一种新型冠状病毒主蛋白酶的抑制剂及其制备方法和用途
文献发布时间:2023-06-19 19:28:50
本申请是对申请号为2020115682827,申请日为2020年12月25日的发明专利提出的分案申请。
技术领域
本发明属于有机合成药物技术领域,具体涉及一种新型冠状病毒主蛋白酶的抑制剂及其制备方法和制药用途。
背景技术
冠状病毒的基因组RNA长约30knt,具有5′帽结构和3′-poly-a尾,至少含有6个开放阅读框(ORF)。第一个ORF(ORF1a/b)约占基因组长度的三分之二,直接翻译两种多蛋白:pp1a和pp1ab,ORF1a和ORF1b之间存在a-1移码。这些多蛋白由一种主蛋白酶(简称M
M
M
因此,亟需研究出一种能够有效抑制SARS-CoV-2病毒的M
发明内容
本发明的目的是提供新型冠状病毒主蛋白酶的抑制剂及其制备方法和制药用途。
本发明提供了式I所示化合物、或其药学上可接受的盐、或其立体异构体、或其旋光异构体、或其同位素替代形式:
/>
其中,X为O或S;
A环选自未取代或被一个或多个R
R
R
R
进一步地,所述化合物的结构如式II、式III或式IV所示:
其中,X为O或S;
n选自0~3的整数,优选为0~2的整数;
R
R
R
R
进一步地,R
R
R
R
进一步地,所述式II如式II-1或式II-2所示:
其中,X为O或S,优选为O;
R
m选自0~3的整数,R
R
R
L
所述卤素优选为氯、氟。
进一步地,所述化合物的结构为以下结构之一:
/>
/>
/>
/>
/>
本发明还提供了一种药物组合物,所述药物组合物是以上述化合物、或其药学上可接受的盐、或其立体异构体、或其旋光异构体、或其同位素替代形式为活性成分,加上药学上可接受的辅料制成的制剂。
本发明还提供了上述化合物、或其药学上可接受的盐、或其立体异构体、或其旋光异构体、或其同位素替代形式在制备冠状病毒蛋白水解酶抑制剂中的用途;优选的,所述冠状病毒蛋白水解酶为冠状病毒主蛋白酶;更优选的,所述冠状病毒蛋白水解酶为SARS-COV-2M
本发明还提供了上述化合物、或其药学上可接受的盐、或其立体异构体、或其旋光异构体、或其同位素替代形式在制备抗冠状病毒的药物中的用途,优选的,所述冠状病毒为新型冠状病毒SARS-CoV-2。
本发明还提供了上述化合物、或其药学上可接受的盐、或其立体异构体、或其旋光异构体、或其同位素替代形式在制备预防和/或治疗与SARS-COV-2M
进一步地,所述冠状病毒蛋白水解酶抑制剂、抗冠状病毒的药物或预防和/或治疗病毒性肺炎的药物能够抑制SARS-COV-2M
关于本发明的使用术语的定义:除非另有说明,本文中基团或者术语提供的初始定义适用于整篇说明书的该基团或者术语;对于本文没有具体定义的术语,应该根据公开内容和上下文,给出本领域技术人员能够给予它们的含义。
碳氢基团中碳原子含量的最小值和最大值通过前缀表示,例如,前缀C
本文“取代”是指分子中的1个、2个或多个氢原子被其它不同的原子或分子所替换,包括该分子中同位原子或异位原子上的1个、2个或多个取代。
“同位素替代形式”指化合物中的一个或两个以上的原子被其对应的同位素替换后得到的化合物,比如化合物中的氢被替换为氕、氘或氚。
“药学上可接受的”是指某载体、运载物、稀释剂、辅料,和/或所形成的盐通常在化学上或物理上与构成某药物剂型的其它成分相兼容,并在生理上与受体相兼容。
“盐”是将化合物或其立体异构体,与无机和/或有机酸和/或碱形成的酸式和/或碱式盐,也包括两性离子盐(内盐),还包括季铵盐,例如烷基铵盐。这些盐可以是在化合物的最后分离和纯化中直接得到。也可以是通过将化合物,或其立体异构体,与一定数量的酸或碱适当(例如等当量)进行混合而得到。这些盐可能在溶液中形成沉淀而以过滤方法收集,或在溶剂蒸发后回收而得到,或在水介质中反应后冷冻干燥制得。
“药学上可接受的盐”可以是化合物的盐酸盐、硫酸盐、枸橼酸盐、苯磺酸盐、氢溴酸盐、氢氟酸盐、磷酸盐、乙酸盐、丙酸盐、丁二酸盐、草酸盐、苹果酸盐、琥珀酸盐、富马酸盐、马来酸盐、酒石酸盐或三氟乙酸盐。
卤素为氟、氯、溴或碘。
“芳基”指具有共轭的π电子体系的全碳单环或稠合多环(也就是共享毗邻碳原子对的环)基团,例如苯基。所述芳基不含有杂原子,如氮,氧,或硫,同时连接母体的点必须在具有共轭的π电子体系的环上的碳原子上。芳基可以是取代的或未取代的。“5~6元芳基”指环碳原子数为5或6的芳基。
“杂芳基”指包含一个到多个杂原子的杂芳族基团。这里所指的杂原子包括氧、硫和氮。例如呋喃基、噻吩基、吡啶基、吡唑基等。所述杂芳基可以是任选取代的或未取代的。“5~6元杂芳基”指环原子数为5或6的杂芳基。
“环烷基”指饱和或不饱和的环状烃取代基;环状烃可以是单环也可以是多环。例如,“3~6元饱和环烷基”指环碳原子数为3~6的饱和的环烷基。
“杂环基”指饱和或不饱和的环状烃取代基;环状烃可以是单环也可以是多环,且携带至少一个环杂原子(包括但不限于O、S或N)。例如,“3~6元饱和杂环基”指环原子数为3~6的饱和的杂环基。
“稠环烷基”指多环的环烷基,且该多环的环烷基中有两个环共用两个相邻的碳原子。
“杂稠环基”指多环的杂环基,其中至少含有1个杂原子,且该多环的杂环基中有两个环共用两个相邻的碳原子或杂原子。
“亚烷基”指烷基失去一个原子后的基团。例如C
“亚烯基”指烯基失去一个原子后的基团。例如C
“亚炔基”指炔基失去一个原子后的基团。例如C
实验结果表明,本发明提供了一种能够有效抑制新型冠状病毒主蛋白酶M
显然,根据本发明的上述内容,按照本领域的普通技术知识和惯用手段,在不脱离本发明上述基本技术思想前提下,还可以做出其它多种形式的修改、替换或变更。
以下通过实施例形式的具体实施方式,对本发明的上述内容再作进一步的详细说明。但不应将此理解为本发明上述主题的范围仅限于以下的实例。凡基于本发明上述内容所实现的技术均属于本发明的范围。
附图说明
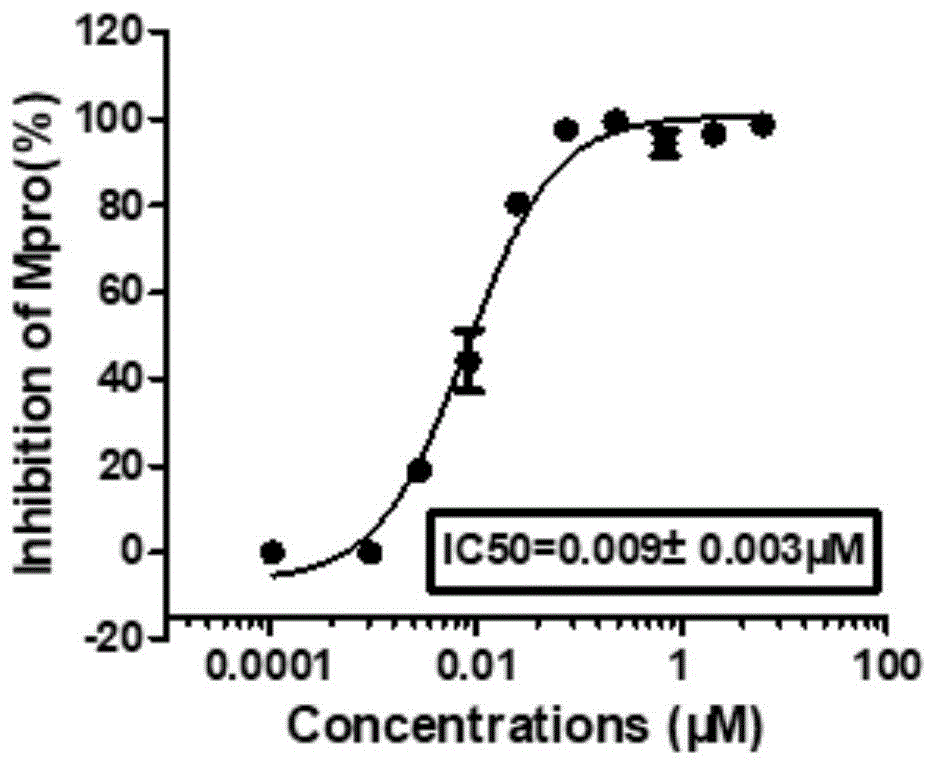
图1为化合物26对SARS-COV-2M
图2为化合物33对SARS-COV-2M
图3为化合物37对SARS-COV-2M
图4化合物对SARS-COV-2在人肺泡上皮细胞中复制的抑制实验。
图5SARS-CoV-2感染小鼠的肺部病毒载量。
图6SARS-CoV-2感染小鼠的肺部病理组织切片(3dpi)。
图7SARS-CoV-2感染小鼠的肺部代表性细胞因子表达水平(3dpi)。
图8SARS-CoV-2感染小鼠的肺部中性粒细胞和巨噬细胞计数(3dpi)。
具体实施方式
本发明所用原料与设备均为已知产品,通过购买市售产品所得。
实施例1:化合物1的制备
按照上述制备路线制得本发明的化合物1,路线中各步骤的反应条件如下:
i、a,2-氟丙二酸二甲酯,苄醇,甲苯,对甲苯磺酸,110℃;b,异丙醇,正己烷,-10℃;
ii、异丙醇,氢氧化钠,水,45℃;
iii、无水四氢呋喃,异丙基氯化镁四氢呋喃溶液,Ar,0℃;
iv、无水四氢呋喃,N,N'-羰基二咪唑,Ar,0℃;
v、乙酸乙酯,10%钯碳,氢气,室温;
vi、二氯甲烷,盐酸二氧六环溶液;
vii、二氯甲烷,N,N,N′,N′-四甲基-O-(7-氮杂苯并三唑-1-基)六氟磷酸脲,N,N-二异丙基乙胺,-20℃;
viii、二氯甲烷,三氟乙酸;
ix、二氯甲烷,N,N,N′,N′-四甲基-O-(7-氮杂苯并三唑-1-基)六氟磷酸脲,N,N-二异丙基乙胺,-20℃;
以下为具体合成步骤:
中间体2:2-氟丙二酸二苄酯的制备
2-氟丙二酸二甲酯(10g,66.6mmol,1.0eq)和苄醇(35mL,338.2mmol,5.0eq)用100mL甲苯溶解,加入1.15g对甲苯磺酸(6.7mmol,0.1eq),回流反应,TLC监控反应,约8小时后反应完毕。冷却至室温,减压蒸去甲苯,加入15mL异丙醇,搅拌均匀,搅拌下慢慢加入30mL正己烷,置于-10℃冷阱中继续搅拌2小时,析出大量白色固体。抽滤,滤饼用10mL×2冷冻正己烷洗涤两次,滤饼30℃真空减压干燥得18.4g产品,收率91.4%。
中间体3:2-氟丙二酸单苄酯的制备
2-氟丙二酸二苄酯(18.4g,60.9mmol,1.0eq)用100mL异丙醇溶解,升温至45℃,氢氧化钠(2.55g,63.9mmol,1.05eq)溶于60mL水后慢慢滴入,滴加时间>1小时。滴加完毕后继续反应30分钟,减压蒸去异丙醇,加入50mL水,用饱和碳酸氢钠溶液调节pH值至9左右。水相用二氯甲烷20mL×2二氯甲烷萃取两次,用6mol/L盐酸调节水相pH值至1-2,用40mL×3异丙醚萃取三次,合并有机相,用30mL饱和盐水洗涤一次。有机相加入无水硫酸镁干燥,过滤,浓缩得粘稠状残留物,加入60mL正己烷搅拌过夜,析出白色固体,过滤,滤饼40℃真空减压干燥得6.5g产品,收率50.3%.
中间体4的制备
2-氟丙二酸单苄酯溶于无水四氢呋喃(2mL/mmol),氩气置换保护,冷却至0℃,慢慢滴加异丙基氯化镁四氢呋喃溶液(2M四氢呋喃溶液,2.0eq),得白色悬浊液。0℃下继续搅拌1小时,产品悬浊液直接用于下步反应。
中间体6:1-苄基6-甲基(4S)-4-(((叔丁氧羰基)氨基)-2-氟-3-氧代己二酸酯的制备
Boc-L-天冬氨酸4-甲酯(2.2g,8.8mmol,1.0eq)溶于50mL无水四氢呋喃,氩气置换保护,冷却至0℃,加入CDI(1.5g,9.3mmol,1.05eq),保温反应1小时。反应液冷却至-20℃,慢慢加入1.5eq中间体4,保温反应1小时后升温至室温反应6小时。冰水浴下将反应液慢慢倒入300mL 2M稀盐酸中,用100mL×3乙酸乙酯萃取三次,合并有机相,用饱和碳酸氢钠溶液洗涤至弱碱性,再用50mL饱和盐水洗涤一次后加入无水硫酸镁干燥,过滤,浓缩,所得粗品直接用于下步反应。
中间体7:(S)-3-(((叔丁氧羰基)氨基)甲基-5-氟-4-氧戊酸甲酯的制备
上步所得中间体6粗品加入50mL乙酸乙酯,加入200mg 10%钯碳,氢气置换,氢气下室温反应过夜,过滤,浓缩,所得粗品用石油醚:乙酸乙酯=10:1流动相柱层析得1.5g无色油状物,收率65%。
中间体8:(S)-3-氨基-5-氟-4-氧戊酸甲酯的制备
500mg中间体7加入5mL二氯甲烷溶解,然后加入5mL盐酸二氧六环,反应完全后旋干,得中间体8,收率为91.2%。
中间体11:甲基(1H-吲哚-2-羰基)-L-脯氨酸的制备
1H-吲哚-2-羧酸(1g,6.21mmol,1.0eq)用二氯甲烷溶解后,于-20℃加入HATU(2.81g,7.40mmol,1.2eq)后加入L-脯氨酸甲酯盐酸盐(1.03g,6.21mmol,1.0eq),最后加入DIEA(3mL,18.51mmol,3.0eq),TLC监控反应。反应完毕后,用水溶液和DCM进行萃取,有机层浓缩后经柱层析分离得到中间体11(1.53g),收率为75.2%。
中间体12:(1H-吲哚-2-羰基)-L-脯氨酸的制备
500mg中间体11加入10mL二氯甲烷溶解,然后加入5mL三氟乙酸,反应完全后旋干,得378mg中间体12,直接作为先一步反应。收率为91.2%。
化合物1:甲基(S)-3-((S)-1-(1H-吲哚-2-羰基)吡咯烷-2-甲酰胺)-5-氟-4-氧代戊酸的制备
中间体12(168mg,0.61mmol,1.0eq)用二氯甲烷溶解后,于-20℃加入HATU(280mg,0.73mmol,1.2eq)后加入中间体8(100mg,0.61mmol,1.0eq),最后加入DIEA(301μL,1.83mmol,3.0eq),TLC监控反应。反应完毕后,用水溶液和DCM进行萃取,浓缩有机层,柱层析分离得到化合物1,收率为34%。
实施例2:化合物3的制备
按照上述制备路线制得本发明的化合物3,路线中各步骤的反应条件如下:
i、Boc-L-谷氨酸二甲酯,LiHMDS四氢呋喃溶液,氩气,无水四氢呋喃,-78℃,;
ii、(2S,4R)-二甲基2-(叔-丁氧基羰基氨基)-4-(氰基甲基)戊二酸酯,无水甲醇,六水氯化钴,硼氢化钠;
iii、(S)-甲基2-(叔-丁氧基羰基氨基)-3-((S)-2-羰基吡咯烷-3-基)丙酸酯,一水合氢氧化锂,四氢呋喃,0℃;
iv、无水四氢呋喃,Ar,N,N'-羰基二咪唑,0℃;
v、乙酸乙酯,10%钯碳,氢气,室温;
vi、二氯甲烷,盐酸二氧六环溶液;
vii、二氯甲烷,N,N,N′,N′-四甲基-O-(7-氮杂苯并三唑-1-基)六氟磷酸脲,N,N-二异丙基乙胺,-20℃;
viii、二氯甲烷,三氟乙酸;
ix、二氯甲烷,N,N,N′,N′-四甲基-O-(7-氮杂苯并三唑-1-基)六氟磷酸脲,N,N-二异丙基乙胺,-20℃;
以下为具体合成步骤:
中间体14:(2S,4R)-2-((叔丁氧羰基)氨基)-4-(氰基甲基)戊二酸二甲酯的制备
Boc-L-谷氨酸二甲酯(12g,43.6mmol,1.0eq)溶于100mL无水四氢呋喃,氩气置换保护,冷却至-78℃,慢慢滴加94mL LiHMDS四氢呋喃溶液(1M四氢呋喃溶液,94mmol,2.2eq),滴加完毕后保温反应1小时。3.24mL溴乙腈(46.6mmol,1.1eq)慢慢滴加入反应液,保温反应6小时后用50mL饱和氯化铵溶液淬灭反应。将淬灭后的反应液升温至室温,用60mL×3乙酸乙酯萃取三次,合并有机相,用50mL饱和盐水洗涤后加入无水硫酸镁干燥,过滤,浓缩,所得粗品用石油醚:乙酸乙酯=4:1流动相柱层析得9.36g浅黄色油状物,收率68.3%。
中间体15:(S)-2-(((叔丁氧羰基)氨基)甲基-3-((S)-2-氧吡咯烷-3-基)丙酸甲酯的制备
(2S,4R)-二甲基2-(叔-丁氧基羰基氨基)-4-(氰基甲基)戊二酸酯(9.36g,29.8mmol,1.0eq)溶于150mL无水甲醇,冷却至0℃。加入六水氯化钴(4.25g,18mmol,0.6eq),再分批加入硼氢化钠(6.76g,180mmol,6.0eq),加完后升温至室温,反应过夜,TLC监控反应完毕。加入50mL饱和氯化铵溶液淬灭反应,减压蒸去甲醇,用100mL×3乙酸乙酯萃取三次,合并有机相,依次用200mL×3饱和氯化铵溶液洗涤三次、200mL×3饱和盐水洗涤三次,有机相加入无水硫酸镁干燥,过滤,浓缩,所得粗品用石油醚:乙酸乙酯=1:1流动相柱层析得3.94g白色固体,收率46.2%。
中间体16:(S)-2-((叔丁氧羰基)氨基)-3-((S)-2-氧吡咯烷-3-基)丙酸的制备
(S)-甲基2-(叔-丁氧基羰基氨基)-3-((S)-2-羰基吡咯烷-3-基)丙酸酯(0.88g,3.1mmol,1.0eq)溶于10mL四氢呋喃,冷却至0℃。一水合氢氧化锂(0.64g,15.4mmol,5.0eq)溶于10mL水后慢慢滴入,滴完后保温反应4小时,TLC监控反应完毕。饱和柠檬酸水溶液调节pH值至中性,减压蒸去四氢呋喃,10mL乙酸乙酯萃取一次,水相用饱和柠檬酸水溶液调节pH值至3-4,20mL×3乙酸乙酯萃取三次,合并有机相,用20mL饱和盐水洗涤后加入无水硫酸镁干燥,过滤,浓缩得0.78g类白色固体,收率93.2%。
中间体17:(4S)-4-((叔丁氧羰基)氨基)-2-氟-3-氧代-5-((S)-2-氧吡咯烷-3-基)戊酸苄酯的制备
(S)-2-((叔-丁氧基羰基)氨基)-3-((S)-2-羰基吡咯烷-3-基)丙酸(2.4g,8.8mmol,1.0eq)溶于50mL无水四氢呋喃,氩气置换保护,冷却至0℃,加入CDI(1.5g,9.3mmol,1.05eq),保温反应1小时。反应液冷却至-20℃,慢慢加入1.5eq中间体4,保温反应1小时后升温至室温反应6小时。冰水浴下将反应液慢慢倒入300mL 2M稀盐酸中,用100mL×3乙酸乙酯萃取三次,合并有机相,用饱和碳酸氢钠溶液洗涤至弱碱性,再用50mL饱和盐水洗涤一次后加入无水硫酸镁干燥,过滤,浓缩,所得粗品直接用于下步反应。
中间体18:((S)-4-氟-3-氧代-1-((S)-2-氧吡咯烷-3-基)丁烷-2-基)氨基甲酸叔丁酯的制备
上步所得中间体17粗品加入50mL乙酸乙酯,加入200mg 10%钯碳,氢气置换,氢气下室温反应过夜,过滤,浓缩,所得粗品用石油醚:乙酸乙酯=1:1流动相柱层析得1.3g白色固体,收率50%。
中间体19:(S)-3-((S)-2-氨基-4-氟-3-氧丁基)吡咯烷-2-酮的制备
500mg中间体18加入5mL二氯甲烷溶解,然后加入5mL盐酸二氧六环,反应完全后旋干,得中间体19,收率为85%。
中间体22:乙基(1S,3aR,6aS)-2-(2-(2,4-二氯苯氧)乙酰基)八氢环戊烯并[c]吡咯-1-羧酸酯的制备.
将2,4-二氯苯氧乙酸(中间体21,0.58g,2.62mmol),2-(7-氧化苯并三氮唑)-N,N,N',N'-四甲基脲六氟磷酸酯(1.2g,3.14mmol),N,N-二异丙基乙胺(1.3mL,7.86mmol),and(1S,3aR,6aS)-八氢环戊烯并[c]吡咯-1-羧酸乙酯盐酸盐(中间体20,0.58g,2.62mmol)溶于15mL超干N,N-二甲基甲酰胺中,反应在25℃氩气保护条件下反应12小时,d反应加入4倍体积水,用二氯甲烷萃取三次,合并有机相用饱和氯化铵溶液、饱和碳酸钠溶液洗涤,无水硫酸钠干燥后过滤,硅胶拌样柱层析(石油醚/乙酸乙酯=1:1)得到中间体22(0.60g,59%)。
中间体23:(1S,3aR,6aS)-2-(2-(2,4-二氯苯氧)乙酰基)八氢环戊烯并[c]吡咯-1-羧酸的制备
将乙基(1S,3aR,6aS)-2-(2-(2,4-二氯苯氧)乙酰基)八氢环戊烯并[c]吡咯-1-羧酸酯(中间体22,200mg,0.52mmol)溶于20mL甲醇中,然后加入2M氢氧化钠溶液(10mL),反应在25℃下搅拌4小时.TLC监控反应结束后,旋干甲醇,用盐酸调pH至弱酸性,二氯甲烷萃取三次,合并有机相无水硫酸钠干燥后选干得粗产品直接进行下一步反应。
化合物3:(1S,3aR,6aS)-2-(2-(2,4-二氯)乙酰基)-N-((S)-4-氟-3-氧-1-((S)-2-羰基-3-基)丁烷-2-基)八氢环戊烯并[c]吡咯-1-甲酰胺的制备
中间体23(168mg,0.61mmol,1.0eq)用二氯甲烷溶解后,于-20℃加入HATU(280mg,0.73mmol,1.2eq)后加入中间体19(100mg,0.61mmol,1.0eq),最后加入DIEA(301μL,1.83mmol,3.0eq),TLC监控反应。反应完毕后,用水溶液和DCM进行萃取,有机层浓缩后经柱层析分离得到化合物3,收率为34%。
实施例3:制备化合物9
按照上述制备路线制得本发明的化合物9,路线中各步骤的反应条件如下:
i、1-羟基苯并三唑,1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐,N,N-二异丙基乙胺,N,N-二甲基甲酰胺,室温。
ii、氢氧化钠,甲醇,水,55度
iii、2-(7-氧化苯并三氮唑)-N,N,N',N'-四甲基脲六氟磷酸酯,N,N-二异丙基乙胺,N,N-二甲基甲酰胺,0℃
iv、硼氢化钠,甲醇,室温
v、戴斯马丁氧化剂,超干二氯甲烷,室温
以下为具体合成步骤:
中间体25:甲基(1R,2S,5S)-6,6-二甲基3-(喹啉-2-羰基)-3-氮杂双环[3.1.0]己烷-2-羧酸的制备
将原料23(喹啉2-羧酸1.0g,11.6mmol)、1-羟基苯并三唑(2.03g,15.08mmoL)、1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(4.43g,23.3mmol),N,N-二甲基甲酰胺30mL置于圆底烧瓶常温搅拌0.5小时,加入N,N-二异丙基乙胺2.5mL,再加入0.59g中间体24,反应8h后,减压蒸馏除去溶剂,用二氯甲烷和氯化铵溶液、碳酸氢钠溶液进行萃取,水、饱和氯化钠溶液进行洗涤,最后用硫酸钠干燥,抽滤,将有机相柱层析得白色固体。收率为85%。
中间体26:(1R,2S,5S)-6,6-二甲基3-(喹啉-2-羰基)-3-氮杂双环[3.1.0]己烷-2-羧酸的制备
将中间体25溶于20mL四氢呋喃,缓慢加入10mL 2mol/L的氢氧化钠溶液,将反应液在逐渐升温至55度搅拌3小时后终止并冷却至常温,浓缩反应液,加水调pH为弱酸性,析出白色固体,抽滤的得中间体5,未经进一步纯化被用作下一步反应
中间体28:甲基((1R,5S)-6,6-二甲基-3-(喹啉-2-羰基)-3-氮杂双环[3.1.0]己烷-2-羰基)-L-苯基丙氨酸的制备
将中间体26 1.0g、2-(7-氧化苯并三氮唑)-N,N,N',N'-四甲基脲六氟磷酸酯1.93g加入20mL N,N-二甲基甲酰胺中,0℃搅拌0.5小时,加入N,N-二异丙基乙胺2.0mL,再加入中间体27 0.89g,氩气保护0℃条件下反应12h后,加入4倍体积的水,用乙酸乙酯进行萃取三次。合并有机相,用氯化铵溶液、碳酸氢钠溶液进行萃取,水、饱和氯化钠溶液进行洗涤,最后用硫酸钠干燥,抽滤,将有机相柱层析得白色固体。收率为65%。
中间体29:(1R,5S)-N-((S)-1-羟基-3-苯丙-2-基)-6,6-二甲基-3-(喹啉-2-羰基)-3-氮杂双环[3.1.0]己烷-2-甲酰胺的制备
将中间体28(((1R,5S)-6,6-二甲基-3-(喹啉-2-羰基)-3-氮杂双环[3.1.0]己烷-2-羰基)-L-苯基丙氨酸)500mg,溶于30mL干燥甲醇中,常温条件下加入硼氢化钠,搅拌3小时后,加水淬灭,旋干甲醇,用乙酸乙酯萃取水相(50mL×3),收集有机相用硫酸钠进行干燥,抽滤后旋干有机相是为中间体29,产率80%。
化合物9:(1R,5S)-6,6-二甲基-N-((S)-1-氧-3-苯丙-2-基)-3-(喹啉-2-羰基)-3-氮杂双环[3.1.0]己烷-2-甲酰胺的制备
将中间体29(1R,5S)-N-((S)-1-羟基-3-苯丙-2-基)-6,6-二甲基-3-(喹啉-2-羰基)-3-氮杂双环[3.1.0]己烷-2-甲酰胺200mg,溶于10mL干燥二氯甲烷中,常温搅拌条件下加入戴斯马丁氧化剂,TLC监控反应进行完毕,抽滤除去氧化剂,滤液柱层析得化合物9,产率65%。
实施例4:制备化合物14
按照上述制备路线制得本发明的化合物14,路线中各步骤的反应条件如下:
i、三氟乙酸,二氯甲烷,25℃
ii、2-(7-氧化苯并三氮唑)-N,N,N',N'-四甲基脲六氟磷酸酯,N,N-二异丙基乙胺,N,N-二甲基甲酰胺,室温。
iii、氢氧化钠,甲醇,水,55度
iv、2-(7-氧化苯并三氮唑)-N,N,N',N'-四甲基脲六氟磷酸酯,N,N-二异丙基乙胺,N,N-二甲基甲酰胺,0℃
v、硼氢化钠,甲醇,室温
vi、戴斯马丁氧化剂,超干二氯甲烷,室温。
以下为具体合成步骤:
中间体30:甲基(S)-2-氨基-3-((S)-2-羰基-3-基)丙酸酯三氟乙酸盐的制备
将(S)-甲基2-(叔-丁氧基羰基氨基)-3-((S)-2-羰基吡咯烷-3-基)丙酸酯(中间体14,2.5g)溶于30mL二氯甲烷中,再加入20mL三氟乙酸,常温搅拌14小时,然后直接旋干得粗产品直接用于下一步反应。
中间体32:甲基(1R,2S,5S)-6,6-二甲基-3-(2-(4-(三氟甲氧基)苯氧)乙酰)-3-氮杂双环并[3.1.0]己烷-2-甲酸的制备
将2-(4-(三氟甲氧基)苯氧)乙酸(商业购得中间体31,0.24g,1.0mmol),2-(7-氧化苯并三氮唑)-N,N,N',N'-四甲基脲六氟磷酸酯(0.49g,1.2mmol)和N,N-二异丙基乙胺(494μL,3mmol)溶于一定N,N-二甲基甲酰胺中,然后加入(1R,2S,5S)-6,6-二甲基-3-氮杂双环[3.1.0]己烷-2-羧酸甲酯盐酸盐(商业购得中间体24,0.21g,1.0mmol).氩气保护常温反应12小时.反应加入4倍体积水,用二氯甲烷萃取三次,合并有机相用饱和氯化铵溶液、饱和碳酸钠溶液洗涤,无水硫酸钠干燥后过滤,硅胶拌样柱层析(石油醚/乙酸乙酯=1:1)得到白色固体中间体32(0.34g,88%)。
中间体33:(1R,2S,5S)-6,6-二甲基-3-(2-(4-(三氟甲氧基)苯氧)乙酰)-3-氮杂双环并[3.1.0]己烷-2-甲酸的制备
用30mL甲醇溶解甲基(1R,2S,5S)-6,6-二甲基-3-(2-(4-(三氟甲氧基)苯氧)乙酰)-3-氮杂双环[3.1.0]己烷-2-甲酸(中间体32,200mg),然后加入2M NaOH溶液20mL.常温搅拌2.5小时.TLC监控反应结束后,旋干甲醇,用盐酸调pH至弱酸性,二氯甲烷萃取三次,合并有机相无水硫酸钠干燥后选干得粗产品直接进行下一步反应。
中间体34:甲基(1R,2S,5S)-6,6-二甲基-3-(2-(4-(三氟甲氧基)苯氧)乙酰)-3-氮杂双环并[3.1.0]己烷--2-酰胺)-3-((S)-2-羰基-3-基)丙酸酯的制备
在0℃反应条件下,往(1R,2S,5S)-6,6-二甲基-3-(2-(4-(三氟甲氧基)苯氧)乙酰)-3-氮杂双环[3.1.0]己烷-2-甲酸(中间体33,0.45g,1.2mmol)的超干DMF溶液中,加入2-(7-氧化苯并三氮唑)-N,N,N',N'-四甲基脲六氟磷酸酯(0.61g,1.6mmol),搅拌30分钟后,加入N.N-二异丙基乙胺(0.59mL,3.6mmol).然后,中间体14粗产品(0.27g1.45mmol)加入反应体系。反应在0℃氩气保护条件下搅拌12小时。TLC监控反应完后,加入4倍体积的水,用乙酸乙酯萃取三次,合并有机相用饱和氯化铵溶液、饱和碳酸氢钠溶液洗涤,无水硫酸钠干燥后过滤,拌样柱层析(乙酸乙酯/甲醇=10:1)得到白色固体是为中间体34.
中间体35:(1R,2S,5S)-N-((S)-1-羟基-3-((S)-2-羰基-3-基)丙酸酯-2-基)-3-(2-(4-(三氟甲氧基)苯氧)乙酰基)-3-氮杂双环并[3.1.0]己烷-2-甲酰胺的制备
将甲基(1R,2S,5S)-6,6-二甲基-3-(2-(4-(三氟甲氧基)苯氧)乙酰)-3-氮杂双环[3.1.0]己烷--2-酰胺)-3-((S)-2-羰基-3-基)丙酸酯(中间体34 0.56mg,1.1mmol)加入50毫升中,低温条件下分批加入硼氢化钠(0.14g,8.8mmol).反应常温条件下搅拌2小时后,加水淬灭,旋干甲醇,用乙酸乙酯(50mL×3)对剩余水相进行萃取.有机相合并后用无水硫酸钠干燥,过滤旋干得白色固体为粗产品,被直接用于下一步反应。
化合物14:(1R,2S,5S)-6,6-二甲基-N-((S)-1-醛基-3-((S)-2-羰基-3-基)丙酸酯-2-基)-3-(2-(4-(三氟甲氧基)苯氧)乙酰基)-3-氮杂双环并[3.1.0]己烷-2-甲酰胺的制备
将(1R,2S,5S)-N-((S)-1-羟基-3-((S)-2-羰基-3-基)丙酸酯-2-基)-3-(2-(4-(三氟甲氧基)苯氧)乙酰基)-3-氮杂双环[3.1.0]己烷-2-甲酰胺(中间体36,(0.38g,0.75mmol)溶于超干二氯甲烷中,然后分批加入戴斯马丁氧化剂(0.95mg,0.79mmol),室温条件下反应3.5小时,TLC监控反应结束,反应体系过滤,用硫代硫酸钠溶液和饱和碳酸氢钠溶液洗涤有机相,浓缩后用制备色谱分离体系(乙腈/水=30:70)分离得化合物14(0.28g,45%)as a white solid.
实施例5:化合物15的制备
按照上述制备路线制得本发明的化合物15,路线中各步骤的反应条件如下:
i、2-(7-氧化苯并三氮唑)-N,N,N',N'-四甲基脲六氟磷酸酯,N,N-二异丙基乙胺,N,N-二甲基甲酰胺,0℃
ii、硼氢化钠,甲醇,室温
iii、戴斯马丁氧化剂,超干二氯甲烷,室温。
具体合成步骤如下:
中间体36:甲基(S)-2-((1S,3aR,6aS)-2-(2-(2,4-二氯苯氧)乙酰基)八氢环戊烯并[c]吡咯-1-甲酰胺)-3-((S)-2-羰基-3-基)丙酸酯的制备
首先,2-(7-氧化苯并三氮唑)-N,N,N',N'-四甲基脲六氟磷酸酯(0.099g,0.26mmol)被加入(1S,3aR,6aS)-2-(2-(2,4-二氯苯氧)乙酰基)八氢环戊烯并[c]吡咯-1-羧酸(中间体23,0.071g,0.20mmol)的超干N,N-二甲基甲酰胺溶液中,反应体系先搅拌30分钟,N,N-二异丙基乙胺(100μL,0.60mmol),甲基(S)-2-氨基-3-((S)-2-羰基-3-基)丙酸酯三氟乙酸盐(中间体30,0.060g,0.32mmol)被依次加入反应体系.反应在0℃氩气保护条件下反应12小时,TLC监控反应结束,加入4倍体积的水,用乙酸乙酯萃取三次,合并有机相用饱和氯化铵溶液、饱和碳酸氢钠溶液洗涤,无水硫酸钠干燥后过滤,拌样柱层析(乙酸乙酯/甲醇=10:1)获得中间体36(0.063g,59%).
中间体37:(1S,3aR,6aS)-2-(2-(2,4-二氯苯氧)乙酰基)-N-((S)-1-羟基-3-((S)-2-羰基-3-基)丙酸酯-2-基)八氢环戊烯并[c]吡咯-1-甲酰胺的制备
将甲基(S)-2-((1S,3aR,6aS)-2-(2-(2,4-二氯苯氧)乙酰基)八氢环戊烯并[c]吡咯-1-甲酰胺)-3-((S)-2-羰基-3-基)丙酸酯(中间体36,1,00g,2.0mmol)溶于无水甲醇中,然后再0℃条件下分批加入硼氢化钠(0.6g,16mmol),然后将温度升至室温,继续搅拌2小时,TLC检测反应结束,加水淬灭,旋干甲醇,用乙酸乙酯(50mL×3)对剩余水相进行萃取.有机相合并后用无水硫酸钠干燥,过滤旋干得白色固体为粗产品,被直接用于下一步反应。
化合物15:(1S,3aR,6aS)-2-(2-(2,4-二氯苯氧)乙酰基)-N-((S)-1-醛基-3-((S)-2-羰基-3-基)丙酸酯-2-基)八氢环戊烯并[c]吡咯-1-甲酰胺的制备
在(1S,3aR,6aS)-2-(2-(2,4-二氯苯氧)乙酰基)-N-((S)-1-羟基-3-((S)-2-羰基-3-基)丙酸酯-2-基)八氢环戊烯并[c]吡咯-1-甲酰胺(中间体37,0.50g,1.0mmol)的超干二氯甲烷溶液中,分批缓慢加入戴斯马丁氧化剂(0.55g,1.3mmol),室温条件下反应3.5小时,TLC监控反应结束,反应体系过滤,用硫代硫酸钠溶液和饱和碳酸氢钠溶液洗涤有机相,浓缩后用制备色谱分离体系(乙腈/水=45:55)得到白色固体化合物30(0.21g,42%)。
实施例6:化合物42的制备
按照上述制备路线制得本发明的化合物42,路线中各步骤的反应条件如下:
i、LDA,ClCH
ii、HCl二氧六环溶液
iii、2-(7-氧化苯并三氮唑)-N,N,N',N'-四甲基脲六氟磷酸酯,N,N-二异丙基乙胺,N,N-二甲基甲酰胺,0℃。
具体合成步骤如下:
中间体38:叔丁基((S)-4-氯-3-氧-1-((S)-2-羰基-3-基)丁烷-2-基)氨基甲酸酯的制备
选取干燥三口瓶,分别准备氩气保护和温度计,加入(S)-甲基2-(叔-丁氧基羰基氨基)-3-((S)-2-羰基吡咯烷-3-基)丙酸酯(中间体16,5g,17.5mmol),四氢呋喃(50mL),氯碘甲烷(5mL,68mmol)在-77℃下进行搅拌.二异丙基氨基锂(70mL,105mmol)被进一步滴入。加完以后进一步反应2小时,然后再低温条件下加入乙酸和四氢呋喃进行淬灭,产生的黑色悬浮物被进一步搅拌10分钟同时升温至室温。反应进一步用乙酸乙酯稀释,用水、饱和碳酸氢钠溶液和饱和食盐水洗涤,无水硫酸钠干燥后,过滤浓缩拌样柱层析得浅黄色固体为中间体38。
中间体39:(S)-3-((S)-2-氨基-4-氯-3-氧丁基)吡咯烷-2-酮盐酸盐的制备
将叔丁基((S)-4-氯-3-氧-1-((S)-2-羰基-3-基)丁烷-2-基)氨基甲酸酯(中间体38)250mg加入20mL二氧六环中,随后加入20mL氯化氢二氧六环溶液20mL,常温搅拌4小时后,TLC检测反应完,旋干反应液得粗产品用于下一步反应。
化合物42:(1S,3aR,6aS)-N-((S)-4-氯-3-氧-1-((S)-2-羰基-3-基)丁烷-2-基)-2-(2-(2,4-二氯苯氧)乙酰基)八氢环戊烯并[c]吡咯-1-甲酰胺的制备
将2-(7-氧化苯并三氮唑)-N,N,N',N'-四甲基脲六氟磷酸酯(1g,2.6mmol)加入(1S,3aR,6aS)-2-(2-(2,4-二氯苯氧)乙酰基)八氢环戊烯并[c]吡咯-1-羧酸(中间体23,0.71g,2.0mmol)的超干N,N-二甲基甲酰胺溶液中,反应体系先搅拌30分钟,N,N-二异丙基乙胺(1mL,6.0mmol),(S)-3-((S)-2-氨基-4-氯-3-氧丁基)吡咯烷-2-酮盐酸盐(中间体39,0.652g,3.2mmol)被依次加入反应体系.反应在0℃氩气保护条件下反应16小时,TLC监控反应结束,加入3倍体积的水,用乙酸乙酯萃取三次,合并有机相用饱和氯化铵溶液、饱和碳酸氢钠溶液洗涤,无水硫酸钠干燥后过滤,制备纯化系统(乙腈/水=30:70)分离获得化合物42.
实施例7:化合物50的制备
按照上述制备路线制得本发明的化合物50,路线中各步骤的反应条件如下:
i、叔丁基乙异腈,乙酸,超干二氯甲烷;
ii、1M氢氧化钠溶液,甲醇;
iii、戴斯马丁氧化剂、超干二氯甲烷。
具体合成步骤如下:
中间体40:(3S)-1-(叔丁氨基)-3-((1S,3aR,6aS)-2-(2-(2,4-二氯)乙酰基)八氢环戊烯并[c]吡咯-1-甲酰胺)-1-氧代-4-((S)-2-羰基-3-基)丁烷-2-基羧酸的制备
首先,化合物15(0.40mmol)被溶于超干二氯甲烷中,然后依次加入乙酸(0.028g,0.47mmol),叔丁基异腈(0.43mmol)。反应在常温条件下搅拌24小时,减压蒸馏柱层析(二氯甲烷/甲醇=15:1)得到中间体40。
中间体41:(1S,3aR,6aS)-N-((2S)-4-(叔丁氨基)-3-羟基-4-氧代-1-((S)-2-羰基-3-基)丁烷-2-基)-2-(2-(2,4-二氯)乙酰基)八氢环戊烯并[c]吡咯-1-甲酰胺的制备
1M的氢氧化钠溶液被加入(0.5mL)中间体40(0.164mmol)的甲醇溶液中,反应在常温条件下搅拌2小时,用1M盐酸调pH到中性.旋干反应液,残渣用二氯甲烷溶解,用水萃取后旋干反应液得粗产品41直接用于下一步反应。
化合物50:(1S,3aR,6aS)-N-((S)-4-(叔丁氨基)-3,4-氧代-1-((S)-2-羰基-3-基)丁烷-2-基)-2-(2-2,4-二氯)乙酰基)八氢环戊烯并[c]吡咯-1-甲酰胺的制备
在中间体41的超干二氯甲烷溶液中,分批缓慢加入戴斯马丁氧化剂,室温条件下反应4小时,TLC监控反应结束,反应体系过滤,用硫代硫酸钠溶液和饱和碳酸氢钠溶液洗涤有机相,浓缩后用制备色谱分离体系(乙腈/水=50:50)得到白色固体化合物50。
根据实施例1~7中的制备路线,改变原料,制得本发明表1所示的剩余化合物。
表1本发明化合物的结构和表征数据
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
以下通过实验例证明本发明化合物的药理效果。
实验例1:本发明化合物对M
(1)实验方法
将重组SARS-CoV-2M
(2)实验结果
表2化合物对SARS-COV-2M
/>
从表2和图1、图2、图3可以看出,本发明的化合物能够有效抑制SARS-CoV-2M
实验例2:本发明的化合物对SARS-COV-2感染Vero E6细胞导致细胞死亡的抑制实验
(1)实验方法
通过检测化合物对SARS-COV-2感染Vero E6细胞导致的细胞死亡的抑制效果,初步评价化合物的抗病毒活性。具体实验方案是:将Vero E6细胞以细胞密度为2×10
(2)实验结果
表3本发明化合物对SARS-COV-2感染Vero E6细胞导致细胞死亡的抑制活性
/>
/>
注:NT代表未测试细胞活性
从表3可以看出,本发明的化合物能够有效抑制SARS-COV-2感染Vero E6细胞导致的细胞死亡,说明本发明的化合物可以有效抑制SARS-COV-2病毒在细胞内的复制。
实验例3:化合物对Vero E6细胞的毒性实验
(1)实验方法
使用Vero E6细胞进行化合物的细胞毒性评估。具体实验方案是:将Vero E6细胞以细胞密度为2×10
(2)实验结果
表4本发明化合物对Vero E6细胞的毒性
/>
/>
从表4可以看出,本发明的化合物对Vero E6细胞的毒性很低。
实验例4:化合物对SARS-COV-2在人肺泡上皮细胞中复制的抑制实验
(1)实验方法
对于RT-qPCR方法,将人肺泡上皮细胞以8×10
(2)实验结果
测试结果如图4所示,化合物14、15、26、43、44和45均在人肺泡上皮细胞中表现出纳摩尔级的抑制SARS-COV-2复制的活性,优于已报道的活性最高的SARS-COV-2M
实验例5:噬斑法评价化合物抗SARS-COV-2活性(Vero E6细胞)
(1)实验方法
使用噬斑法评价化合物3和39在Vero E6细胞中的抗SARS-COV-2活性。Vero E6以每孔1.0×10
计算公式:抑制率(%)=(病毒对照孔空斑数-样品孔空斑数)/病毒对照孔空斑数*100
计算所得的细胞活性和抑制率,用Graphpad Prism8计算EC
(2)实验结果
表5小分子化合物对SARS-CoV-2的抑制作用
实验结果如表5所示,本发明化合物可以有效抑制Vero E6细胞中SARS-COV-2感染;特别是化合物3,EC
实验例6:化合物对大鼠的体内药代动力学特性评估
(1)给药方案
雄性Sprague-Dawley(SD)大鼠60只,体重200-230g,随机分成3组,每组3只。按照如下表6方案分别灌胃(p.o.)、静脉(i.v.)和腹腔(i.p.)给予系列受试化合物。实验前禁食12h,自由饮水。给药后2h统一进食。
灌胃、静脉和腹腔给药溶液以DMSO/HS15/NaCl(5/3/92,v/v/v)配制。按表6所示给药剂量给予药物,记录给药时间,并在以上设定的时间点经颈静脉采血或其他合适方式,每个样品采集约0.20mL,肝素钠抗凝,采集后放置冰上。并于1小时之内离心分离血浆(离心条件:6800g,6分钟,2-8℃)。血浆样本在分析前存放时则放于-80℃冰箱内。分组及采血时间点见表6,每个时间点3只动物。
表6化合物对大鼠的体内药代动力学特性评估实验方案
(2)实验结果
表7化合物的主要药动学参数
/>
结果如表7所示。本发明对化合物3,14,15,26,39,40,43,44和45进行了药代动力学研究。其中,化合物3的口服暴露量为2293h*ng/mL,生物利用度为55.1%。化合物14的腹腔暴露量为11581h*ng/mL,生物利用度为78.0%;口服暴露量为1665h*ng/mL,生物利用度为11.2%。化合物15的腹腔注射给药暴露量为12166h*ng/mL,生物利用度为62.3%;口服暴露量为2843h*ng/mL,生物利用度为14.6%。化合物26的口服暴露量为842h*ng/mL,生物利用度为7.2%。化合物39的口服暴露量为14586h*ng/mL,生物利用度为14.7%。化合物40的口服暴露量为2888h*ng/mL,生物利用度为22.1%。化合物43的口服暴露量为258h*ng/mL,生物利用度为4.8%。化合物44的口服暴露量为381h*ng/mL,生物利用度为4.1%。化合物45的口服暴露量为968h*ng/mL,生物利用度为5.1%。
实验结果表明,本发明化合物在大鼠的体内具有良好的药代动力学性质。
实验例7:化合物对大鼠的体内安全性的初步评价
(1)实验方法
将化合物溶解在5%(v/v)DMSO(Sigma-Aldrich),3%(v/v)HS15(GLPBIO)和92%生理盐水中。SPF SD大鼠(年龄:7-11周)雌190-220克)雄(体重200-230克)各半。按表8的给药方案进行试验,并对所有动物进行临床观察。并在实验结束时,收集心脏,肝,脾,肺,肾和给药部位的样品。试验结果如表8所示。
(2)实验结果
表8对大鼠的体内安全性的初步评价
实验结果显示,本发明化合物对大鼠的体内安全性良好。
实验例8:化合物对转基因小鼠的体内抗SARS-COV-2感染的活性研究
(1)实验方案
人源化血管紧张素转化酶2(ACE2)转基因小鼠(年龄:8-10周)购自江苏集萃药康生物科技有限公司(#T037659。将化合物溶于5%(v/v)DMSO(Sigma-Aldrich),3%(v/v)HS15(GLPBIO)和92%生理盐水中。按表9的试验方案进行SARS-CoV-2(stain107)滴鼻感染和给药。观察所有小鼠并每天监测其体重直至处死。病毒感染后第1(1dpi)、3(3dpi)和5(5dpi)天,收集肺组织(n=3,每个dpi组)用于病毒载量检测、H&E组织病理学分析、代表性炎性细胞因子和趋化因子测定及炎性细胞(中性粒细胞和巨噬细胞)计数。
表9化合物对转基因小鼠体内抗SARS-COV-2感染的活性研究方案
肺部病毒载量检测的具体实验方案是:使用TRIzol
肺部组织病理学分析的具体实验方案是:将肺部组织用4%多聚甲醛固定至少7天,包埋在石蜡中并切成3μm的切片。将切片用苏木精和曙红(H&E)染色,并通过光学显微镜进行分析。根据组织学特征(肺泡间隔增厚,出血,炎性细胞浸润等)评估肺损伤。
肺部代表性炎性细胞因子和趋化因子测定的具体实施方案是:使用PrimeScript
表10测定代表性炎性细胞因子和趋化因子的引物序列
肺部测定炎性细胞(中性粒细胞和巨噬细胞)数量的具体实施方案是:将小鼠肺部组织在4%多聚甲醛中固定至少7天,然后按照标准程序将石蜡包埋并切成4μm切片。在二甲苯中进行去石蜡,抗原回收和封闭后,将肺切片与大鼠单克隆抗体F4/80(Huabio,1:100)或兔多克隆抗体Ly6G(Servicebio,1:300)在4℃孵育过夜,然后与辣根过氧化物酶(HRP)偶联的山羊抗大鼠二抗或HRP偶联的山羊抗兔二抗在室温下反应1小时,以根据酪酰胺信号放大(TSA)催化Cy3-酪胺和Cy5-酪胺并放大染色信号。用DAPI对细胞核染色后,所有切片均使用LEICA DMI 4000B显微镜(德国)拍照,并通过ImageJ软件(美国NIH)和FlowJo软件(美国BD)进行分析。为了半定量地测量巨噬细胞和嗜中性白细胞的浸润,通过光学显微镜检查每个肺切片中5个任意选择的肺实质区域,以观察嗜中性白细胞或巨噬细胞的存在。该评估以盲法进行。每只动物的累积得分表示为每100个视野的阳性视野数(%)。
上述实验以安慰剂作为对照。该安慰剂为与受试药物剂型相同、但是不含药物活性成分的制剂。
(2)实验结果
肺部病毒载量检测实验结果如图5所示,口服和腹腔给予化合物14及腹腔给予化合物15均可以有效降低SARS-COV-2感染的转基因小鼠肺部的病毒载量。
肺部组织病理学分析实验结果如图6所示,口服和腹腔给予化合物14及腹腔给予化合物15均可以有效改善SARS-COV-2感染的转基因小鼠肺部的病理损伤。
肺部代表性炎性细胞因子和趋化因子测定实验结果如图7所示,口服和腹腔给予化合物14及腹腔给予化合物15可以有效降低肺部趋化因子配体10(CXCL10)和β型干扰素(IFN-β)的基因表达水平。
肺部测定炎性细胞(中性粒细胞和巨噬细胞)数量的实验结果如图8所示,口服和腹腔给予化合物14及腹腔给予化合物15均可以有效降低SARS-COV-2感染的转基因小鼠肺部的中性粒细胞(NEU)和巨噬细胞(MAC)的数量。
实验结果表明,本发明化合物能够有效抵抗转基因小鼠的体内SARS-COV-2感染。
综上,本发明提供了一种式I所示的新型冠状病毒主蛋白酶抑制剂及其制备方法和用途。式I所示化合物能够有效抑制SARS-CoV-2M