一种辅酶A半胱胺化杂质的精制方法及其合成方法
文献发布时间:2023-06-19 19:28:50
技术领域
本发明涉及一种辅酶A半胱胺化杂质的精制方法及其合成方法。
背景技术
辅酶A分子是由泛酸、β-巯基乙胺和3’,5’-二磷酸腺苷组成。分子式C
辅酶A,是生物体内乙酰化酶的辅酶,参与三羧酸循环,起着传递乙酰基的作用,对糖、蛋白质和脂类的代谢起重要作用。它广泛地存在于动物、植物、微生物组织和细胞中,在动物的肝脏含量最高,心脏、肾上腺次之,临床上主要应用在脂肪肝、肝炎、肝昏迷、冠状动脉粥样硬化、心肌梗死、肾病综合征、尿毒症、新生儿缺氧、糖尿病和酸中毒等疾病的辅助治疗,也可以用于放射性伤害的保护,减慢肌萎缩的发展进程等。
就一个药品而言,其中所含的少量杂质是引发药品副作用最重要的原因,因此对其纯度的检查是保证药品安全有效性的重要基础之一,而纯度检查的内容,根据各个药物的性质和特点有些不同,但基本上均要涉及各自的“有关物质”检查研究。辅酶多种有关物质主要来自生产过程中的工艺杂质和由于其不稳定而产生的降解产物,尽管目前辅酶A的纯化工艺已经有了很大进步,但工艺杂质仍是辅酶A有关物质的重要来源,这主要是由于辅酶A制造过程主要依靠微生物合成方法,菌体发酵代谢后的成分复杂,而且辅酶A分子中的巯基易被空气、过氧化氢、碘等氧化成无活性的二硫化物,这些产物与药物本身的性质可能非常近似,从而给纯化造成了一定的难度。目前市场上可以购买获得的辅酶A杂质只有氧化型辅酶A和3’-脱磷辅酶A,而辅酶A半胱胺化杂质还未有生产,该杂质是由于工艺过程中会加入半胱氨酸,同时由于辅酶A生物体内合成途径中半胱氨酸的存在,也会增加半胱氨酸的积累,其脱羧后的产物半胱胺与辅酶A反应后生成有关物质,该杂质属于工艺杂质,在氧、热、碱的破坏下会增加该杂质。在辅酶A原料药以及注射用辅酶A中都可能存在该杂质,目前没有辅酶A原料药及其制剂中该种杂质合成方法分析的研究,缺少为辅酶A的生产和用药安全提供检测方法及判定依据,因此这对于辅酶A相关产品的质量研究具有重要意义。
发明内容
本发明提供一种辅酶A半胱胺化杂质的精制方法及其合成方法。本发明首次采用成本低廉和简单的工艺反应条件,快速制得纯度高和收率高的产物;采用本发明的精制方法可以得到HPLC纯度高和收率高的辅酶A半胱胺化杂质。
本发明通过以下技术方案解决上述技术问题:
本发明第一方面提供了一种辅酶A半胱胺化杂质的精制方法,其包括下述步骤:采用高效液相色谱方法,将待测物在色谱柱上进行梯度洗脱,即可;
所述的待测物包括辅酶A半胱胺化杂质;
所述的梯度洗脱中所用的流动相为流动相A和流动相B;
所述的流动相A为醋酸铵水溶液和甲醇;
所述的醋酸铵水溶液与所述的甲醇的体积比为(8~12):1;
所述的醋酸铵水溶液的摩尔浓度为10~60mM;
所述的流动相B为甲醇或乙腈;
本发明第一方面中,所述的辅酶A半胱胺化杂质的HPLC纯度可为60%~90%,优选70%~85%,更优选85%~90%。
所述的辅酶A半胱胺化杂质优选以浓度≤10mg/mL的溶液进行进样。当所述的辅酶A半胱胺化杂质的浓度>10mg/mL时,可进行稀释后再进样。所述的稀释的溶剂为水,例如注射用水。
本发明第一方面中,所述的待测物还可包括辅酶A、半胱胺和辅酶A有关物质中的一种或多种。
所述的辅酶A有关物质可以选自如下化合物中的一种或多种:
经过所述的梯度洗脱,所述的待测物中的杂质的去除率可为10%以上。
本发明第一方面中,所述的辅酶A半胱胺化杂质优选通过如下合成方法制得:
所述的辅酶A半胱胺化杂质的合成方法包括如下步骤:在催化剂的存在下,将辅酶A与半胱胺在溶剂中进行反应,即可;
所述的辅酶A半胱胺化杂质的制备方法中,所述的辅酶A可为常规的辅酶A,优选纯度为50%以上的辅酶A。
所述的溶剂可为此类反应常规的溶剂,优选水,例如纯化水或注射用水。
所述的溶剂的用量只要不影响反应进行即可。所述的辅酶A与所述的溶剂的质量体积比可为1.2~5mg/mL,优选1.3~3.5mg/mL,例如2.5mg/mL。
所述的半胱胺与所述的溶剂的质量体积比可为1.5~2.5mg/mL,例如2.5mg/mL。
所述的半胱胺与所述的辅酶A的摩尔比可为此类反应常规的摩尔比,优选(8~12):1,例如10:1。
所述的催化剂可为此类反应常规的催化剂,优选过氧化氢,更优选浓度为1%的过氧化氢。
所述的催化剂与所述的辅酶A的摩尔比可为此类反应常规的摩尔比,优选(18~22):1,例如20:1。
所述的反应的温度可为此类反应常规的温度,优选37℃~60℃,例如38℃。
所述的反应优选以振荡方式进行。所述的振荡的频率可为100~150次/min,例如150次/min。所述的振荡的冲程可为15mm。所述的反应所用的仪器优选水浴往复摇床。所述的反应的进程可以采用本领域常规的方式进行监测(例如TLC、HPLC),一般以原料辅酶A消失或者不再反应作为反应的终点。所述的反应的时间可为20min以上,例如1小时。
本发明第一方面中,所述的梯度洗脱可以为五段式梯度洗脱。
当所述的梯度洗脱为五段式梯度洗脱时,在第一梯度中,所述的流动相A的体积百分比可为100%至100%,所述的流动相B的体积百分比可为0%至0%;
在第二梯度中,所述的流动相A的体积百分比可为100%至94%,所述的流动相B的体积百分比可为0%至6%;
在第三梯度中,所述的流动相A的体积百分比可为94%至94%,所述的流动相B的体积百分比可为6%至6%;
在第四梯度中,所述的流动相A的体积百分比可为94%至75%,所述的流动相B的体积百分比可为6%至25%;
在第五梯度中,所述的流动相A的体积百分比可为75%至50%,所述的流动相B的体积百分比可为25%至50%。
当所述的梯度洗脱为五段式梯度洗脱时,在第一梯度中,所述的洗脱时长可为4-8min,例如6min;
在第二梯度中,所述的洗脱时长可为3~7min,例如5min;
在第三梯度中,所述的洗脱时长可为4~8min,例如6min;
在第四梯度中,所述的洗脱时长可为15~22min,例如18min;
在第五梯度中,所述的洗脱时长可为3~7min,例如5min。
所述的洗脱时长是指该梯度洗脱段中洗脱时间起点与终点的差值。
所述的梯度洗脱的程序优选为如下的程序:
。
本发明第一方面中,所述的流动相A中,所述的醋酸铵水溶液与所述的甲醇的体积比可为(8~11):1,例如10:1。
本发明第一方面中,所述的醋酸铵水溶液的摩尔浓度可为10~30mM,例如20mM。
本发明第一方面中,所述的流动相A的pH可为6.0~7.5,例如6.5。
本发明中所述的流动相A的pH为6.0~7.5时,所述的辅酶A半胱胺化杂质的峰形为对称的峰型。
本发明第一方面中,所述的流动相B可为甲醇。
本发明第一方面中,较佳地,所述的梯度洗脱的程序开始之前,先使用10%甲醇水溶液冲洗平衡色谱柱,再用流动相A冲洗平衡色谱柱。
其中,所述的10%甲醇水溶液冲洗的时间可为5~10min;所述的10%甲醇水溶液冲洗平衡色谱柱的时间长短可依高效液相色谱仪厂家型号的不同进行相应调整。
所述的梯度洗脱中,优选收集保留时间为15~25min的洗脱液;更优选收集保留时间为10~13min的洗脱液。
所述的梯度洗脱的程序结束后,还包括清洗和保存色谱柱的步骤。
本发明第一方面中,将所述的待测物进行所述的梯度洗脱前,优选采用10%甲醇冲洗色谱柱10~15min,流速为1~2mL/min;再采用85%甲醇冲洗色谱柱10min,流速2mL/min。
本发明第一方面中,所述的色谱柱可为制备色谱柱。
本发明第一方面中,所述的高效液相色谱所采用的仪器优选为Varian SD-1高压液相制备系统。
其中,所述的色谱柱的填料可为耐水性填料。
所述的耐水性填料优选十八烷基硅烷键合硅胶。
所述的色谱柱的填料的孔径可为7~10nm。
所述的色谱柱的填料的粒径可为10μm。
所述的色谱柱的长度可为25cm。
所述的色谱柱的直径可为20mm。
所述的色谱柱优选填料为十八烷基硅烷键合硅胶的色谱柱,填料的粒径为10μm,长度为25cm,直径为20mm;例如SHIM-PACK PRC-ODS(H)(20mm*25cm,10μm)。
所述的流动相的流速可为8~12mL/min,例如10mL/min。
所述的高效液相色谱中所用的检测器可为紫外检测器。所述的紫外检测器的检测波长可为259nm。
本发明第二方面提供了一种辅酶A半胱胺化杂质的合成方法,其包括如下步骤:在催化剂的存在下,将辅酶A与半胱胺在溶剂中进行反应,即可;
本发明第二方面所述制备方法中,所述的辅酶A半胱胺化杂质的合成方法中的条件和操作均同本发明第一方面中辅酶A半胱胺化杂质的合成方法所述。
本发明中,“制备色谱柱”是指长度为50mm~500mm,直径为10mm~100mm的色谱柱。
在不违背本领域常识的基础上,上述各优选条件,可任意组合,即得本发明各较佳实例。
本发明所用试剂和原料均市售可得。
本发明的积极进步效果在于:本发明首次采用成本低廉和简单的工艺反应条件,快速制得纯度和收率高的辅酶A半胱胺化杂质;采用本发明的精制方法可以得到HPLC纯度高和收率高的辅酶A半胱胺化杂质。
附图说明
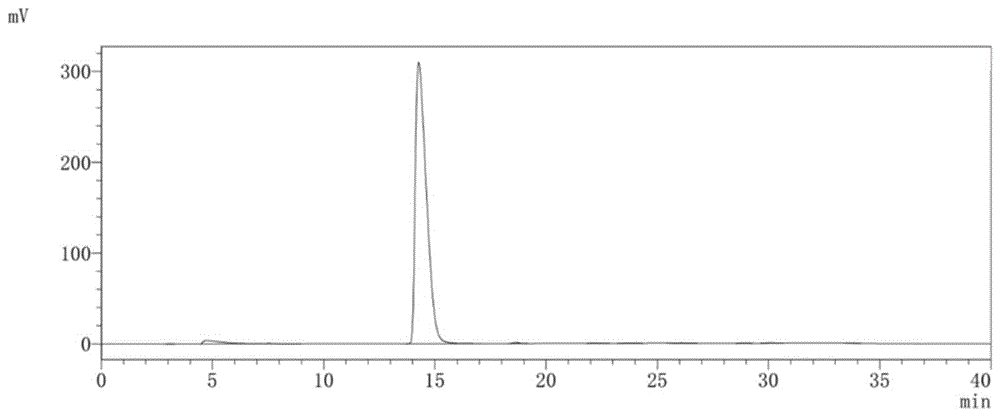
图1为辅酶A半胱胺化杂质粗品溶液的液相图。
图2为辅酶A半胱胺化杂质制备纯化后的液相图。
图3为辅酶A半胱胺化杂质的一级质谱图。
图4为辅酶A半胱胺化杂质的二级质谱图。
具体实施方式
下面通过实施例的方式进一步说明本发明,但并不因此将本发明限制在所述的实施例范围之中。下列实施例中未注明具体条件的实验方法,按照常规方法和条件,或按照商品说明书选择。
实施例中,产品的来源和规格如下:
辅酶A水合物(Sigma,≥85%HPLC);甲醇(HPLC,Fisher);乙酸铵(HPLC级,Fluka);30%双氧水(AR,国药);半胱胺(Sigma TCI,>95.0%)。
实施例中,所使用的仪器型号及来源如下:
高效液相色谱仪(LC2030C,日本岛津科技有限公司);制备液相系统(PrepStarSD-1);Waters超高效液相色谱-离子淌度-四极杆飞行时间质谱(G2-XS);WNE水浴摇床;SHIM-PACK PRC-ODS(H)制备柱(20mm*25cm,10μm);Dikma色谱柱(250*4.6mm,5μm);pH计(FE28,Mettler Toledo);电子天平(T1000Y,常熟市双杰测试仪器厂);超声波清洗机(深圳市钰洁清洗设备有限公司);真空冷冻干燥机(上海东富龙科技股份有限公司)。
实施例中,所使用的数据处理软件为LabSolutions化学工作站。
实施例1
辅酶A半胱胺化杂质的制备方法:
起始原料采用辅酶A水合物,纯度≥85%,为HPLC级别,10mL反应体系:25mg辅酶A和25mg半胱胺于8mL水中,再加入2mL 1%过氧化氢溶液混匀并充分溶解。水浴往复摇床设定温度为38℃,振荡频率150次/min,1小时后停止。
实施例2
HPLC法检测辅酶A半胱胺化杂质粗品溶液和纯化后产品溶液中半胱胺化杂质纯度:
实验条件如下所示:
仪器:LC2030C高效液相色谱仪
色谱柱:Dikma C18(250*4.6mm,5μm)
流动相:流动相A为20mM醋酸铵:甲醇=94:6,流动相B为甲醇
流速为1.0mL/min,检测波长为259nm,柱温:25℃,进样量:10μL,检测器:紫外检测器。洗脱梯度见下表,百分比为体积百分比。
采用以上方法测定的辅酶A半胱胺化杂质粗品溶液纯度达84%以上。HPLC图如图1所示。
实施例3
对实施例1得到的辅酶A半胱胺化杂质粗品溶液进行纯化,具体步骤如下:
仪器:Varian SD-1高压液相制备系统
色谱柱:SHIM-PACK PRC-ODS(H)(20mm*25cm,10μm)
流动相:流动相A为20mM醋酸铵:甲醇=91:9,pH=6.5,流动相B为甲醇,C相为辅酶A半胱胺化杂质粗品溶液(含有未反应完全的原料)。
流速为10mL/min,检测波长为259nm。洗脱梯度见下表,百分比为体积百分比。
收集保留时间为10~13min的洗脱液,采用实施例2的方法测得辅酶A半胱胺化杂质HPLC的纯度为97%,经过两次冷冻干燥后,收率为95.8%。HPLC图如图2所示。
实施例4
对辅酶A半胱胺化杂质进行质谱检测:
采用Waters超高效液相色谱-离子淌度-四极杆飞行时间质谱(G2-XS)对得到的辅酶A半胱胺化杂质进行一级和二级质谱分析,测定条件为:采用电喷雾电离(ESI)源,正离子电离模式,四级杆扫描范围为50~1000m/z,二级质谱碰撞能量25~30eV。
检测的结果为:一级质谱分子离子峰[M+H]