多个探针的至少80、85、90、92、95、98%在多个CpG检测位置上仅具有CpG或仅具有CpA。因此,在一些实施例中,多个含有多核苷酸的探针具有一核酸序列(1)与一标靶基因组区域(例如,列表1至列表8中所列的多个标靶基因组区域)中的一序列在序列上相同,或者(2)相对于所述基因组区域中的一序列仅一个或多个转换(transition)(例如,由于亚硫酸氢盐转化或其他转化技术而在一位点处碱基组成的改变)变化,其中,一个或多个转换中的每一个相应的转换发生在与所述基因组区域中一CpG位点相对应的一核苷酸处。 在一些实施例中,所述化验板上的多个探针包含少于20、15、10、8或6个CpG检测位置。在一些实施例中,所述化验板上的多个探针包含5、6、7、8、9或10个以上的CpG检测位置。 在一些实施例中,多个探针接合到一标记(tag)(例如,一非核酸亲和部分),例如一生物素部分。在一些实施例中,多个探针附在一固体支称件上,例如一阵列(array)。 询问甲基化状态的一癌症化验板也可以被设计用于询问癌症和健康样本中cfDNA之间不同的其他遗传或表观遗传标记。在一些实施例中,多个探针被设计用于扩增来自一特定标靶区域的所有cfDNA,而不管cfDNA分子的甲基化状态如何。这可能是因为标靶基因组区域不是高度甲基化或未甲基化的,或者是为了标靶多个小突变或多个SCNA而不是甲基化变化。 所述多个探针的长度可选地范围为10s到100s的核苷酸。所述多个探针可包含至少20、30、50、75、100或120个核苷酸。所述多个探针的长度可以小于300、250、200或150个核苷酸。在一些实施例中,所述多个探针包含100至150个核苷酸。在一个特定实施例中,所述多个探针长度为120个核苷酸。 在一些实施例中,所述多个探针设计为一“2倍平铺(2×tiled)”的方式(fashion)来覆盖一标靶区域的多个重叠部分。每个探针可选地在覆盖范围上至少部分地与文库(1ibrary)中的另一个探针相重叠。在这些实施例中,所述化验板包含多对探针,一对探针中的每一个探针彼此至少25、30、35、40、45、50、60、70、75或100个核苷酸相重叠。在一些实施例中,所述重叠序列可被设计为与一标靶基因组区域(或从中衍生的cfDNA)相互补或与一标靶区域或cfDNA具有同源性的一序列相互补。因此,在一些实施例中,至少两个探针与一标靶基因组区域内的相同的序列互补,及与所述标靶基因组区域相对应或衍生自所述标靶基因组区域的一核苷酸片段可被所述多个探针中的至少一个结合并下拉。可以使用其他级别的平铺,例如3倍平铺(3×tiling)、4倍平铺(4×tiling)等,其中一标靶区域中的每个核苷酸可与两个以上的探针相结合。 在一个实施例中,一标靶基因组区域中的每个碱基正好由两个探针重叠,如图1所示。如果所述两个探针之间的重叠比所述标靶基因组区域长并且延伸到所述标靶基因组区域的两端以外,那么单一对的探针就足以下拉一基因组区域。在某些情况下,甚至相对较小的多个标靶区域也可以用三个探针作为标靶(参见图1A)。一探针集合包含三个或更多个探针可选地用于捕获一更大的基因组区域。参见图1B。在一些实施例中,探针的多个子集将共同延伸到整个基因组区域(例如,可与来自所述基因组区域的未转化或转化的片段互补)。一平铺探针集合可选地包含多个探针,所述多个探针共同包含至少两个与所述基因组区域中的每个核苷酸相重叠。图1A和图1B。这样做是为了确保包含一标靶基因组区域的一小部分的多个cfDNA在一端处将具有延伸到相邻的非标靶基因组区域以至少一个探针实质性重叠,以提供有效的捕获。 例如,100个碱基(bp)的cfDNA片段包含一30个核苷酸(nt)的标靶基因组区域可保证与重叠的多个探针的至少一个具有至少65bp的重叠。可以使用其他级别的平铺。例如,为了在一化验板中增加标靶尺寸及添加更多探针,可以将探针设计为将30bp的标靶区域至少扩展为70bp、65bp、60bp、55bp或50bp。为了捕获与标所述靶区域完全重叠的任何片段(即使只有1bp),所述多个探针可以设计为延伸到所述标靶区域两端的两侧。 在一个特定实施例中,最小的标靶基因组区域为30bp。当一个新的CpG位点被添加到所述化验板中时(基于如上所述的贪婪选择),一个30nt的新标靶被集中在感兴趣的CpG位点上。然后,检查这个新标靶的每一边缘是否足够接近其他标靶,以便它们可以合并。合并避免了一化验板包含接近但不同的标靶跟多个重叠探针。这是基于一“合并距离”的参数,该参数在每个标靶区域的任一侧延伸约100nt、150nt、200nt、250nt或300nt。合并创建了一个更大的标靶基因组区域。如果一个新的CpG位点被合并到多个标靶的两侧,标靶基因组区域的数量就会减少。 在一些实施例中,本文提供的一化验板包含多个多核苷酸探针,配置为与从多个cfDNA分子的处理中获得的一修饰片段杂交,其中所述多个cfDNA分子中的每一个对应于或衍生自一个或多个基因组区域。在一些实施例中,所述多个基因组区域的至少15%、20%、30%或40%是外显子或内含子。在一些实施例中,所述多个基因组区域的至少5%、10%、15%、20%、30%或40%是外显子。在一些实施例中,所述多个基因组区域的少于5%、10%、15%、20%、25%或30%是基因间区域。 在一些实施例中,所述化验板上的多个探针的一整体一起被配置为与多个修饰的片段杂交,所述多个修饰的片段从对应于或衍生自列表1至列表8中的至少20%、30%、40%、50%、60%、70%、80%、90%或95%的所述多个基因组区域的所述多个cfDNA分子获得。在一些实施例中,所述化验板上的多个探针的一整体一起被配置为与多个修饰的片段杂交,所述多个修饰的片段从对应于或衍生自列表1中的至少20%、30%、40%、50%、60%、70%、80%、90%或95%的所述多个基因组区域的所述多个cfDNA分子获得。在一些实施例中,所述化验板上的多个探针的一整体一起被配置为与多个修饰的片段杂交,所述多个修饰的片段从对应于或衍生自列表2中的至少20%、30%、40%、50%、60%、70%、80%、90%或95%的所述多个基因组区域的所述多个cfDNA分子获得。在一些实施例中,所述化验板上的多个探针的一整体一起被配置为与多个修饰的片段杂交,所述多个修饰的片段从对应于或衍生自列表3中的至少20%、30%、40%、50%、60%、70%、80%、90%或95%的所述多个基因组区域的所述多个cfDNA分子获得。在一些实施例中,所述化验板上的多个探针的一整体一起被配置为与多个修饰的片段杂交,所述多个修饰的片段从对应于或衍生自列表5中的至少20%、30%、40%、50%、60%、70%、80%、90%或95%的所述多个基因组区域的所述多个cfDNA分子获得。在一些实施例中,所述化验板上的多个探针的一整体一起被配置为与多个修饰的片段杂交,所述多个修饰的片段从对应于或衍生自列表7中的至少20%、30%、40%、50%、60%、70%、80%、90%或95%的所述多个基因组区域的所述多个cfDNA分子获得。在一些实施例中,所述化验板上的多个探针的一整体一起被配置为与多个修饰的片段杂交,所述多个修饰的片段从对应于或衍生自列表8中的至少20%、30%、40%、50%、60%、70%、80%、90%或95%的所述多个基因组区域的所述多个cfDNA分子获得。在一些实施例中,所述化验板上的多个探针的一整体一起被配置为与多个修饰的片段杂交,所述多个修饰的片段从对应于或衍生自列表4中的至少20%、30%、40%、50%、60%、70%、80%、90%或95%的所述多个基因组区域的所述多个cfDNA分子获得。在一些实施例中,所述化验板上的多个探针的一整体一起被配置为与多个修饰的片段杂交,所述多个修饰的片段从对应于或衍生自列表6中的至少20%、30%、40%、50%、60%、70%、80%、90%或95%的所述多个基因组区域的所述多个cfDNA分子获得。在一些实施例中,将所述化验板上的多个探针的一整体一起被配置为与多个修饰的片段杂交,所述多个修饰的片段从对应于或衍生自列表1至列表8中的一个或多个中的至少500、1000、5000、10000或15000个基因组区域的cfDNA分子获得。 6.4.选择标靶基因组区域的方法 在另一方面,提供了用于从一样本中识别异常甲基化的多个DNA分子的多种方法。在一些实施例中,本文公开的多种方法可用于选择用于检测癌症、评估癌症的起源组织、或癌症类型的多个标靶基因组区域。所述多个标靶基因组区域可用于设计和制造用于一癌症化验板的探针。所述多个标靶基因组区域的甲基化状态可以使用所述癌症化验板进行筛选。在其他实施例中,这些方法可以用作一过滤程序的一部分,以限制一数据集(例如,一序列数据集)以减少后续处理或分析需求。例如,本文公开的多种方法可用于设置多个异常甲基化片段的一阈值,所述多个异常甲基化片段很可能衍生自癌症或癌细胞,及所述阈值用作滤除不符合所述阈值的多个序列读数或片段,因此,更可能来自健康细胞。 6.4.1.数据结构生成 图2是根据一个实施例描述对于一健康的对照组生成一数据结构的程序200的流程图。为了创建一健康的对照组的数据结构,分析系统接收来自多个健康对象的多个DNA片段(例如cfDNA和/或ctDNA)中的多个序列。对于每个片段识别一甲基化状态向量(例如,通过程序100)。 所述分析系统细分210每个DNA片段的所述甲基化状态向量为多个字符串(strings)的多个CpG位点。在一个实施例中,分析系统细分210所述甲基化状态向量,使得所产生的多个字符串都小于一给定长度。例如,划分长度11的一甲基化状态向量可细分为长度小于或等于3的多个字符串,从而导致长度3的9个字符串、长度2的10个字符串和长度1的11个字符串。在另一个示例中,长度7的一甲基化状态向量细分为长度小于或等于4的多个字符串,从而导致长度4的4个字符串、长度3的5个字符串、长度2的6个字符串和长度1的7个字符串。如果从一DNA片段所得的所述甲基化状态向量小于或与指定字符串长度相同,则所述甲基化状态向量可以转换为单个字符串,所述单个字符串包含所述向量的所有CpG位点。 分析系统通过计算运算(tallies)220所述多个字符串,对于每个可能的CpG位点和在向量中多个甲基化状态的可能性,在对照组中具有特定CpG位点作为字符串中第一个CpG位点并具有甲基化状态可能性的字符串数量。例如,对于在一给定的CpG位点处,一字符串长度为3,有2^3或8个可能的字符串配置。对于每个给定的CpG位点,分析系统运算220在对照组中出现的每一个可能的甲基化状态向量的发生次数。这可能涉及运算以下数量: 设置字符串长度的一上限具有几个好处。首先,取决于一字符串的最大长度,通过分析系统创建的数据结构的大小可以在大小上急遽地增加。例如,一最大字符串长度为4意味着,最多有2^4个数字用于在每个CpG处运算。将最大字符串长度增加到5加倍了甲基化状态的可能数目。减小字符串大小有助于减少数据结构的计算和数据存储负担。在某些实施例中,字符串尺寸为3。在某些实施例中,字符串尺寸为4。限制最大字符串长度的第二个理由是避免过度拟合下游模型。如果对结果(例如,预测癌症的存在的预测异常)没有很强的影响,则基于多个CpG位点的大字符串的概率可能会有问题,因为它需要大量的数据,而这些数据可能不可用,因此,对于一个模型适当地执行来说过于稀疏。例如,计算以前100个CpG位点为条件的异常/癌症的一概率将需要长度100的数据结构中的多个字符串计数,理想情况下,某些与前面100个甲基化状态完全匹配。如果只有长度100的字符串的稀疏计数可用,则没有足够的数据来确定测试样本中给定的长度为100的字符串是否异常。 6.4.1.数据结构验证 一旦创建了数据结构,分析系统可能会试图验证240数据结构和/或使用数据结构的任何下游模型。 第一类验证确保从健康对照组中移除潜在的多个癌性样本,以便在健康数据结构中引入偏差。这种类型的验证检查控制组的数据结构内的一致性。例如,健康对照组可包含来自具有未被诊断癌症的一个体的样本,所述个体包含多个异常甲基化片段。分析系统可以执行各种计算,以确定是否排除明显未被诊断癌症的一对向的数据。 一第二类型的验证检查用于使用数据结构本身(即来自健康对照组)的计数来计算多个p值的概率模型。下面结合图5描述p值计算的一程序。一旦所述分析系统针对验证组中的多个甲基化状态向量生成一p值,所述分析系统使用多个p值构建一累积密度函数(cumulative density function,CDF)。使用所述CDF,所述分析系统可以对所述CDF执行各种计算,以验证对照组的数据结构。一个测试使用了所述CDF在理想情况下应为或低于一恒等函数(identity function)的事实,使得CDF(x)≤x。相反的,高于所述恒等函数则表明用于对照组数据结构的概率模型中存在一些缺陷。例如,如果1/100的片段具有一p值得分为1/1000,意味着CDF(1/1000)=1/100>1/1000,则所述第二类型的验证失败,表明概率模型存在问题。参见例如美国专利申请案第16/352,602号公开为美国专利申请案公开第2019/0287652号,其全部内容通过引用并入本文。 一第三类型的验证使用了从用于构建所述数据结构的那些分离的多个验证样本的一健康集合。这测试所述数据结构是否构建正确,所述模型是否有效。下面结合图3描述了执行此类型的验证的示例程序。所述第三类型的验证可以量化所述健康对照组对多个健康样本分布的概括(generalizes)程度。如果所述第三类型的验证失败,则所述健康对照组就不能很好地概括健康分布。 一第四类型的验证测试,多个样本来自一非健康验证组。所述分析系统计算多个p值并对所述非健康验证组构建CDF。对于一非健康验证组,所述多个分析系统预计看到至少一些样本的CDF(x)>x,或者,换句话说,在所述健康对照组和所述健康验证组的所述第二类型的验证和所述第三类型的验证中预期的结果相反。如果所述第四种类型的验证失败,则这表示模型没有适当地识别设计用来识别的异常。 图3是根据一个实施例描述验证图2的对照组的数据结构的附加步骤240的流程图。在验证所述数据结构的这个步骤240中,所述分析系统执行如上所述的第四类型的验证测试,其利用一验证组,所述验证组具有假设与对照组相似组合的多个对象、多个样本和/或多个片段。例如,如果所述分析系统为对照组选择不具有癌症的多个健康对象,则所述分析系统也使用所述验证组中不具有癌症的多个健康对象。 所述分析系统采用所述验证组并生成100多个甲基化状态向量的一集合,如图2所示。所述分析系统对来自所述验证组的每个甲基化状态向量执行一p值计算。所述p值计算程序将结合图4和图5进一步描述。对于每个甲基化状态向量的可能性,所述分析系统从对照组的数据结构计算320一概率。一旦对于甲基化状态向量的多个可能性被计算出概率,所述分析系统基于计算出的多个概率计算330该甲基化状态向量的一p值得分。所述p值得分代表了发现特定甲基化状态向量和其他可能甲基化状态向量在对照组中具有更低概率的期望值。因此,一低的p值得分通常对应于一甲基化状态向量,所述甲基化状态向量与对照组内其他甲基化状态向量相比相对非期望的,其中一高p值得分通常对应于一甲基化状态向量,所述甲基化状态向量与在对照组中发现的其他甲基化状态向量相比相对更期望。一旦所述分析系统对所述验证组中的多个甲基化状态向量生成一p值得分,所述分析系统将使用来自所述验证组的所述p值得分构建340一累积密度函数(CDF)。所述分析系统验证370上述第四类型的验证测试中CDF的一致性。 6.4.2.异常甲基化片段 根据图4中概述的一个实施方案,选择具有多个异常甲基化模式的多个异常甲基化片段作为多个标靶基因组区域。选定的多个异常甲基化片段440的一个示例性程序在图5中视觉上地示出,并且在图4的描述在下面进一步描述。在程序400中,所述分析系统从所述样本的多个cfDNA片段生成100多个甲基化状态向量。所述分析系统如下处理每个甲基化状态向量。 在一些实施例中,所述分析系统在一个或多个CpG位点处过滤405具有不确定状态的多个片段。在这些实施例中,所述分析系统执行一预测模型以识别不太可能具有一异常甲基化模式的多个片段以用于过滤。对于一样本片段,所述预测模型计算与健康对照组的数据结构相比样本片段的甲基化状态向量发生的一样本概率。所述预测模型随机抽样多个可能甲基化状态向量的一子集,所述多个可能甲基化状态向量的所述子集包含所述样本片段的甲基化状态向量中的多个CpG位点。所述预测模型计算对应于多个抽样的可能甲基化状态向量中的每一个的一概率。对于所述片段的甲基化状态向量和所述多个抽样的可能甲基化状态向量的多个概率计算可以根据一马尔可夫链(Markov chain)模型计算,如下6.4.2.1节“P值得分计算”中所述。所述预测模型计算所述多个抽样的可能甲基化状态向量对应于小于或等于样本概率的多个概率的一比例。所述预测模型基于计算出的比例为所述片段生成一估计的p值得分。所述预测模型可以过滤对应于高于一阈值的多个p值得分的多个片段,及保留对应于低于所述阈值的多个p值得分的多个片段。同样,参见例如美国申请案第16/352,602号,公开为美国专利申请案公开第2019/0287652号,其通过引用将整体并入本文。 在其他实施例中,所述预测模型可以计算一置信概率,所述置信概率是所述预测模型用于确定何时继续或何时终止采样。所述置信概率描述了片段的真实p值得分低于基于估计的p值得分和多个抽样的可能甲基化状态向量的概率的一阈值的可能性。所述预测模型可以在迭代计算所述估计的p值得分和所述置信概率的同时,对另外一个或多个可能的甲基化状态向量进行抽样。当所述置信概率高于一置信阈值时,所述预测模型可以终止抽样。 对于一给定的甲基化状态向量,所述分析系统列举410具有相同的起始CpG位点和相同的长度(即,多个CpG位点的一集合)的多个甲基化状态向量的所有可能性作为所述甲基化状态向量中。每个CpG位点只有两个可能的状态,甲基化或未甲基化,因此甲基化状态向量的不同可能性的计数取决于2的幂次,因此长度为n的一甲基化状态向量将与甲基化状态向量的2 所述分析系统通过访问健康对照组数据结构,计算420观察已识别起始CpG位点/甲基化状态向量长度的甲基化状态向量的每个可能性的概率。在一个实施例中,计算观察一给定的可能性的概率使用马尔可夫链概率来建模联合概率计算,将关于图5更详细地描述所述联合概率计算。在其它实施例中,使用马尔可夫链概率以外的计算方法来确定观察甲基化状态向量的每个可能性的概率。 所述分析系统使用计算出的对于每种可能性的多个概率计算430对于甲基化状态向量的一p值得分。在一个实施例中,这包含确定与匹配所讨论的所述甲基化状态向量的可能性相对应的计算概率。具体地说,这是具有多个CpG位点的相同集合,或具有相同的起始CpG位点、长度及甲基化状态作为甲基化状态向量的可能性。所述分析系统将具有概率小于或等于识别概率的任何可能性的计算概率相加,以生成p值得分。 此p值代表观察片段的甲基化状态向量,或者其他甲基化状态向量在健康对照组中甚至更低的可能性的概率。因此,一低的p值得分通常对应于在一健康对象中是罕见的一甲基化状态向量,并且相对于健康对照组,这会导致片段被标记为异常甲基化。一高的p值得分通常与在一个健康的对象中预期存在(在相对意义上)的一甲基化状态向量有关。如果健康对照组是非癌症组,例如,一低的p值表示所述片段相对于所述非癌症组是异常甲基化,因此可能表示测试对象中存在癌症。 如上所述,所述分析系统计算多个甲基化状态向量中的每一个的p值得分,每个代表测试样本中的一cfDNA和/或ctDNA片段。为了辨识哪些片段是异常甲基化,所述分析系统可以基于它们的p值得分过滤440所述多个甲基化状态向量的所述集合。在一个实施例中,通过将所述多个p值得分与一阈值进行比较并仅保留在所述阈值以下的那些片段来执行滤波。此阈值p值得分可以是0.1、0.01、0.001、0.0001或类似的值。 6.4.2.1.P值得分计算 图5是根据一个实施例的示例p值得分计算的图示500。为了计算给定的一测试甲基化状态向量505的一p值得分,所述分析系统采用该测试甲基化状态向量505并列举410多个甲基化状态向量的多个可能性。在这个说明性示例中,所述测试甲基化状态向量505是 分析系统计算420多个甲基化状态向量列举的多个可能性的概率515。由于甲基化是有条件地取决于邻近的多个CpG位点的甲基化状态,计算观察一给定的甲基化状态向量的可能性的概率的一种方法是使用马尔可夫链模型。通常,一甲基化状态向量,例如 P( 马尔可夫链模型可以被用于更有效地计算每种可能性的条件概率。在一个实施例中,所述分析系统选择一马尔可夫链阶(Markov chain order)k,所述马尔可夫链阶k对应于在条件概率(conditional probability)计算中需考虑在向量(或窗口)中先前的CpG位点数量,使得条件概率被建模为P(S 为了计算甲基化状态向量的可能性的每个马尔可夫建模的概率,所述分析系统访问对照组的数据结构,特别是不同字符串的CpG位点和状态的计数。为了计算P(M
所述计算还可以另外通过应用一先验分布来实现计数的平滑。在一个实施例中,所述先验分布是一均匀的先验,例如拉普拉斯平滑(Laplace smoothing)。例如,在上述方程式的分子上加一常数,在上述方程的分母上加上另一个常数(例如分子中常数的两倍)。在其它实施例中,使用例如Knesser-Ney平滑的算法技术。 在图示中,上述表示的公式应用于覆盖位点23-26的测试甲基化状态向量505。一旦计算的概率515完成,所述分析系统计算430一p值得分525,其加总小于或等于甲基化状态向量与测试甲基化状态向量505匹配的可能性的概率。 在具有多个不确定状态的多个实施例中,所述分析系统可计算出在一片段的甲基化状态向量中具有多个不确定状态的多个CpG位点的一p值得分。所述分析系统识别具有和甲基化状态向量的所有甲基化状态一致的所有的可能性,不包含不确定状态。所述分析系统可以将概率分配给甲基化状态向量,作为多个已识别的可能性的概率的一总和。作为一示例,所述分析系统计算一甲基化状态向量 在一个实施例中,计算多个概率和/或多个p值得分的运算负担可以通过缓存至少一些计算来进一步降低。例如,所述分析系统可以在临时或持久内存中缓存针对多个甲基化状态向量(或其窗口)的多个可能性的概率计算。如果其他片段具有相同的多个CpG位点,则缓存多个可能性概率可以有效率的计算p值得分,而无需重新计算潜在的可能性概率。同样,所述分析系统可以从向量(或其窗口)与多个CpG位点的一集合相关联的多个甲基化状态向量的多个可能性中的每一个计算多个p值得分。所述分析系统可以缓存所述多个p值得分,以用于确定包含相同CpG位点的其他片段的p值得分。通常,具有相同CpG位点的多个甲基化状态向量的可能性的p值得分可用于确定来自同一集合的多个CpG位点的不同CpG位点的可能性的p值得分。 6.4.2.2.滑动窗口 在一个实施例中,所述分析系统使用435一滑动窗口来确定多个甲基化状态向量的可能性并计算p值。不是针对整个甲基化状态向量列举可能性和计算p值,所述分析系统而是仅针对连续的(sequential)多个CpG位点的一窗口列举可能性并计算p值,其中所述窗口的长度(CpG位点的长度)比至少一些片段的长度短(否则,窗口毫无意义)。窗口长度可以是静态的、用户确定的、动态的或以其他方式选择的。 在计算大于所述窗口的一甲基化状态向量的p值时,所述窗口从向量中的第一个CpG位点开始,在窗口内从向量识别多个CpG位点的序列集合。所述分析系统计算对于所述窗口(包含第一个CpG位点)的一p值得分。然后,所述分析系统将所述窗口“滑动(slides)”到向量中的第二个CpG位点,并计算第二个窗口的另一个p值得分。因此,对于一窗口大小l和甲基化向量长度m,每个甲基化状态向量将生成m-l+1个p值得分。在完成对于向量的每个部分的p值计算后,所有滑动窗口中的最低p值得分被作为甲基化状态向量的整体的p值得分。在另一实施例中,所述分析系统总合多个甲基化状态向量的多个p值得分以生成一整体的p值得分。 使用所述滑动窗口有助于减少甲基化状态向量列举的可能性的数量及其相对应的概率计算,否则将需要执行这些操作。示例概率计算如图5所示,但通常甲基化状态向量的可能性的数量随着甲基化状态向量的大小成指数地增加2倍。在一个现实的示例,对于多个片段有可能有54个以上的CpG位点。作为对2^54(~1.8×10^16)种可能性计算概率来生成单个p得分的替代,所述分析系统可以改为使用大小为5的一窗口(例如),从而对该片段的甲基化状态向量的50个窗口中的每个窗口进行50个p值计算。50个计算中的每一个都列举了甲基化状态向量的2^5(32)个可能性,总的结果是50×2^5(1.6×10^3)个概率计算。这导致要执行的计算大大减少,而对异常片段的准确识别没有任何意义。当用验证组的甲基化状态向量验证240对照组时,此附加步骤也可应用。 6.4.3.高甲基化或低甲基化片段的分析 在一些实施例中,执行一附加过滤步骤以识别多个基因组区域,所述多个基因组区域可被标靶以用于癌症检测、一癌症的起源组织、或癌症的一类型。 分析系统可以对多个异常甲基化片段的集合执行任何变异和/或额外分析的可能性。一个附加的分析从经过滤的集合中识别450多个低甲基化片段或多个高甲基化片段。低甲基化或高甲基化的多个片段可被定义为一定长度的多个CpG位点(例如,超过3、4、5、6、7、8、9、10个等)的多个片段,分别具有一高百分比的甲基化的多个CpG位点(例如,超过80%、85%、90%或95%,或50%至100%范围内的任何其他百分比)或一高百分比的未甲基化的多个CpG位点(例如超过80%、85%、90%或95%或50%至100%范围内的任何其他百分比)。下文描述图6示出了基于异常甲基化的多个片段的一集合来识别一基因组的这些异常高甲基化或低甲基化部分的示例程序。 一交替分析应用460一训练的分类模型在多个异常片段的集合上。训练的分类模型可以被训练以识别可以从所述多个甲基化状态向量中被识别的任何感兴趣的条件。在一个实施例中,所述训练的分类模型是基于从具有癌症的一对象群体中获得的多个cfDNA片段的甲基化状态训练的二元分类器,及可选地基于从不具有癌症的一健康对象群体中获得的多个cfDNA片段的甲基化状态,然后,基于多个异常甲基化状态向量,用于分类具有癌症或不具有癌症的一测试对象概率。在进一步实施例中,可以使用已知具有特定癌症(例如乳腺癌、肺癌、前列腺癌等)的多个对象群体来训练不同的分类器,以预测一测试对象是否患有那些特定癌症。 在一个实施例中,基于来自程序450关于高/低甲基化区域的信息以及下文对于图6的描述,对分类器进行训练。 图6是描述根据一个实施例基于多个cfDNA片段的甲基化状态训练一分类器的程序600的流程图。一分析系统可用于执行所述程序600。所述程序访问两个训练群组的样本(一非癌症群组和一癌症群组),并获得400一非癌症的甲基化状态向量集合和一癌症的甲基化状态向量集合,包含每群组中多个样本的多个异常片段。例如,可以根据图4的程序来识别所述多个异常片段。 所述分析系统确定610(对于每个甲基化状态向量)甲基化状态向量是否是高甲基化或低甲基化。此处,如果至少一些数量的CpG位点具有一特定状态(分别为甲基化或未甲基化)和/或具有所述特定状态的多个位点的一阈值百分比(再次分别为甲基化或未甲基化),则高甲基化或低甲基化的标签被分配。在一个示例中,如果片段与至少5个CpG位点重叠,并且其CpG位点的至少80%、90%、或100%是甲基化或至少80%、90%、或100%是未甲基化,则多个cfDNA片段分别被识别为低甲基化或高甲基化。 在一个替代的实施例中,所述分析系统考虑甲基化状态向量的多个部分并确定所述部分是否低甲基化还是高甲基化,并且可以区分该部分是低甲基化还是高甲基化。此替代方法解决了丢失的多个甲基化状态向量,所述丢失的多个甲基化状态向量的尺寸很大,但含有至少一个区域的密集的低甲基化或高甲基化。这种定义低甲基化和高甲基化的程序可应用于图4的步骤450中。 在一个实施例中,所述分析系统在基因组中的每个CpG位点产生620一低甲基化得分(P 在一个实施例中,在一给定的CpG位点处的低甲基化得分被定义为(1)与(3)的一比率的一对数。同样地,高甲基化得分被计算为(2)与(4)的一比率的一对数。此外,可以使用如上所述的附加平滑技术计算这些比率。 在另一个实施例中,低甲基化得分被定义为(1)与(1)和(3)加总的一比率。高甲基化得分被定义为(2)与(2)和(4)加总的一比率。与上面的实施例类似,平滑技术可以实现在这些比率中。 所述分析系统为每个异常甲基化状态向量生成630一总合的低甲基化得分和一总合的高甲基化得分。所述总合的高甲基化和低甲基化得分,是基于甲基化状态向量中所述多个CpG位点的高甲基化和低甲基化得分来确定。在一个实施例中,总合的高甲基化得分和低甲基化得分分别被分配为每个状态向量中所述多个位点的最大高甲基化和低甲基化得分。然而,在替代的实施例中,所述多个总合的得分可基于平均值、中位数或使用每个向量中所述多个位点的高/低甲基化得分的其他计算。在一个实施例中,所述分析系统将总合的低甲基化得分和总合的高甲基化得分中的较大者分配给所述异常甲基化状态向量。 所述分析系统然后通过其总合的低甲基化得分和总合的高甲基化得分对该对象的多个甲基化状态向量进行排名640,结果每个对象有两个排名。所述程序从低甲基化排名中选择总合的低甲基化得分,从高甲基化排名中选择总合的高甲基化得分。根据所选的多个得分,所述分类器为每个对象生成650单个特征向量。在一个实施例中,从任一排名中选择的多个得分以一固定顺序选择,对于多个训练组中的每一个的每个对象的每个生成的特征向量是相同的。作为一示例,在一个实施例中,所述分类器从每个排名中选择第一、第二、第四和第八、第十六、第三十二、第六十四总合的高甲基化得分,及类似地对于每个总合的低甲基化得分,以及将这些得分写入该对象的特征向量中(特征向量中共有14个特征)。在附加实施例中,为了调整样本的测序深度,所述分析系统以与相对的样本深度成线性比例的方式调整排名。例如,如果相对的样本深度为x,则在x*原始排名处取插值得分(即x=1.1,取排名1.1、2.2、...、x*2i处计算的得分)。然后,所述分析系统可以基于调整后的排名来定义特征向量以用于进一步分类。 所述分析系统训练660二进制分类器,以区分癌症和非癌症训练群组之间的多个特征向量。所述分析系统可以将所述多个训练样本分组成一个或多个训练样本的多个集合,用于所述二元分类器的迭代批次训练。在输入所有训练样本的集合(包含其训练特征向量)并调整分类参数后,对二值分类器进行充分训练,以在一定的误差范围内根据所述多个训练样本的特征向量对多个测试样本进行标记。 在一个实施例中,所述分类器是非线性分类器。在一个具体实施例中,所述分类器是利用具有一高斯径向基函数(RBF)核(Gaussian radial basis function kernel)的一L2正则化核逻辑回归(L2-regularized kernel logistic regression)的一非线性分类器。具体地说,以各向同性径向基函(isotropic radial basis function)(幂指数2)为核,具有尺度参数(scale parameter)为伽马(gamma)及L2正则化参数(L2regularizationparameter)为拉姆达(lambda)训练一正则化核逻辑回归分类器(regularized kernel logistic regression classifier,KLR)。伽马(gamma)和拉姆达(lambda)为在指定的训练数据中使用内部交叉验证优化抵抗对数损失(log-loss),并使用网格搜索(grid-search)以乘法步骤进行优化,从最大值开始,每一步骤将参数减半。在其它实施例中,所述分类器可包含其它类型的分类器,例如一随机森林分类器、一混合模型、一卷积神经网络和一自动编码器模型。 在一些实施例中,对每个CpG位点进行计算。具体地,一第一计数被确定是包含与该CpG重叠的一片段的多个癌性样本的数量(癌症_计数),及一第二个计数被确定是包含在集合中含有与该CpG重叠的多个片段的多个样本的总数(总数)。多个基因组区域可以根据所述数量来选择,例如,基于与多个癌性样本的数量(癌症_计数)呈正相关的标准,所述多个癌性样本包含与该CpG重叠的一片段,及与在该集合中含有与该CpG重叠的多个片段的多个样本的总数(总数)呈负相关。具体地,在一个实施例中,具有一片段与一CpG位点重叠的非癌症样本的数量(n 在某些情况下,附加的分析计算出来自一对象的多个异常片段通常表示癌症的对数比(log-odds ratio)。所述对数比可以通过取癌性的一概率与非癌性的一概率的一比率的对数(即1减去为癌性的概率)的对数来计算,两者均由所应用的分类模型确定。 6.4.4.脱靶基因组区域 在一些实施例中,标靶所选的多个基因组区域的多个探针基于其脱靶区域的数量被进一步过滤465。这是为了筛选能下拉太多含有多个脱靶基因组区域的多个cfDNA片段的多个探针。排除具有许多脱靶区域的多个探针可能是有价值的,因为它降低了脱靶率,并增加了一给定数量的测序的标靶覆盖率。 一脱靶基因组区域是指与一标靶基因组区域具有显着同源性的一基因组区域,以通过设计用于标靶所述标靶基因组区域的一探针结合并下拉。一非靶标基因组区域可以是一基因组区域(或该相同的区域的一转化的序列),沿着至少35bp、40bp、45bp、50bp、60bp、70bp或80bp以具有至少为80%、85%、90%、95%或97%的匹配率与一探针对齐。在一个实施例中,一脱靶基因组区域是一基因组区域(或该相同的区域的一转化的序列)沿着至少45bp以至少90%的匹配率与一探针对齐。可以采用本领域已知的各种方法来筛选出多个标靶基因组区域。 彻底搜索基因组以找到所有脱靶基因组区域可能在计算上具有挑战性。在一个实施例中,一k-mer(k个碱基的子字符串)接种策略(可允许一个或多个不匹配)组合到多个种子位置处的局部对齐。在这种情况下,可以基于k-mer长度、允许的不匹配数量和一特定位置的k-mer种子命中数来保证对良好对齐的彻底搜索。这需要在大量位置进行动态编程局部对齐,因此此方法被高度优化以使用多个向量CPU指令(例如,AVX2、AVX512),并且可以在一机器内的多个核心上并行,也可以在通过一网络连接在多台机器上并行。本领域普通技术人员将认识到可以对这种方法进行多种修改和多种变型,以识别出脱靶的多个基因组区域。 在一些实施例中,具有多个脱靶基因组区域的多个探针大于一阈值数量的被排除。例如,具有多于30个、多于25个、多于20个、多于18个、多于15个、多于12个、多于10个或多于5个的探针被排除。 在一些实施例中,取决于脱靶区域的数量,将探针分为2、3、4、5、6或更多个单独的群组。例如,具有无脱靶区域的多个探针被分配到高质量组,具有1至19个脱靶区域的多个探针被分配到低质量组,具有19个以上的脱靶区域的多个探针被分配到劣质组。其他截止值可用于分组。 在一些实施例中,最低质量组中的多个探针被排除。在一些实施例中,除了最高质量组之外的组中的多个探针被排除。在一些实施例中,为每组中的多个探针制作单独的化验板。在一些实施例中,所有探针使用在相同化验板上,但是基于分配的组执行单独的分析。 在一些实施例中,一化验板包含的数量较多的高质量探针较低的组中的探针的数量多。在一些实施例中,一化验板包含的较少数量的劣质探针少于其他组中的数量。在一些实施例中,一化验板中超过95%、90%、85%、80%、75%或70%的探针是高质量探针。在一些实施例中,一化验板中少于35%、30%、20%、10%、5%、4%、3%、2%或1%的探针是低质量探针。在一些实施例中,一化验板中少于5%、4%、3%、2%或1%的探针是劣质探针。在一些实施例中,一化验板中不包含劣质的探针。 在一些实施例中,低于50%、低于40%、低于30%、低于20%、低于10%或低于5%的探针被排除。在一些实施例中,一化验板中选择性地包含具有30%以上、40%以上、50%以上、60%以上、70%以上、80%以上或90%以上的探针。 6.5.使用癌症化验板的方法 在另一方面,提供了使用一癌症化验板的多种方法。所述多种方法可包含步骤:亚硫酸氢盐处理多个cfDNA片段,以将未甲基化的胞嘧啶转化为尿嘧啶,将多个样本应用于所述癌症化验板并对所述化验板中结合到多个探针的所述多个cfDNA片段进行测序。多个序列读数可以进一步与一参考基因组进行比较。这种化验可以识别在所述多个片段中的多个CpG位点处的甲基化状态,从而提供与癌症诊断相关的信息。 6.5.1.样本处理 图7A是根据一个实施例制备用于分析的一核酸样本的方法的流程图。所述方法包含但不限于以下步骤。例如,所述方法的任何步骤可包含用于质量控制的定量子步骤或本领域技术人员已知的其他实验室化验程序。 在步骤105中,从一对象中提取一核酸样本(DNA或RNA)。在本发明的公开中,除非另有说明,否则DNA和RNA可以互换使用。也就是说,本文所描述的实施例可以同时适用于核酸序列的DNA和RNA类型。然而,为了清楚和解释的目的,本文描述的示例可以集中于DNA。所述样本可以包含人类基因组的任何子集,包含整个基因组。所述样本可包含血液、血浆、血清、尿液、粪便、唾液、其他类型的体液或其任何组合。在一些实施例中,用于提取一血液样本的多种方法(例如,注射器或手指刺)可比用于获取组织活检的程序(可能需要手术)具有更小的侵入性。提取的样本可以包含cfDNA和/或ctDNA。对于多个健康的个体,人体可能会自然清除cfDNA和其他细胞片段。如果一对象患有一癌症或疾病,提取样本中的cfDNA或ctDNA可能在可检测水平上以用于诊断。 在步骤110中,所述多个cfDNA片段被处理以将未甲基化胞嘧啶转化为尿嘧啶。在一个实施例中,所述方法使用DNA的亚硫酸氢盐处理,将未甲基化胞嘧啶转化为尿嘧啶,而不转化甲基化胞嘧啶。例如,用于亚硫酸氢盐转化的商用试剂盒,如EZ-DNAMethylationTM-Gold、EZ-DNA MethylationTM-Direct或EZ-DNA MethylationTM-Lightning试剂盒(可从Zymo Research Corp公司(加利福尼亚州尔湾市))获得。在另一实施例中,利用一酶反应来完成未甲基化的胞嘧啶到尿嘧啶的转化。例如,所述转化可以使用商业上可买到的试剂盒将未甲基化的胞嘧啶转化为尿嘧啶,例如APOBEC-Seq(NEBiolabs公司,马萨诸塞州伊普斯威奇市)。 在步骤115,制备一测序文库。在一第一步骤中,使用一ssDNA连接反应(1igationreaction)将一ssDNA适配器(adapter)添加到一亚硫酸氢盐转化的ssDNA分子的3′-OH端。在一个实施例中,所述ssDNA连接反应使用环化连接酶(CircLigase)II(Epicentre公司)将所述ssDNA适配器连接到一亚硫酸氢盐转化的ssDNA分子的3′-OH端,其中所述适配器的5′-端被磷酸化并且所述亚硫酸氢盐转化的ssDNA已经被脱磷(即,3′端具有一羟基)。在另一实施例中,所述ssDNA连接反应使用热稳定5′AppDNA/RNA连接酶(Thermostable 5′AppDNA/RNA ligase)(可从New England BioLabs公司(马萨诸塞州伊普斯威奇市)获得)将所述ssDNA适配器连接到一亚硫酸氢盐转化的ssDNA分子的3′-OH端。在本示例中,第一UMI适配器在5′端被腺苷酸化(adenylated)并且在3′端被阻断(blocked)。在另一实施例中,所述ssDNA连接反应使用T4 RNA连接酶(T4 RNA ligase)(可从New England BioLabs公司获得)将所述ssDNA适配器连接到一亚硫酸氢盐转化的ssDNA分子的3′-OH端。在一第二步骤中,在一延伸反应(extension reaction)中合成一第二链DNA。例如,在一引物延伸反应中使用与ssDNA适配器中包含的一引物序列杂交的一延伸引物,以形成一双链亚硫酸氢盐转化的DNA分子。可选地,在一个实施例中,所述延伸反应使用能够读取亚硫酸氢盐转化模板链中的多个尿嘧啶残基(residues)的酶。可选地,在一第三步骤中,将一dsDNA适配器添加到所述双链亚硫酸氢盐转化的DNA分子中。最后,所述双链亚硫酸氢盐转化的DNA被扩增(amplified)以添加多个测序适配器。例如,使用包含一P5序列的一正向引物和包含一P7序列的一反向引物的PCR扩增用于将P5和P7序列添加到所述亚硫酸氢盐转化的DNA中。可选地,在文库制备期间,多个唯一分子标识符(UMI)可通过适配器连接添加到多个核酸分子(例如多个DNA分子)中。所述多个唯一分子标识符(UMIs)是多个短的核酸序列(例如,4-10个碱基对),在适配器连接期间添加到多个DNA片段的末端。在一些实施例中,UMIs是多个简并(degenerate)碱基对,其作为一唯一标记以可用于识别源自一特定DNA片段的多个序列读数。在PCR扩增过程后适配器连接期间,所述多个唯一分子标识符与附加的多个DNA片段一起复制,以在下游分析中提供了一种方法以识别来自相同原始片段的多个序列读数。 在步骤120中,多个标靶DNA序列可从文库中扩增。例如,在多个样本上执行标靶化验板化验。在扩增中,多个杂交探针(在本文中也被称为“多个探针”)用于标靶并下拉多个核酸片段,这些片段可提供关于癌症(或疾病)的存在与否、或癌症状态的信息。对于一给定的工作流程,所述多个探针可以设计成与DNA或RNA的一标靶(互补的)链进行退火(或杂交)。所述标靶链可以是“正”链(例如,转录成mRNA的链,随后转化为蛋白质)或互补的“负”链。所述多个探针的长度可以是10s、100s或1000s的碱基对。此外,所述多个探针可以覆盖一标靶区域的多个重叠部分。 在一杂交步骤120之后,杂交的多个核酸片段被捕获及可以使用PCR放大(扩增125)。例如,可以对多个标靶序列进行扩增以获得可随后测序的多个扩增序列。一般而言,本领域任何已知方法均可用于分离并扩增探针杂交的多个标靶核酸。例如,如本领域已知,可将一生物素部分添加至探针的5′端(即,生物素化),以便于分离使用链霉亲和素涂层表面(例如,链霉亲和素包披珠)与探针杂交的多个标靶核酸。 在步骤130中,多个序列读数从扩增的多个DNA序列(例如,多个扩增序列)生成。测序数据可以通过本领域已知的手段从扩增的多个DNA序列获得。例如,所述方法可包含下一代测序(NGS)技术,包含合成技术(Illumina公司)、焦磷酸测序(454 Life Sciences公司)、离子半导体技术(Ion Torrent测序)、单分子实时测序(Pacific Biosciences公司)、边连接边测序(SOLiD测序),纳米孔测序(Oxford Nanopore Technologies公司),或双端测序(paired-end sequencing)。在一些实施例中,通过使用具有多个可逆染料终止子(reversible dye terminators)的边合成边测序(sequencing-by-synthesis)来执行大规模平行测序。 6.5.2.序列读数的分析 在一些实施例中,所述多个序列读数可以使用本领域已知的方法与一参考基因组对齐以确定对齐位置信息。所述对齐位置信息可表示所述参考基因组中的一起始位置和一结束位置,所述参考基因组中的所述起始位置和所述结束位置对应于一给定序列读数的一起始核苷酸碱基和一结束核苷酸碱基。对齐位置信息还可以包含序列读数长度,其可从所述起始位置和所述结束位置确定。所述参考基因组中的一区域可能与一基因或一基因的一片段相关联。 在各种实施例中,一序列读数包含表示为R 根据所述序列读数,每个CpG位点的位置和甲基化状态可以基于与一参考基因组的对齐来确定。此外,每个片段的甲基化状态向量可以被生成以指定所述片段在所述参考基因组中的一位置(例如,通过每个片段中第一CpG位点的位置或另一个类似的度量来指定)、所述片段中的多个CpG位点的一数量,以及所述片段中每个CpG位点的甲基化状态是甲基化(例如,表示为M)、未甲基化(例如,表示为U)或不确定(例如,表示为I)。所述多个甲基化状态向量可以被存储在临时或持久的计算机存储器中以供之后使用和处理。此外,来自一单一对象的多个重复读数或多个重复甲基化状态向量可以被移除。在另一实施例中,可以确定某个片段具有一个或多个具有不确定甲基化状态的CpG位点。这种片段可以从以后的处理中排除,也可以选择性地包含在下游数据模型说明此类不确定的甲基化状态。 图7B是根据一个实施例图7A的程序100的对一cfDNA片段测序以获得一甲基化状态向量的图示。作为一个示例,所述分析系统取用一cfDNA片段112。在本示例中,所述cfDNA片段112包含三个CpG位点。如图所示,cfDNA片段112的第一和第三CpG位点被甲基化114。在处理步骤120期间,所述cfDNA片段112被转换以产生一转换后的cfDNA片段122。在处理120期间,第二未甲基化的CpG位点使其胞嘧啶转化为尿嘧啶。然而,所述第一和第三CpG位点没有被转换。 转换后,准备一测序文库130并测序140生成一序列读数142。所述分析系统将序列读数142与一参考基因组144对齐150。所述参考基因组144提供了所述cfDNA片段起源于一人类基因组中的哪个位置。在这个简化的示例中,所述分析系统对齐150所述序列读数,使得三个CpG位点与CpG位点23、24和25(为了便于描述而使用的任意参考标识符)相关。所述分析系统因此产生了cfDNA片段112上所有CpG位点的甲基化状态的信息,以及在人类基因组中CpG位点地图定位的信息。如图所示,序列读数142上被甲基化的CpG位点被读作胞嘧啶。在本示例中,胞嘧啶出现在序列读数142中,仅出现在第一和第三CpG位点,这允许推断原始cfDNA片段中的第一和第三CpG位点被甲0基化。然而,第二CpG位点被读作胸腺嘧啶(在测序程序中U被转换成T),因此,可以推断第二CpG位点在原始cfDNA片段中是未甲基化的。利用这两个信息,甲基化状态和位置,所述分析系统为所述片段cfDNA 112生成160一甲基化状态向量152。在此示例中,所得的甲基化状态向量152是 图9A和9B示出了验证来自一对照组的测序一致性的数据的三个图示。第一图示170示出了不同癌症阶段(阶段I、阶段II、阶段III、阶段IV和非癌症)的多个对象从一测试样本获得的cfDNA片段上未甲基化胞嘧啶转化为尿嘧啶(步骤120)的转化精度。如图所示,将多个cfDNA片段上的未甲基化胞嘧啶转化为尿嘧啶具有均一的一致性。总的转化准确率为99.47%,具有一精密度为±0.024%。第二图示180示出了癌症不同阶段的平均覆盖率。所有组的平均覆盖率为多个DNA片段的基因组覆盖率约34倍(~34×),仅使用确信地映射到所述基因组的那些进行计数。第三图示190示出了不同癌症阶段每个样本的cfDNA浓度。 6.5.3.癌症检测 通过本文提供的方法获得的多个序列读数或多个片段可以通过一医学专家分析或通过一自动化算法进一步处理。例如,所述分析系统用于从一测序器接收测序数据并执行如本文所述的各个方面的处理。所述分析系统可以是个人电脑(PC)、台式电脑、膝上型计算机、笔记本电脑、平板电脑、移动装置中的一种。一计算装置可以通过无线、有线或无线和有线通信技术的组合通信地耦合到所述测序器。通常,所述计算装置配置有一处理器和存储器,用于存储多个计算机指令,当由所述处理器执行时,所述多个计算机指令使处理器执行本文档其余部分中描述的步骤。一般来说,遗传数据和由此产生的数据量足够大,而计算能力又要求如此之大,以致于不可能在纸上或仅凭人类的头脑来执行。 多个标靶基因组区域的甲基化状态的临床解释是一程序,所述程序包含对每个甲基化状态或甲基化状态组合的临床效果进行分类,并以对医学专业人员有意义的方式报告结果。所述临床解释可以是基于多个序列读数与特定于癌症或非癌症对象的数据库的比较,和/或基于从一样本中识别出的具有癌症特异性甲基化模式的多个cfDNA片段的数量和类型。在一些实施例中,多个标靶基因组区域基于在多个癌性样本中的差异甲基化的相似性被排名或分类,并且在解释程序中使用多个等级或多个分类。所述多个等级和所述多个分类可以包含(1)临床疗效的类型,(2)疗效证据的强度,以及(3)疗效的大小。临床分析和基因组数据解释的各种方法可用于序列读数的分析。在一些其它实施例中,这种多个差异甲基化区域的多个甲基化状态的临床解释可以基于机器学习方法,所述机器学习方法基于一分类或回归方法来解释一当前样本,所述机器学习方法使用来自癌症和非癌性样本的患者具有已知癌症状态、癌症类型、癌症阶段、起源组织等的多个差异甲基化区域的多个甲基化状态进行训练。 具有临床意义的信息通常可以包含癌症存在或不存在,某些类型的癌症存在或不存在,癌症的阶段,或者其他类型的疾病存在或不存在。在一些实施例中,所述信息涉及一种或多种癌症类型的存在或不存在,所述癌症类型选自由以下组成的群组:肛门直肠癌、膀胱癌和尿路上皮癌、血癌、乳腺癌(激素受体阳性和激素受体阴性)、胆道癌、子宫颈癌、结直肠癌、子宫内膜癌、食管癌、头颈癌、胃癌、肝胆癌、肝癌、肺癌、淋巴肿瘤,胆囊癌、黑色素瘤、多发性骨髓瘤、卵巢癌、胰腺癌、上消化道癌、前列腺癌、肾癌、肉瘤、甲状腺癌、胆道癌、尿路上皮癌和子宫癌。 癌症分类器 为了训练一癌症类型分类器,所述分析系统获得多个训练样本,每个训练样本具有表示癌症的低甲基化和高甲基化的多个片段的一集合,例如,通过程序400中的步骤450识别,以及训练样本的癌症类型的一标签。所述分析系统基于表示癌症的低甲基化和高甲基化的多个片段的所述集合确定(对于每个训练样本的)一特征向量。所述分析系统计算多个标靶基因组区域中每个CpG位点的一异常得分。在一个实施例中,所述分析系统基于来自包含所述CpG位点的所述集合中是否存在一低甲基化或高甲基化片段,将所述特征向量的所述异常得分定义为二进制得分。一旦确定了一训练样本的所有异常得分,所述分析系统将所述特征向量确定为多个元素的一向量,包含对于每个元素,所述异常得分中的一个与其中所述多个CpG位点中的一个相关联。所述分析系统可以基于样本覆盖率(即所有CpG位点的中位数或平均排序深度)标准化所述特征向量的所述多个异常得分。 使用多个训练样本的多个特征向量,所述分析系统可以对所述癌症分类器进行训练。在一个实施例中,所述分析系统基于所述多个训练样本的所述多个特征向量,训练一二进制癌症分类器,以区分多个标签、癌症和非癌症。在本实施例中,所述分类器输出表示癌症存在或不存在的似然性的一预测得分。在另一实施例中,所述分析系统训练一多类癌症分类器(multiclass cancer classifier),以在多种癌症类型之间进行区分(例如,在头颈癌、肝癌/胆道癌、上消化道癌、胰腺癌/胆囊癌、结直肠癌、卵巢癌、肺癌、多发性骨髓瘤、淋巴肿瘤、黑色素瘤、肉瘤、乳腺癌和子宫癌)。在此多类癌症分类器实施例中,对所述癌症分类器进行训练,以确定一癌症预测,所述癌症预测包含对被分类的多个癌症类型的每一种的一预测值。多个所述预测值可能对应于一给定样本具有所述多种癌症类型中的每一种的一似然性。例如,所述癌症分类器返回一癌症预测,包含对于乳腺癌、肺癌和非癌症的一预测值。例如,所述癌症分类器可以返回对于一测试样本的癌症预测,包含对于乳腺癌、肺癌和/或无癌症的一预测得分。在任一实施例中,所述分析系统通过将多个训练样本的多个集合及其多个特征向量输入到所述癌症分类器中并调整多个分类参数来训练所述癌症分类器,从而所述分类器的一功能能够将所述多个训练特征向量准确地关联到他们相应的标签。所述分析系统可以将所述多个训练样本分组为一个或多个训练样本的多个集合,以用于所述癌症分类器的迭代批次训练。在输入包含其多个训练特征向量的多个训练样本的所有集合并调整多个分类参数后,对所述癌症分类器进行充分训练,以在一定误差范围内根据其特征向量标记多个测试样本。所述分析系统可以根据多种方法中的任何一种来训练所述癌症分类器。例如,二进制癌症分类器可以是使用一对数损失函数(log-loss function)训练的L2正则化逻辑回归分类器(L2-regularized logistic regression classifier)。作为另一个示例,多癌症分类器可以是一多类逻辑回归(multinomial logisticregression)。在实践中,可以使用其他技术训练任何类型的癌症分类器。这些技术有很多,包含潜在使用的多种内核方法(kernel method)、多种机器学习算法(如多层神经网络等),特别是PCT专利申请案第PCT/US2019/022122号和美国专利申请案第16/352602号中所述的方法,其通过引用将其整体并入本文可以用于各种实施例。 在特定实施例中,一癌症分类器通过一程序来训练,所述程序包含以下步骤:a.从多个训练对象中获取多个训练片段的序列信息;b.对于每一个训练片段,确定所述训练片段是低甲基化或高甲基化,其中所述多个低甲基化和高甲基化的训练片段中的每一个包含至少一个阈值数的多个CpG位点,所述多个CpG位点分别具有至少一个阈值百分比为未甲基化或甲基化;c.对于每一个训练对象,基于所述多个低甲基化的训练片段及所述多个高甲基化的训练片段产生一训练特征向量;及d.利用来自不具有癌症的一个或多个训练对象的所述多个训练特征向量和来自具有癌症的一个或多个训练对象的所述多个训练特征向量训练所述模型。所述训练方法还可以包含以下步骤:a.从多个训练对象中获取多个训练片段的序列信息;b.对于每一个训练片段,确定所述训练片段是低甲基化或高甲基化,其中所述多个低甲基化和高甲基化的训练片段中的每一个包含至少一个阈值数的多个CpG位点,所述多个CpG位点分别具有至少一个阈值百分比为未甲基化或甲基化;c.对于在一参考基因组中的多个CpG位点中的每一个:量化与所述CpG位点重叠的多个低甲基化的训练片段的一数量和与所述CpG位点重叠的多个高甲基化的训练片段的一数量;及基于多个低甲基化的训练片段和多个高甲基化的训练片段的所述数量,生成一低甲基化得分和一高甲基化得分;d.对于每一个训练片段,基于所述训练片段中所述多个CpG位点的所述低甲基化得分生成一总合的低甲基化得分,以及基于所述训练片段中所述多个CpG位点的所述高甲基化得分生成一总合的高甲基化得分;e.对于每一个训练对象:基于总合的低甲基化得分对所述多个训练片段进行排名,并基于总合的高甲基化得分对所述多个训练片段进行排名;及基于所述多个训练片段的所述排名生成一特征向量;f.获取不具有癌症的一个或多个训练对象的多个训练特征向量,以及具有癌症的一个或多个训练对象的多个训练特征向量;以及g.利用不具有癌症的所述一个或多个训练对象的所述多个特征向量及具有癌症的所述一个或多个训练对象的所述多个特征向量训练所述模型。在一些实施例中,所述模型包含一内核逻辑回归分类器、一随机森林分类器、一混合模型、一卷积神经网络和一自动编码器模型中的一种。 在一些实施例中,量化与所述CpG位点重叠的多个低甲基化的训练片段的一数量以及与所述CpG位点重叠的多个高甲基化的训练片段的一数量进一步包含步骤:a.量化来自与所述CpG位点重叠的具有癌症的一个或多个训练对象的多个低甲基化的训练片段的一癌症数量,以及量化来自与所述CpG位点重叠的不具有癌症的一个或多个训练对象的多个低甲基化的训练片段的一非癌症数量;以及b.量化来自与所述CpG位点重叠的具有癌症的一个或多个训练对象的多个高甲基化的训练片段的一癌症数量,以及量化来自与所述CpG位点重叠的不具有癌症的一个或多个训练对象的多个高甲基化的训练片段的一非癌症数量。在一些实施例中,基于多个低甲基化的训练片段和多个高甲基化的训练片段的所述数量来生成一低甲基化得分和一高甲基化得分还包含步骤:a.对于生成所述低甲基化得分,计算多个低甲基化的训练片段的所述癌症数量与多个低甲基化的训练片段的所述癌症数量及多个低甲基化的训练片段的所述非癌症数量的一低甲基化总合的一低甲基化比率;以及b.对于生成所述高甲基化得分,计算多个高甲基化的训练片段的所述癌症数量与多个高甲基化的训练片段的所述癌症数量及多个高甲基化的训练片段的所述非癌症数量的一高甲基化总合的一高甲基化比率。 在部署期间,所述分析系统从一对象收集的一测试样本中获取多个序列读数。本领域可用的各种测序方法可用于获得多个序列读数。在一些实施例中,所述多个序列读数从全基因组测序或标靶测序获得。在一些实施例中,所述多个序列读数包含修饰的多个测试片段的多个序列读数的一集合,其中所述修饰的多个测试片段是通过多个核酸片段的一集合的处理获得,其中所述多个核酸片段中的每一个对应于或衍生自从列表1至8中的任何一个中选择的多个基因组区域。在一些实施例中,所述多个序列读数来自使用如本文描述的化验板扩增的多个DNA样本。 所述分析系统处理所述多个序列读数,以获得与多个训练样本描述的类似程序中一测试特征向量。在一些实施例中,所述测试特征向量通过一程序获得,所述程序包含步骤:a.对于所述多个核酸片段中的每一个,确定所述核酸片段是低甲基化或高甲基化,其中低甲基化和高甲基化的所述多个核酸片段中的每一个包含至少一个阈值数的多个CpG位点,所述多个CpG位点分别具有至少一个阈值百分比为未甲基化或甲基化;b.对于一参考基因组中的多个CpG位点中的每一个:量化与所述CpG位点重叠的低甲基化的多个核酸片段的一数量和与所述CpG位点重叠的高甲基化的多个核酸片段的一数量;及基于低甲基化的多个核酸片段和高甲基化的多个核酸片段的所述数量,生成一低甲基化得分和一高甲基化得分;c.对于每一个核酸片段,基于所述核酸片段中所述多个CpG位点的所述低甲基化得分生成一总合的低甲基化得分和基于所述核酸片段中所述多个CpG位点的所述高甲基化得分生成一总合的高甲基化得分;d.基于总合的低甲基化得分对所述多个核酸片段进行排名,及基于总合的高甲基化得分对所述多个核酸片段进行排名;以及e.基于所述多个核酸片段的所述排名生成所述测试特征向量。 然后,所述分析系统将所述测试特征向量输入到经过训练的癌症分类器中,以产生一癌症预测,例如,二元预测(癌症或非癌症)或多类癌症预测(多种癌症类型中的每一种的预测得分)。在一些实施例中,所述分析系统输出测试样本的一癌症概率。所述癌症概率可以与一阈值概率进行比较,以确定来自一对象的所述测试样本具有癌症或不具有癌症。 示例性测序器和分析系统 图8A是根据一个实施例用于测序多个核酸样本的多种系统和装置的流程图。此说明性的流程图包含多个装置,例如一测序器820和一分析系统800。所述测序器820和所述分析系统800可串联工作以执行本文所述程序中的一个或多个步骤。 在各种实施例中,所述测序器820接收一扩增的核酸样本810。如图8A所示,所述测序器820可以包含一图形用户界面825,所述图形用户界面825允许用户与多个特定任务互动(例如,启动测序或终止测序)以及一个或多个加载站830,用于加载一测序药盒(包含多个扩增片段样本)和/或用于加载执行测序化验所需的多种缓冲液。因此,一旦所述测序器820的一用户已向所述测序器820的所述加载站830提供了所需的试剂和测序药盒,所述用户就可以通过与所述测序器820互动的所述图形用户界面825来启动测序。一旦启动,所述测序器820执行测序并输出来自所述核酸样本810的多个扩增片段的多个序列读数。 在一些实施例中,所述测序器820与所述分析系统800通信地耦合。所述分析系统800包含一些用于处理各种应用的序列读数的计算装置,例如评估一个或多个CpG位点的甲基化状态、识别变体(variant calling)或质量控制。测序器820可以将BAM文件格式的多个序列读数提供给所述分析系统800。所述分析系统800可以通过无线、有线或无线和有线通信技术的组合以通信方式耦合到所述测序器820。通常,所述分析系统800配置有 一处理器和存储多个计算机指令的非临时计算机可读存储介质,当由所述处理器执行时,所述多个计算机指令导致处理器处理所述多个序列读数或执行本文公开的任何方法或程序的一个或多个步骤。 在一些实施例中,所述多个序列读数可以使用本领域已知的方法与一参考基因组对齐以确定对齐位置信息,例如,图3A中程序100的步骤140的一部分。对齐位置通常可以描述所述参考基因组中一区域的一起始位置和一结束位置,其对应于一给定序列读数的一起始碱基和一结束碱基。与甲基化测序相对应,所述对齐位置信息可概括为表示根据与所述参考基因组的对齐的所述序列读数中包含的一第一CpG位点和一最后CpG位点。所述对齐位置信息可以进一步表示在一给定的序列读数中多个甲基化状态和所有CpG位点的位置。所述参考基因组中的一区域可与一基因或一基因的一片段相关联;因此,所述分析系统800可使用一个或多个基因与所述序列读数对齐来标记一序列读数。在一个实施例中,从所述起始位置和所述结束位置来确定片段长度(或大小)。 在各种实施例中,例如当使用一成对的结束测序程序时,一序列读数由表示为R_1和R_2的一读数对组成。例如,一第一读数R_1可从一双链DNA(dsDNA)分子的一第一端测序,而一第二读数R_2可从所述双链DNA(dsDNA)的一第二端测序。因此,所述第一读数R_1和所述第二读数R_2的核苷酸碱基对可与所述参考基因组的多个核苷酸碱基一致地(例如,在相反方向上)对齐。衍生自读数对R_1和R_2的对齐位置信息可以包含所述参考基因组中的一起始位置,所述起始位置对应于所述第一读数(例如,R_1)的终点及所述参考基因组中的一结束位置,所述结束位置对应于一第二读数(例如,R_2)的终点。在一个实施例中,读数对R 现在参考图8B,图8B是根据一个实施例的用于处理多个DNA样本的一分析系统800的方框图。所述分析系统实现了一个或多个用于分析多个DNA样本的计算装置。所述分析系统800包含一序列处理器840、序列数据库845、模型数据库855、多个模型850、参数数据库865和评分引擎860。在一些实施例中,所述分析系统800执行在图3A的程序100、图3B的程序340、图4的程序400、图5的程序500、图6A的程序600或图6B的程序680和本文描述的其他程序中的一个或多个步骤。 所述序列处理器840从一样本的多个片段生成多个甲基化状态向量。在一片段上的每个CpG位点处,所述序列处理器840通过图3A的程序100为每个片段生成一甲基化状态向量,其指定在参考基因组中所述片段的位置、所述片段中的多个CpG位点的一数量以及所述片段中每个CpG位点的甲基化状态,是甲基化、未甲基化还是不确定。所述序列处理器840可将多个片段的多个甲基化状态向量存储在所述序列数据库845中。所述序列数据库845中的数据可以被组织以使得来自一样本的多个甲基化状态向量彼此相关联。 此外,多个不同的模型850可以存储在所述模型数据库855中,或者检索以用于多个测试样本。在一个示例中,一模型是一经过训练的癌症分类器,用于使用来自多个异常片段的一特征向量来确定测一试样本的一癌症预测。所述癌症分类器的训练和使用在本文其他地方讨论。所述分析系统800可以训练一个或多个模型850并将各种被训练参数存储在所述参数数据库865中。所述分析系统800将所述多个模型850与多个函数一起存储在所述模型数据库855中。 在推断期间,所述评分引擎860使用一个或多个850模型返回多个输出。所述评分引擎860访问所述模型数据库855中的所述多个模型850以及所述参数数据库865中的多个被训练参数。根据每个模型,所述评分引擎接收所述模型的一适当输入,并基于接收到的输入、所述多个参数以及与输入和输出相关的每个模型的一函数来计算一输出。在一些使用的情况中,所述评分引擎860进一步计算与所述模型计算的多个输出的一置信度相关的多个度量。在其他使用的情况中,所述评分引擎860用于所述模型中计算其他中间值。 应用 在一些实施例中,本发明的方法、分析系统和/或分类器可用于检测癌症的存在(或不存在)、监测癌症的进展或复发、监测治疗反应或有效性、确定最小残留疾病灶(MRD)的存在或监测最小残留疾病灶或其任何组合。在一些实施例中,所述分析系统和/或分类器可用于识别癌症的组织或来源。例如,所述系统和/或分类器可用于将癌症识别为以下癌症类型中的任何一种:头颈癌、肝癌/胆道癌、上消化道癌、胰腺癌/胆囊癌、结直肠癌、卵巢癌、肺癌、多发性骨髓瘤、淋巴肿瘤、黑色素瘤、肉瘤、乳腺癌和子宫癌。例如,如本文所述,一分类器可用于生成一样本特征向量是来自一癌症患者的一似然或概率得分(例如,从0到100)。在一些实施例中,将所述概率得分与一阈值概率进行比较以确定一对象是否患有癌症。在其它实施例中,可在不同时间点(例如,治疗前或治疗后)评估所述似然性或概率得分以监视疾病进展或监视治疗有效性(例如,治疗疗效)。在其他实施例中,所述似然性或概率得分可用于作出或影响一临床决策(例如,癌症诊断、治疗选择、治疗效果评估等)。例如,在一个实施例中,如果所述似然性或概率得分超过一阈值,则医生可以开出适当的治疗方案。 癌症的早期检测 在一些实施例中,本发明的方法和/或分类器用于检测怀疑患有癌症的一对象中癌症存在或不存在。例如,一分类器(如本文所述)可用于确定一样本特征向量是来自具有癌症的一对象的一似然性或概率得分。 在一个实施例中,大于或等于60的概率得分可表示所述对象患有癌症。在其他实施例中,概率得分大于或等于65、大于或等于70、大于或等于75、大于或等于80、大于或等于85、大于或等于90、或大于或等于95,表示所述对象患有癌症。在其它实施例中,一概率得分可表示疾病的严重性。例如,与低于80分(例如,70分)相比,概率得分为80分可能表示癌症的更严重程度或晚期。类似地,随着时间的推移(例如,在一第二、更晚的时间点)概率得分的增加可以表示疾病的进展,或者随着时间的推移(例如,在一第二、更晚的时间点)概率得分的减少可以表示治疗成功。 在另一个实施例中,可以通过取一癌性概率与一非癌性概率(即1减去癌性概率)的比值的对数来计算一测试对象的一癌症对数概率比,如本文所述。根据本实施例,大于1的癌症对数概率比可表示所述对象患有癌症。在其他实施例中,癌症对数概率比大于1.2、大于1.3、大于1.4、大于1.5、大于1.7、大于2、大于2.5、大于3、大于3.5或大于4,表示对象患有癌症。在其它实施例中,一癌症对数概率比可表示疾病的严重程度。例如,与得分低于2(例如,得分为1)相比,一癌症对数概率比大于2可能表示癌症的更严重程度或晚期。同样,随着时间的推移(例如,在第二、更晚的时间点)癌症对数概率比的增加可以表示疾病的进展,或者随着时间的推移(例如,在第二、更晚的时间点)癌症对数概率比的减少可以表明治疗成功。 根据本发明的各个方面,本发明的方法和系统可以被训练来检测或分类多个癌症适应症。例如,本发明的方法、系统和分类器可用于检测一种或多种、两种或多种、三种或多种、五种或多种不同类型癌症的存在。 在一些实施例中,所述癌症是以下的一种或多种:头颈癌、肝癌/胆道癌、上消化道癌、胰腺癌/胆囊癌、结直肠癌、卵巢癌、肺癌、多发性骨髓瘤、淋巴肿瘤、黑色素瘤、肉瘤、乳腺癌和子宫癌。在一些实施例中,所述癌症是肛门直肠癌、膀胱癌或尿路上皮癌或子宫颈癌中的一种或多种。 癌症与治疗监测 在一些实施例中,可在不同的时间点(例如,或在治疗之前或之后)评估似然性或概率得分,以监测疾病进展或监测治疗效果(例如,治疗疗效)。例如,本发明提供的方法涉及在一第一时间点从一癌症患者获得一第一样本(例如,一第一血浆cfDNA样本)、从中确定一第一似然性或概率得分(如本文所述)、在第二时间点从所述癌症患者获得一第二测试样本(例如,一第二血浆cfDNA样本),并从中确定一第二似然性或概率得分(如本文所述)。 在某些实施例中,所述第一时间点在一癌症治疗之前(例如,在切除手术或治疗干预之前),并且所述第二时间点在一癌症治疗之后(例如,在切除手术或治疗干预之后),以及用于监测治疗的有效性的方法。例如,如果所述第二似然性或概率得分比所述第一似然性或概率得分降低,则认为治疗成功。然而,如果所述第二似然性或概率得分比所述第一似然性或概率得分增加,则认为治疗不成功。在其它实施例中,所述第一和所述第二时间点都在一癌症治疗之前(例如,在切除手术或治疗干预之前)。在其他实施例中,所述第一和所述第二时间点都是在一癌症治疗之后(例如,在切除手术或治疗干预之前),以及用于监视治疗的有效性或治疗的有效性的损失的方法。在其它实施例中,多个cfDNA样本可在第一和第二时间点从一癌症患者获得并分析。例如,用于监测癌症进展,确定癌症是否处于缓解期(例如,治疗后),监测或检测残留疾病或疾病复发,或监测治疗(treatment)(例如,治疗(therapeutic))的疗效。 本领域技术人员将容易理解,可以在任何期望的时间点从一癌症患者获得测试样本,并根据本发明的方法进行分析以监测患者的癌症状态。在一些实施例中,所述第一时间点和第二时间点被范围从大约15分钟到大约30年的时间量分开,例如大约30分钟,例如大约1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、21、22、23或大约24小时,例如大约1、2、3、4、5、10、15、20、25或大约30天,或例如约1、2、3、3、4、5、6、7、8、9、10、11、或12个月,或例如约1、1.5、2、2.5、3、3.5、4、4.5、5、5、5.5、6.5、6、6.5、7、7.5、8、8.5、9、9.5、10、10.5、11、11.5、12、12.5、13、13.5、14、14.5、15、15.5、16、16.5、17、17.5、18、18.5、19、19.5、20、20.5、21、21.5、22、22.5、23、23.5、24、24.5、25、25.5、26、26.5、27、27.5、28、28.5、29,29.5年或约30年。在其它实施例中,可至少每3个月一次、至少每6个月一次、至少一年一次、至少每2年一次、至少每3年一次、至少每4年一次或至少每5年一次从所述患者处获取多个测试样本。 治疗 在另一实施例中,从本文描述的任何方法获得的信息(例如,似然性或概率得分)可用于作出或影响一临床决策(例如,癌症诊断、治疗选择、治疗效果评估等)。例如,在一个实施例中,如果似然性或概率得分超过一阈值,医生可以开出适当的治疗方案(例如,切除手术、放射治疗、化疗和/或免疫疗法)。在一些实施例中,例如一似然性或概率得分之类的信息可以作为一输出数值提供给医生或对象。 一分类器(如本文所述)可用于确定来自具有癌症的一对象的一样本特征向量的似然性或概率得分。在一个实施例中,当所述似然性或概率超过一阈值时,处方一适当的治疗(例如,切除手术或治疗)。例如,在一个实施例中,如果所述似然性或概率得分大于或等于60,则处方一个或多个适当的治疗。在另一实施例中,如果所述似然性或概率得分大于或等于65、大于或等于70、大于或等于75、大于或等于80、大于或等于85、大于或等于90、或大于或等于95,则处方一个或多个适当的治疗。在其它实施例中,一癌症对数概率比可表示癌症治疗的有效性。例如,随着时间的推移(例如,在治疗后的一第二时间)癌症对数概率比的增加可以表示治疗无效。同样,随着时间的推移(例如,在治疗后的一第二时间)癌症对数概率比的降低可以表示治疗成功。在另一实施例中,如果癌症对数概率比大于1、大于1.5、大于2、大于2.5、大于3、大于3.5或大于4,则处方一种或多种适当的治疗。 在一些实施例中,所述治疗是选自由以下组成的群组的一种或多种的癌症治疗剂:一化学治疗剂、一靶向的癌症治疗剂、一分化治疗剂、一激素治疗剂和一免疫治疗剂。例如,所述治疗可以是选自由以下组成的群组的一种或多种的化学治疗剂:烷基化剂、抗代谢物、蒽环类、抗肿瘤抗生素、细胞骨架破坏剂(紫杉醇)、拓扑异构酶抑制剂、有丝分裂抑制剂、皮质类固醇、激酶抑制剂、核苷酸类似物和铂基药物及其任何组合。在一些实施方案中,所述治疗是选自由以下组成的群组的一种或多种的靶向癌症治疗剂:信号转导抑制剂(例如酪氨酸激酶和生长因子受体抑制剂)、组蛋白脱乙酰基酶(HDAC)抑制剂、视黄酸受体激动剂、蛋白质组抑制剂、血管生成抑制剂和单克隆抗体结合物。在一些实施例中,所述治疗是一种或多种的分化治疗剂包含维甲酸类,例如维甲酸、阿利维甲酸和贝沙罗汀。在一些实施例中,所述治疗是选自由以下组成的群组的一种或多种的激素治疗剂:抗雌激素、芳香化酶抑制剂、孕激素、雌激素、抗雄激素和GnRH激动剂或类似物。在一个实施例中,所述治疗是选自由以下组成的群组的一种或多种的免疫治疗剂:单克隆抗体疗法,如利妥昔单抗(RITUXAN)和阿仑单抗(CAMPATH),非特异性免疫疗法和佐剂,如卡介苗、白细胞介素2(IL-2)和干扰素α,免疫调节药物,例如沙利度胺和来那度胺(REVLIMID)。熟练的医生或肿瘤学家有能力基于诸如肿瘤的类型、癌症阶段、先前接受过癌症治疗或治疗剂的暴露以及癌症的其他特征的特征来选择合适的癌症治疗剂。 6.6.示例 为了向本领域技术人员提供关于如何制作和使用本说明书的完整公开和描述,提出以下示例,并且不打算限制发明人所认为的描述的范围,也不打算表示下面的实验是全部或唯一进行的实验。已经努力确保所用数字(如数量、温度等)的准确性,但应考虑一些实验误差和偏差。 6.6.1.示例1:癌症和非癌症对照的个体的cfDNA测序 循环无细胞基因组图谱研究(“CCGA”;Clinical Trial.gov识别符:NCT02889978)是一项前瞻性、多中心、病例对照、纵向随访的观察性研究。从142个地点约15000名参与者中收集未鉴定的生物样本。样本被选择为了确保每个群体中多个癌症类型和非癌症的预先指定分布,并且癌症和非癌症样本的频率与性别年龄匹配。STRIVE研究是一项前瞻性、多中心、观察性群体研究,旨在验证乳腺癌和其他浸润性癌症的早期检测的化验(参见ClinicalTrial.gov识别符:NCT03085888(clinicaltrials.gov/ct2/show/NCT03085888))。如下所述,从STRIVE研究中选择额外的非癌症样本并用于分类器训练。 在一些实施例中,从CCGA多个研究对象血浆中分离的cfDNA的全基因组亚硫酸氢盐测序(WGBS;30倍(30×)深度)被用于cfDNA的分析。在其它实施例中,如下文所示,一标靶的亚硫酸氢盐测序程序用于样本的分析。在标靶亚硫酸氢盐测序程序下,使用一探针集合扩增衍生自多个标靶基因组区域的cfDNA分子。对于这两种方法,每位患者从两支血浆管中提取cfDNA(总体积不超过10毫升)使用改良的QIAamp循环核酸试剂盒(modified QIAampCirculating Nucleic Acid kit)(Qiagen公司;杰曼敦,马里兰州)。使用EZ-96DNA甲基化试剂盒(Accel-NGS Methyl-Seq DNA library preparation kits)(Zymo Research公司,D5003)对高达75纳克(ng)的血浆cfDNA进行亚硫酸氢盐转化。转化后的cfDNA用Accel-NGS-Methyl-Seq-DNA文库制备试剂盒(Swift-BioSciences公司;密歇根州安娜堡)。使用Illumina平台(IlluminaPlatforms)的KAPA文库定量试剂盒(KAPALibraryQuantification Kit)(Kapa Biosystems公司;马萨诸塞州威尔明顿)对构建的多个文库进行量化。将4个文库以及10%PhiX v3文库(Illumina公司,FC-110-3001)组合并聚集在Illumina NovaSeq 6000 S2流动细胞上,然后进行150bp配对末端测序(30倍(30×)). 6.6.2.示例2:健康个体甲基化状态的建模 多个典型cfDNA片段的一统计模型和一数据结构是使用来自CCGA研究的108名非吸烟无癌症的参与者(年龄:58岁±14年,79名(73%)女性)(即参考基因组)的一独立参考集合产生。这些样本用于训练一马尔可夫链模型(3阶),以估计一片段内一给定序列的多个CpG甲基化状态的可能性,如上文第6.4.2.1节P值得分计算所述。此模型被证明在正常片段范围内(p值>0.001)被校准,并被用于从所述马尔可夫模型中丢弃p值≥0.001的多个片段,这是非常不常见的。 6.6.3.示例3:标靶基因组区域的选择 通过对亚硫酸氢盐转化的多个cfDNA片段进行测序产生的数据库选择多个标靶基因组区域,所述亚硫酸氢盐转化的多个cfDNA片段来自CCGA研究中1548名已被诊断为具有癌症的个体和1163名未被诊断为具有癌症的个体。 选择多个标靶基因组区域的程序开始于从每个个体的亚硫酸氢盐转化cfDNA片段的多个序列中识别多个异常cfDNA片段。所述多个片段与hg19参考基因组对齐,每个片段中的每个CpG位点被评分为甲基化或未甲基化、或不确定。多个异常cfDNA片段有三个特征:(1)根据马尔可夫链模型,一亚硫酸氢盐转化的甲基化模式具有p值<0.001,表明所述甲基化模式不会发生在无癌症的个体中;(2)五个或五个以上的CpG位点;以及(3)一高甲基化或低甲基化状态,其中至少80%、90%或100%的CpG位点分别为甲基化或未甲基化。当计算甲基化或未甲基化的多个CpG位点的百分比时,不确定位点包含在CpG位点的总数中。 标靶选择的第二步骤是确定GRCh37/hg19参考基因组中约3000万个CpG位点的一排名得分。对于每个CpG位点,一计数由具有与所述CpG位点重叠的至少一个异常cfDNA片段的癌症和非癌症样本组成。高甲基化的多个异常片段和高甲基化的多个异常片段被分开地计数。这些计数用于根据以下公式分别计算高甲基化和低甲基化片段的得分:(n 标靶选择的第三步骤是定义基因组内的多个标靶区域。添加到标靶列表中的第一区域是一30个碱基的区域,在排名最高的CpG位点的中心任一侧延伸15个碱基。然后在一迭代程序中扩展多个标靶区域的列表。一30个碱基的区域在排名次高的CpG位点的中心任一侧延伸15个碱基被识别。如果新区域在现有标靶位点的200个碱基内,则将所述新区域与现有区域合并,以形成一个更大的区域,包含新的30个碱基与旧的30个碱基之间的所有碱基。如果新区域不在现有标靶位点的200个碱基内,则只需将其添加到标靶区域的列表中。标靶区域的列表迭代地扩展,直到达到所需的化验板尺寸:化验板1为0.59MB(百万碱基)、化验板2为1.19MB、化验板3为2.38MB、化验板4为4.96MB、及化验板5为8.53MB。 6.6.4.示例4:探针设计 生物素化探针设计用于收集衍生自上述是别的基因组区域的多个亚硫酸氢盐转化的异常cfDNA片段。每个探针有120个碱基长,5’端有一生物素部分。一般而言,多个探针集合被设计成包含多个探针,所述多个探针标靶来自包含在列表1至列表8中任何一个中的多个标靶区域的任何的多个起始/终止范围内的多个CpG位点中的每一个的多个片段。 为了增加捕获衍生自一标靶区域(或一标靶区域的一部分)的多个异常cfDNA片段的概率,包含在其5’或3’端具有部分标靶序列的多个异常cfDNA片段,以平铺的方式排列多个探针,使得两个探针对齐一标靶区域内的每个碱基。相邻的120个碱基的探针设计为通过60个碱基重叠。60个或更少个碱基的多个标靶区域最初由三个探针标靶,如图1A所示。额外的多个探针用于较大尺寸的多个标靶区域(如参见图1B)。对于两种尺寸等级的标靶区域,用于询问每个区域的探针共同延伸到标靶区域之外(例如,由于平铺的排列,在标靶区域的任一侧包含至少60个碱基的非标靶序列;参见图1A)。此外,多个探针被设计用于扩增衍生自每个标靶区域的两条DNA链的多个DNA分子,使得单个标靶基因组区域的单个碱基对可以通过四个探针标靶:每条链两个。 多个探针序列被设计为与来自多个基因组区域中的每一个的亚硫酸氢盐转化的多个异常cfDNA片段互补,其中一cfDNA片段中的每个CpG位点是甲基化或未甲基化。因此,如果与一标靶序列对齐的多个异常cfDNA片段是低甲基化的,则所述片段内的所有或大部分的CpG位点将预期转化为UpG序列,并且相应的多个探针将具有标靶中的所有或大部分的CpG的CpA序列。相反地,如果与一标靶序列对齐的多个异常cfDNA序列是高甲基化的,则所述片段内的所有或大部分的CpG位点预计将被保护以防止转换为UpG序列,并且相应的多个探针将具有标靶中的所有或大部分CpG的CpG序列。不位于一CpG位点的胞嘧啶通常未甲基化。因此,无论相邻的CpG位点的甲基化状态如何,这些旁观者胞嘧啶都会转化为尿嘧啶。与一标靶位点匹配的多个探针被设计用来下拉多个低甲基化cfDNA片段或多个高甲基化cfDNA片段,但通常不能两者兼之。这种“半二元(semibinary)”设计与“二元(binary)”设计形成了对比,在这种设计中,来自相同标靶区域的多个cfDNA片段被两组探针标靶,一组探针被设计用来下拉低甲基化cfDNA,另一组探针被设计用来下拉相同区域的高甲基化cfDNA。 6.6.5.示例5:探针质量控制 从所述多个癌症化验板中排除了可能下拉大量的标靶cfDNA片段的劣质探针。 为了确定劣质探针的特性,进行了一项实验,以测试cfDNA片段和探针之间需要多少重叠才能达到不可忽略的下拉量。 纯化的亚硫酸氢盐转化的cfDNA与生物素化探针在杂交捕获缓冲液(Hybridization Capture Buffer)(Argonaut Technologies公司,加利福尼亚州红木市,Cat#310450)中混合,在96孔板中补充1mM的三羟甲基氨基甲烷(Tris),pH值8.0,并在汉密尔顿星液体处理器(Hamilton Star Liquid Handler)(Hamilton公司,内华达州里诺市,Cat#STARAL 8/96 iSWAP)和96孔BioRad C1000接触式热循环器(BioRad C1000 TouchThermocycler)(BiRad公司,加利福尼亚州赫拉克勒斯市,Cat#1851196)。杂交混合物的温度提高到95℃持续10分钟,然后逐渐下降2℃/分钟至62℃、在93℃和91℃时暂停1分钟。在62℃杂交持续15.5-17.5小时。加入第二份的探针,重复热循环程序。捕获的cfDNA被捕获在链霉亲和素磁珠(Streptavidin Magnetic Beads)(Illumina公司,加利福尼亚州圣地亚哥市,Cat#20014367)上,通过PCR放大,然后测序。 使用多个初步化验板(V1D3、V1D4、V1E2)测试了不同的重叠长度,所述多个化验板设计为包含具有范围从0到120个碱基的不同重叠的多个探针。将包含175bp标靶多个DNA片段的样本施加到所述化验板上并洗涤,然后收集结合到所述多个探针的多个DNA片段。收集的多个DNA片段的数量被测量,然后绘制为重叠的多个尺寸与密度,如图10所示。 当重叠小于45bp时,多个标靶DNA片段没有显着的结合和下拉。这些结果表明,在特定的多个杂交条件下,通常需要至少45bp的片段-探针重叠,以最大限度地实现探针组与衍生自多个标靶基因组区域的多个核酸之间的杂交,从而提高下拉效率。 此外,当重叠区域中所述多个探针和多个片段序列之间的不匹配百分比大于10%时,结合以及由此产生的下拉效率会受到极大破坏。因此得出结论,对于序列与至少45bp的一脱靶基因组区域的匹配率至少为90%的多个探针,可能发生脱靶下拉。 对于每个探针,本申请的发明人对整个参考基因组进行了详尽、亚硫酸氢盐明白的搜索,以寻找具有45bp具有一匹配率为90%以上的多个脱靶区域。具体来说,本申请的发明人将k-mer接种策略(允许一个或多个不匹配)结合在多个接种位置处的局部对位。这保证不会丢失任何基于k-mer长度、允许的不匹配数量和特定位置的k-mer种子命中数的良好对齐。所述搜索涉及到在大量位置执行动态编程本地对齐。这涉及在大量位置执行动态编程局部对齐,因此对实现进行了优化以使用向量CPU指令(例如AVX2、AVX512)并在计算机内部的许多内核以及通过网络连接的许多计算机之间并行化。此实施可以进行详尽的搜索,这对于设计高性能化验板时非常有价值(即,对于给定数量的测序的低脱靶率和高标靶覆盖率)。 在彻底搜索之后,基于其脱靶区域的数量对每个探针进行评分。最好的探针(高)具有一得分为1,这意味着它们仅在一基因组区域-所述标靶匹配。得分在2-19之间的中间得分(中间)的探针被接受,但得分20或超过20(劣)的不佳得分的探针被丢弃。表1给出了化验板3至5中探针的总结,包含在消除具有潜在脱靶效应的低质量探针后保留的探针的数量和百分比。探针区域大于标靶区域,因为一些探针延伸超出标靶区域的末端。 表1、探针质量控制前后的探针数量和标靶区域尺寸
此外,在多个探针标靶的多个高甲基化基因组区域或多个低甲基化基因组区域中对高质量、中间质量和劣质量的探针的数量进行计数。如图11所示,标靶多个高甲基化基因组区域的多个探针显着地具有较少的脱靶匹配。 6.6.6.示例4:标靶基因组区域的特征 列表1至列表8(见下表2)所示的各种尺寸的多个化验板,代表多个标靶基因组区域的部分,其中多个探针被设计用于扩增衍生自这些标靶基因组区域的多个DNA分子。根据用于选择他们的标靶基因组区域的尺寸预算,从小到大排列化验板1至5。这些化验板被过滤,以排除某些CpG位点和基因组区域。化验板6定义了比化验板1至5中的任何一个更大的基因组区域,包含所有的CpG位点和基因组区域,其被滤除以产生化验板1至5中的每一个。化验板3A和4A分别显示在化验板3和4中多个标靶基因组区域的子集,排除在PCT专利申请案第PCT/US2019/025358号中也描述的多个基因组区域。 表2、化验板、列表和序列号(SEQ ID NO)之间的对应关系
序列表包含以下信息:(1)序列号(SEQ ID NO),(2)序列标识符,用于识别(a)CpG位点所在的染色体或重叠群(contig),以及(b)所述区域的起始和终止位置,(3)与(2)和(4)相对应的序列,基于其高甲基化或低甲基化得分是否包含所述区域。染色体的数目以及起始和终止位置是相对于已知的人类参考基因组GRCh37/hg19提供的。GRCh37/hg19的序列可从国家生物技术信息中心(National Center for Biotechnology Information,NCBI)、基因组参考联盟(Genome Reference Consortium)和圣克鲁斯基因组研究(Santa CruzGenomics Institute)所提供的基因组浏览器获得。 一般而言,一探针可设计为标靶包含在列表1至列表8中多个标靶区域中的任一个的开始/停止范围内的多个CpG位点中的任何一个。或者,在一些实施例中,多个探针可设计成与衍生自多个标靶区域中的任一个的多个片段中的任何CpG位点杂交,例如来自标靶区域的转化片段。 所述多个化验板被分析以了解其特征。在滤除多个脱靶探针之前,所述多个化验板的多个标靶基因组区域的尺寸如表10所示。对于各种化验板(在滤除多个脱靶探针之前)中的每个,每个标靶基因组区域的CpG位点的数量,被绘制在图12A中作为密度的函数。图12.B显示了各种化验板的每个探针的G/C含量。 表10。标靶基因组区域尺寸
接下来将多个化验板内的多个标靶基因组区域与一参考基因组对齐以评估它们的位置。多个标靶基因组区域被定位在内含子、外显子、基因间区域、5’UTRs、3’UTRs或对照区域,如启动子或增强子。对于化验板3至5,落在每个基因组注释内的多个标靶基因组区域的数量被计数并绘制在图13中提供的图示中。图13还比较了落入每个基因组注释内的所选的多个靶标基因组区域的数量(黑色条)和随机选择的多个基因组区域的数量(灰色条)。 分析表明,所选的多个标靶基因组区域在其基因组分布上并非随机的。与随机选择的区域相比,例如启动子、5’UTRs、外显子和内含子/外显子边界等功能元素在化验板中的代表度较高,而基因间区域的代表不足。 6.6.7.示例7:混合模型分类器的生成 为了最大限度地提高性能,本示例中描述的预测癌症的多个模型是使用从CCGA子研究(CCGA1和CCGA22)中的多个已知癌症类型和非癌症的多个样本中、从CCGA1获得的多个已知癌症的多个组织样本、以及来自STRIVE研究的多个非癌症样本(参见ClinicalTrail.gov识别符:NCT03085888(//clinicaltrials.gov/ct2/show/NCT03085888))获得的序列数据进行训练的。STRIVE研究是一项前瞻性、多中心、观察性群体的研究,以验证早期检测乳腺癌和其他侵袭性癌症的试验,从中获得额外的非癌症训练样本,以训练本文描述的分类器。已知的癌型包含:乳腺癌、肺癌、前列腺癌、结肠直肠癌、肾癌、子宫癌、胰腺癌、食管癌、淋巴瘤、头颈癌、卵巢癌、肝胆癌、黑色素瘤、子宫颈癌、多发性骨髓瘤、白血病、甲状腺癌、膀胱癌、胃癌和肛门直肠癌。因此,一模型可以是用于检测一个或多个、两个或更多、三个或更多、四个或更多、五个或更多、十个或更多、或20个或更多不同类型的癌症的多癌症模型(或多癌症分类器)。 以下所示的化验板1至6的分类器性能数据,报告了一个针对来自CCGA子研究(CCGA2)和STRIVE研究获得的癌症和非癌样本训练的锁定分类器。CCGA2子研究中的个体与CCGA1子研究中的个体不同,其cfDNA被用于选择多个标靶基因组。从CCGA2研究中,收集了诊断具有未经治疗的癌症(包含20种肿瘤类型和所有癌症阶段)的多个个体和没有癌症诊断的健康个体(对照组)的血液样本。对于STRIVE,在筛查乳房X光检查后28天内,从女性身上收集血液样本。从每个样本中提取无细胞DNA(cfDNA),并用亚硫酸氢盐处理,将未甲基化胞嘧啶转化为尿嘧啶。使用多个杂交探针扩增亚硫酸氢盐处理的cfDNA,以用于获取多个cfDNA分子的信息,所述多个杂交探针设计用于扩增衍生自一化验板中多个标靶基因组区域中的每一个的亚硫酸氢盐转化的多个核酸,所述化验板包含化验板1至6的所有基因组区域。在Illumina平台(Illumina platform)(加利福尼亚州圣地亚哥市)上使用配对末端测序(paired-end sequencing)对扩增的亚硫酸氢盐转化的多个核酸分子进行测序,以获得所述多个训练样本中的每一个的多个序列读数的一集合,并将所得的读数对与参考基因组对齐,组装成多个片段,并识别甲基化和未甲基化CpG位点。 对化验板1至6的分类器性能进行了评估,并在本示例和图14至27中报告了多个训练样本的一集合,其中包含3053个样本(1532个癌症样本和1521个非癌症样本)。用于评估分类器性能的所有样本类型如下表3所示。 表3、个体的癌症诊断的cfDNA用于训练分类器
基于混合模型的特征化 对于每种癌症类型(包含非癌症),概率混合模型被训练并利用于基于在一给定的样本类型中观察到片段的可能性有多大为来自每个癌症及非癌症样本的每个片段分配一概率。 简而言之,对于每个样本类型(癌症和非癌症样本),对于每个区域(其中每个区域长度为1kb,相邻区域之间具有50%重叠),一概率模型适合于衍生自每种类型或癌症和非癌症的训练样本的多个片段。针对每种样本类型训练的所述概率模型是混合模型,其中三种混合组分中的每一种是一独立位点模型(independent-sites model),其中假定每个CpG处的甲基化独立于其他CpG处的甲基化。如果以下情况将多个片段从模型中排除:它们具有一p值大于0.01,标记为重复片段,所述多个片段的一袋尺寸(bag size)大于1;他们没有覆盖至少一个CpG位点;或者如果所述片段的长度大于1000个碱基。 使用最大似然估计对每个概率模型进行拟合,以识别多个参数的一集合,其最大化衍生自每个样本类型的所有片段的对数似然性,受到一正则化惩罚。 特征化 针对每个区域中的每种癌症类型和非癌症样本,从每个训练样本中提取每个片段的多个特征。提取的多个特征是多个异常(outlier)片段(即多个异常甲基化片段)的多个计数(tallies),其被定义为在第一癌症模型下的对数似然超过第二癌症模型或非癌症模型下的对数似然至少一阈值等级数值(threshold tier value)的那些。对每个基因组区域、样本模型(即癌症类型)和等级(1、2、3、4、5、6、7、8和9级)分别计数多个异常片段,每个样本类型每个区域产生9个特征。 特征排名和分类器训练 对于成对特征的每个集合,使用交互信息基于它们区分第一癌症类型(定义特征来源的对数似然模型)与第二癌症类型或非癌症的能力对所述多个特征进行排名。每个成对比较中排名最高的256个特征被识别,并将其添加到每种癌症类型和非癌症的最终特征集合中。对每个样本(癌症类型和非癌症)的最终特征集合中的所述多个特征进行二值化(大于0的任何特征值被设置为1,使得所有特征都是0或1)。然后将多个训练样本分成不同的5倍交叉验证训练集合并用于训练一逻辑回归分类器(用于检测癌症的存在)和多类逻辑回归分类器(用于确定癌症起源组织)。在分类器训练程序中,分配给训练集的交叉验证得分被收集。 一旦二元癌症/非癌症分类器被训练,就从多个训练样本中的每一个从分类器确定一概率得分。概率得分超过一阈值的多个训练样本被称为癌症阳性。基于分配给训练集的多个交叉验证分数的分布,预先指定此阈值以标靶期望的特异性水平。例如,如果期望的特异性水平为99.4%,则所述阈值将被设定为分配给训练集中的多个非癌症样本的交叉验证的多个癌症检测概率得分为99.4%。随后对来自多类分类器确定为癌症阳性的每个训练样本进行起源组织或癌症类型评估。首先,多类逻辑回归分类器为每个样本分配了多个概率得分的一集合,一个概率得分为一种预期的癌症类型。接下来,将这些得分的置信度被评估作为最高和第二高得分之间的差异。将一阈值预先指定为最小值,使得训练集中前两个得分差异超过一阈值的多个癌症样本中,有90%被分配了正确的TOO标签作为其最高得分。为了预测多个新样本,前两个得分差异低于此阈值的多个样本被分配为“不确定的癌症”;那些得分差异超过所述阈值的被分配了癌症标签,其中多类分类器分配了最高得分。 6.6.8.示例8:分类器性能 作为测试不同探针集合(诱饵组)的代理,通过约束数据集并且仅考虑多个片段内多个CpG位点的甲基化状态来确定化验板1至6中的每一个的分类器性能,所述多个片段将通过被指定的化验板扩增。 分类器性能分析的结果在表11至18中提供。对于多个基因组区域(即化验板1至6)中的每个集合,表的检测表示分类器在指定化验板上的性能。在此处,检测是包含在CCGA2研究中通过癌症阶段以99%的特异性分层的所有癌症类型的敏感度。起源组织表示起源组织分类的准确度,仅对诊断为癌症的多个个体具有99%的特异性及当有足够的甲基化数据可用于分配起源组织时。多个数值表示平均准确度,括号内为95%置信区间,正确分配的数字除以括号内类别的总数。 表11、使用化验板1的基因组区域的分类准确度
表12、使用化验板2的基因组区域的分类准确度
表13、使用化验板3的基因组区域的分类准确度
表14、使用化验板4的基因组区域的分类准确度
表15、使用化验板5的基因组区域的分类准确度
表16、使用化验板6的基因组区域的分类准确度
表17、使用化验板3A的基因组区域的分类准确度
表18、使用化验板4A的基因组区域的分类准确度
这些结果表明,后期癌症的检测和来源组织的确定的准确度高于早期癌症。此外,较大的化验板显然为阶段I和阶段II的癌症提供了更准确的癌症检测。 图14至图21的部分A表示了使用上述基于特征化的混合模型从二元逻辑回归分类器的化验板1至6和3A和4A的接收者操作特性曲线(ROC曲线)。所有化验板的ROC曲线结果相似,显示曲线下面积(AUC)落在0.80至0.83的窄范围内。如上所述,图14至图21的B部分表示了多个混淆矩阵,显示了为使用基于混合模型的特征训练的多类逻辑回归分类器确定的起源组织(TOO)的准确度。如图14至图21所示,在化验板1至6中,TOO精确度范围为89.9%至91.1%。 表19至24列出了化验板3至5中标记的随机子集的分类器性能,特异性为99%。 表19、使用化验板3(子集a)的40%的基因组区域的随机选择子集的分类准确度
表20、使用化验板3(子集b)的40%的基因组区域的随机选择子集的分类准确度
表21、使用化验板3(子集c)的40%的基因组区域的随机选择子集的分类准确度
表22、使用化验板3的60%的基因组区域的随机选择的子集的分类准确度
表23、使用化验板4的50%的基因组区域的随机选择子集的分类准确度
表24、使用化验板5的50%的基因组区域的随机选择子集的分类准确度
如上所述,图22至图27的A部分表示使用基于混合模型的特征化从训练的二元逻辑回归分类器中随机选择的化验板3至4的子集的接收者操作特性曲线(ROC曲线)。相同的随机子集的分类器性能结果标记也显示在表19至24中。如图所示,这些接收者操作特性曲线(ROC曲线)的曲线下面积(AUC)彼此相似并且类似于完整的化验板的曲线下面积(AUC)。表25中表示每个化验板的曲线下面积(AUC)的总结。图22至图27的B部分表示混淆矩阵,显示了如上所述的多类逻辑回归分类器确定的起源组织(TOO)的准确度。如图22至27所示,对于随机选择的化验板3至4的子集,TOO精确度范围为90.4%至90.7%。 表25、癌症分类的曲线下面积
表26显示了对于化验板1、3A和5,特异性为0.994,每种癌症类型的起源组织的准确度。每列显示TOO精确度,括号内为95%置信区间,括号内为正确分配的数字除以如上所述在二元逻辑回归分类器下正确检测为具有癌症的样本总数。 表26、化验板1、3A和5的TOO精确度
表27至29示出了如上所述对于化验板1、3A和5的二元逻辑回归分类器的分类器性能。每个表示出了每种癌症类型的敏感度,通过阶段分层,特异性为0.994。每列还示出括号中95%置信区间,括号中正确检测到的数字除以每种癌症类型在阶段I至IV的数据集中的样本总数。 表27、化验板1通过癌症类型和阶段的分类器性能
表28、化验板3A通过癌症类型和阶段的分类器性能
表29、化验板5通过癌症类型和阶段的分类器性能
7.通过引用合并 在本申请中引用的所有出版物、专利、专利申请和其他文件,出于所有目的通过引用全文并入本文,其程度与每个单独的出版物、专利、专利申请或其他文件分别指出通过引用并入的程度相同。用于所有目的。 8.同义词 应当理解,本发明的附图和描述已经被简化,以说明与清楚理解本发明相关的元素,同时为了清晰起见,消除了在典型系统中发现的许多其他元素。本领域的普通技术人员可以认识到在实施本发明公开时需要和/或需要其他元件和/或步骤。然而,由于这些元素和步骤在本领域中是公知的,并且由于它们不会便于更好地理解本公开,因此本文不提供对此类元素和步骤的讨论。本文公开的内容可以针对本领域技术人员已知的元件和方法进行变更和修改。 上述描述的一些部分以算法和信息操作的符号表示来描述实施例。这些算法的描述和表示通常由数据处理领域的技术人员用于将其工作的实质有效地传达给本领域技术人员。这些操作,虽然在功能上、计算上或逻辑上描述,但应理解为通过计算机程序或等效电路、微代码等来实现。所描述的操作及其相关联的模块可以体现在软件、固件、硬件或其任何组合中。 如本文所用,对“一个实施例(one embodiment)”或“一实施例(an embodiment)”的任何引用意味着结合该实施例描述的特定元件、特征、结构或特性包含在至少一个实施例中。在说明书的不同位置出现的短语“在一个实施例中”不一定都指同一实施例,从而为所描述的实施例的各种可能性提供了共同工作的框架。 如本文所用,术语“包含(comprises、comprising、includes、including)”、“具有(has、having)”或其任何其他变体旨在涵盖非排他性包含。例如,包含多个元件列表的程序、方法、物品或设备不一定仅限于这些元件,而是可以包含未明确列出或此类程序、方法、物品或设备固有的其他元素。此外,除非另有明确的相反规定,“或”指的是包含或,而不是排他或。例如,条件A或B由以下任一条件满足:A为真(或存在)且B为假(或不存在),A为假(或不存在),B为真(或存在),及A和B均为真(或存在)。 此外,使用“一(a、an)”来描述本文实施例的元件和组件。这样做仅仅是为了方便和给人一个大致的描述。本说明应理解为包含一个或至少一个,单数也包含复数,除非很明显地其另有含义。 虽然已经说明和描述了特定的实施例和应用,但是应该理解,所公开的实施例不限于本文所公开的精确结构和组件。在不脱离所附权利要求所限定的精神和范围的情况下,可以对本文公开的方法和装置的布置、操作和细节进行对本领域技术人员来说显而易见的各种修改、改变和变化。 虽然已经示出并描述了各种具体实施例,但上述规范并不具有限制性。应理解的是,可以在不偏离描述的精神和范围的情况下进行各种更改。本领域技术人员在审查本规范后,许多变化将变得明显。
- 甲基化标记和标靶甲基化探针板
- 甲基化标记和标靶甲基化探针板