人小脑蛋白配体和包含其的双官能化合物
文献发布时间:2023-06-19 19:28:50
本申请是申请日为2018年1月31日、申请号为201880022865.4、发明名称为“人小脑蛋白配体和包含其的双官能化合物”的中国发明专利申请的分案申请。
相关申请的交叉引用
本公开要求2017年1月31日提交的美国临时申请号62/452,972的优先权,该临时申请以引用方式整体并入本文。
以引用方式并入
作为美国专利申请公布号2017/0065719公布的2016年8月5日提交的美国专利申请序列号15/230,354;2017年11月1日提交的美国专利申请序列号15/801,243;2016年7月11日提交的美国专利申请15/206,497;2016年7月13日提交的美国专利申请15/209,648;2017年10月11日提交的美国专利申请序列号15/730,728;2017年12月1日提交的美国专利申请序列号15/829,541;2018年1月26日提交的美国专利申请序列号15/881,318;作为美国专利申请公布号2015/0291562公布的2015年4月14日提交的美国专利申请序列号14/686,640;作为美国专利申请公布号2016/0058872公布的2015年7月6日提交的美国专利申请序列号14/792,414;作为美国专利申请公布号2014/0356322公布的2014年7月11日提交的美国专利申请序列号14/371,956;以及作为美国专利申请公布号2016/0272639公布的2016年3月18日提交的美国专利申请序列号15/074,820以引用方式整体并入本文。此外,本文引用的所有参考文献都以引用的方式整体并入本文。
技术领域
本说明书提供了基于酰亚胺的化合物,包括包含其的双官能化合物,以及相关使用方法。双官能化合物可用作靶向泛素化的调节剂,尤其是关于多种多肽和其他蛋白质,它们被根据本公开的双官能化合物降解和/或以其他方式抑制。
背景技术
大多数小分子药物在紧密和明确界定的口袋中结合酶或受体。另一方面,由于其大接触表面以及所牵涉的浅沟或平坦界面,蛋白质-蛋白质相互作用众所周知难以使用小分子靶向。E3泛素连接酶(其中数百种在人中已知)赋予关于泛素化的底物特异性,并且因此,由于其对于某些蛋白质底物的特异性,是比一般蛋白酶体抑制剂更有吸引力的治疗靶。E3连接酶配体的开发已证明是挑战性的,部分是由于它们必须破坏蛋白质-蛋白质相互作用的事实。然而,最近的发展已提供了与这些连接酶结合的特异性配体。例如,自从发现第一个小分子E3连接酶抑制剂nutlin以来,已报道了靶向E3连接酶的另外化合物,但该领域仍是发展不完全的。
一种具有治疗潜力的E3连接酶是希佩尔-林道(Hippel-Lindau,VHL)肿瘤抑制因子。VHL包含底物识别亚基/E3连接酶复合物VCB(其包含延伸蛋白B和C)以及包含Cullin-2和Rbx1的复合物。VHL的主要底物是缺氧诱导因子1α(HIF-1α),其是响应低氧水平上调基因例如促血管生成生长因子VEGF和红血细胞诱导细胞因子促红细胞生成素的转录因子。我们生成了针对E3连接酶的底物识别亚基的希佩尔-林道蛋白(VHL)的第一个小分子配体VCB(癌症、慢性贫血和局部缺血中的重要靶标),并且获得晶体结构,证实该化合物模拟转录因子HIF-1α(VHL的主要底物)的结合模式。
人小脑蛋白(cereblon)是人中由CRBN基因编码的蛋白质。CRBN直向同源物从植物到人都是高度保守的,这强调了它的生理重要性。人小脑蛋白与受损的DNA结合蛋白1(DDB1)、Cullin-4A(CUL4A)和cullin调节剂1(ROC1)形成E3泛素连接酶复合物。这种复合物使许多其他蛋白质泛素化。通过尚未完全阐明的机制,靶蛋白的人小脑蛋白泛素化导致成纤维细胞生长因子8(FGF8)和成纤维细胞生长因子10(FGF10)的水平增加。FGF8依次又调节许多发育过程,例如肢体和听泡形成。净结果是这种泛素连接酶复合物对于胚胎中的肢体生长是重要的。在不存在人小脑蛋白的情况下,DDB1与DDB2形成复合物,所述DDB2充当DNA损伤结合蛋白。
已被批准用于治疗多种免疫适应症的沙立度胺也已被批准用于治疗某些赘生疾病,包括多发性骨髓瘤。除多发性骨髓瘤之外,沙立度胺和其若干类似物当前还处于用于治疗多种其他类型癌症的研究中。虽然沙立度胺抗肿瘤活性的确切机理仍在探明,但已知其抑制血管生成。最近讨论酰亚胺生物学的文献包括Lu等人,Science 343,305(2014)和
重要的是,已知沙立度胺和其类似物(例如泼莫灵胺(pomolinamiode)和来纳灵胺(lenalinomide))结合人小脑蛋白。这些药剂结合到人小脑蛋白,从而改变复合物诱导Ikaros(IKZF1)和Aiolos(IKZF3)(多发性骨髓瘤生长所必需的转录因子)泛素化和降解的特异性。事实上,人小脑蛋白的表达较高与酰亚胺药物治疗多发性骨髓瘤的功效提高有关。
BRD4因其作为多种疾病、尤其是癌症的新型靶标的巨大潜力而受到学术界和制药业的广泛关注。BRD4属于溴结构域和额外末端结构域(BET)家族,其特征在于N末端的两个溴结构域(BD结构域)和C末端的额外末端结构域(ET结构域)(J.Shi等人,Molecular cell,54(2014)728-736和A.C.Belkina等人,Nat.Rev.Cancer,12(2012)465-477)。两个BD结构域识别并与组蛋白N末端尾部的乙酰化赖氨酸残基相互作用;ET结构域尚未完全表征,并且在很大程度上被认为在募集多种转录调节因子时起到支架功能的作用。因此,BRD4通过将相关转录调节剂募集到特定基因组位点而在调节基因表达中发挥关键作用。一些研究已确定BRD4优先定位于超级增强子区域,这些区域通常位于重要致癌基因(例如c-MYC、Bcl-xL和BCL-6)的上游,并且在调节它们的表达方面起关键作用(J.Loven等人,Cell,153(2013)320-334和B.Chapuy等人,Cancer Cell,24(2013)777-790)。由于其在调节重要致癌基因表达中的关键作用,BRD4在多种癌症类型(包括中线癌、AML、MM、BL和前列腺癌)中成为有前景的治疗靶标(J.Loven等人,Cell,153(2013)320-334;J.Zuber等人,Nature,478(2011)524-528;J.E.Delmore等人,Cell,146(2011)904-917;J.A.Mertz等人,PNAS,108(2011)16669-16674;A.Wyce等人,Oncotarget,4(2013)2419-2429;I.A.Asangani等人,Nature,510(2014)278-282;以及C.A.French等人,Oncogene,27(2008)2237-2242)。BRD4在特定致癌基因附近的基因组位点的独特高占据率提供了潜在的治疗窗口,该治疗窗口允许特异性靶向肿瘤细胞,同时保留正常组织。具体地讲,BRD4可作为靶向c-MYC的替代策略,其有助于大多数人类癌症的发展和维持,但仍然是无成药性的(J.E.Delmore等人,Cell,146(2011)904-917;J.A.Mertz等人,PNAS,108(2011)16669-16674;M.G.Baratta等人,PNAS,112(2015)232-237;以及M.Gabay等人,Cold Spring Harb Perspect Med.(2014)4:a014241)。
小分子BRD4抑制剂如JQ1、iBET和OTX15的开发已在各种癌症(包括BL)的临床前模型中被证实具有有前景的治疗潜力(J.Loven等人,Cell,153(2013)320-334;B.Chapuy等人,Cancer Cell,24(2013)777-790;J.E.Delmore等人,Cell,146(2011)904-917;J.A.Mertz等人,PNAS,108(2011)16669-16674;I.A.Asangani等人,Nature,510(2014)278-282;M.G.Baratta等人,PNAS,112(2015)232-237;M.Boi等人,Clin.Cancer Res.,(2015)21(7):1628-38;以及A.Puissant等人,Cancer discovery,3(2013)308-323)。实际上,BRD4抑制剂在不同的小鼠肿瘤模型中显示出具有良好耐受性的各种抗肿瘤活性,并且不足为奇的是,对BRD4抑制剂如JQ1的高度敏感性与不同肿瘤类型(包括c-MYC驱动的BL)中高水平的c-MYC和N-MYC相关。几乎所有BL病例都含有c-myc基因易位,使其处于位于IgH上游的超级增强子的控制下,从而驱动异常高水平的c-MYC表达、肿瘤发展和维持(K.Klapproth等人,British journal of haematology,149(2010)484-497)。
目前,四种BET溴结构域抑制剂处于I期临床试验中,主要集中于中线癌和血液恶性肿瘤(CPI-0610、NCT01949883;GSK525762、NCT01587703;OTX015、NCT01713582;TEN-010、NCT01987362)。使用BRD4抑制剂的临床前研究证实了其在抑制BL细胞系中c-MYC和增殖方面的价值,但IC
本领域持续需要有效治疗疾病,尤其是增生和癌症,例如多发性骨髓瘤。然而,非特异性作用以及完全没有能力靶向和调节某些类别的蛋白质(例如转录因子)仍然是开发有效抗癌剂的障碍。因此,非常适用作治疗剂的将是利用或增强人小脑蛋白底物特异性并且同时“能调节”的小分子治疗剂,以便能够靶向各种各样的蛋白质并且调节其而具有特异性。
发明内容
本公开描述了双官能化合物及其使用方法,所述双官能化合物用于将内源蛋白质募集到E3泛素连接酶上以进行降解。确切地说,本公开提供了双官能或蛋白分解靶向嵌合(PROTAC)化合物,其适用作多种多肽和其他蛋白质的靶向泛素化调节剂,所述多肽和其他蛋白质在靶向泛素化之后被如本文所述的双官能化合物降解和/或以其他方式抑制。本文所提供化合物的优势在于,与来自几乎任何蛋白质类别或家族的靶向多肽的降解/抑制一致,可以存在广谱药理学活性。此外,本说明书提供了使用有效量的如本文所述化合物治疗或改善疾病病状如癌症(例如,多发性骨髓瘤)的方法。
因此,在一个方面,本公开提供了如本文所述的基于酰亚胺的新型化合物。
在另一方面,本公开提供了双官能或PROTAC化合物,该化合物包含E3泛素连接酶结合部分(即,E3泛素连接酶的配体或“ULM”基团)和靶蛋白结合部分(即,蛋白质/多肽靶向配体或“PTM”基团),使得靶蛋白/多肽被置于泛素连接酶附近以实现该蛋白的降解(和抑制)。在一个优选的实施方案中,ULM是人小脑蛋白E3泛素连接酶结合部分(即,“CLM”)。例如,双官能化合物的结构可以描绘为:
如本文中所图示的PTM和CLM部分的相应位置以及其数目仅为了举例而提供,并非旨在以任何方式限制所述化合物。如本领域技术人员所理解的,可以合成如本文所述的双官能化合物,使得各个官能部分的数目和位置可以根据需要而变。
在某些实施方案中,双官能化合物还包含化学接头(“L”)。在该实例中,双官能化合物的结构可以描绘为:
其中PTM是蛋白质/多肽靶向部分,L是接头,并且CLM是人小脑蛋白E3泛素连接酶结合部分。
在某些优选的实施方案中,E3泛素连接酶是人小脑蛋白。因此,在某些其他实施方案中,双官能化合物的CLM包含化学部分,例如酰亚胺、酰胺、硫代酰胺、硫代酰亚胺衍生部分。在其他实施方案中,CLM包含邻苯二酰亚胺基团或其类似物或衍生物。在另外的实施方案中,CLM包含邻苯二酰亚胺-戊二酰亚胺基团或其类似物或衍生物。在另外的实施方案中,CLM包含由沙立度胺、来那度胺、泊马度胺和其类似物或衍生物组成的组的成员。
在某些实施方案中,如本文所述的化合物包含多个CLM、多个PTM、多个化学接头或它们的组合。
在本文所述的任何方面或实施方案中,ULM(泛素化连接酶调节剂)可以是希佩尔-林道E3泛素连接酶(VHL)结合部分(VLM),或人小脑蛋白E3泛素连接酶结合部分(CLM),或小鼠双微体2同源物(MDM2)E3泛素连接酶结合部分(MLM),或IAP E3泛素连接酶结合部分(即“ILM”)。在本文所述的任何方面或实施方案中,双官能化合物包含至少一个另外的E3连接酶结合部分,其选自VLM、VLM’、CLM、CLM’、MLM、MLM’、ILM、ILM’或它们的组合。例如,可存在至少1个、2个、3个、4个或5个另外的E3连接酶结合部分。
在另一方面,本说明书提供了治疗组合物,其包含有效量的如本文所述化合物或其盐形式和药学上可接受的载体。治疗组合物调节患者或受试者(例如动物,例如人)中的蛋白质降解,并且可以用于治疗或改善通过降解的蛋白质调节的疾病状态或状况。在某些实施方案中,如本文所述的治疗组合物可用于实现所关注蛋白质的降解,以治疗或改善疾病,例如癌症。在又一方面,本公开提供了泛素化/降解细胞中的靶蛋白的方法。在某些实施方案中,该方法包括施用如本文所述的双官能化合物,该双官能化合物包含优选通过如本文另外所述的接头部分连接的CLM和PTM,其中CLM与PTM偶联且其中CLM识别泛素路径蛋白质(例如,泛素连接酶,优选E3泛素连接酶,如人小脑蛋白)并且PTM识别靶蛋白,使得当靶蛋白被置于泛素连接酶附近时,将发生靶蛋白的降解,从而实现靶蛋白的降解/靶蛋白效应的抑制及蛋白质水平的控制。通过本公开提供的蛋白质水平的控制提供了疾病状态或状况的治疗,其通过降低患者细胞中该蛋白质的水平而通过靶蛋白调节。
在另一方面,本说明书提供了一种用于评估(即,测定和/或测量)CLM结合亲和力的方法。在某些实施方案中,该方法包括提供目的测试剂或化合物,例如具有酰亚胺部分的药剂或化合物,例如邻苯二酰亚胺基团、邻苯二酰亚胺-戊二酰亚胺基团、衍生化沙立度胺、衍生化来那度胺或衍生化泊马度胺,以及对测试剂或测试化合物的人小脑蛋白结合亲和力和/或抑制活性与已知结合和/或抑制人小脑蛋白活性的药剂或化合物进行比较。
在另一方面,本说明书提供了用于治疗或改善受试者或患者(例如动物,例如人类)的疾病、病症或其症状的方法,该方法包括向有需要的受试者施用组合物,该组合物包含有效量(例如治疗有效量)的本文所述化合物或其盐形式,和药学上可接受的载体,其中所述组合物能够有效治疗或改善受试者的疾病或病症或其症状。
在另一方面,本说明书提供了使用根据本公开的化合物鉴别生物系统中的所关注蛋白质降解的影响的方法。
前述一般实用领域仅为了举例而给出,并且不希望对本公开的范围和所附权利要求书构成限制。根据本权利要求、说明书和实例,本领域普通技术人员将了解与本公开的组合物、方法和方法相关的另外目的和优点。例如,本发明的各个方面和实施方案可以众多组合利用,所有这些组合都由本说明书明确地加以考虑。这些额外的优势目标和实施方案明确地包含在本公开的范围内。本文用于阐明本发明的背景并且在特定情况下提供关于实践的另外细节的出版物和其他材料以引用方式并入。
附图说明
并入本说明书中且构成本说明书的一部分的附图示出了本公开的若干实施方案,并且连同本说明书一起用于解释本发明的原理。附图仅用于示出本发明的实施方案的目的,并且不应理解为对本发明的限制。根据与附图结合的下述详细描述,本发明的另外的目的、特征和优点将变得显而易见,所述附图示出了本发明的例示性实施方案,其中:
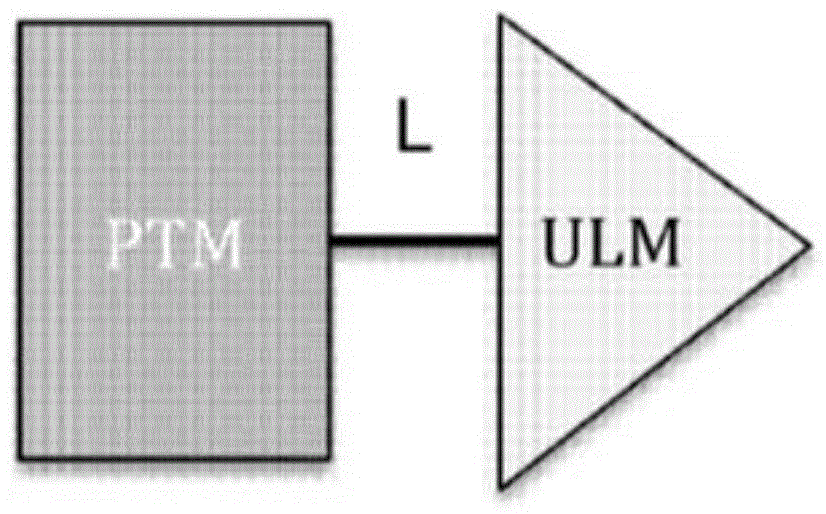
图1A和图1B。PROTAC功能的一般原理的图示。图1A示例性PROTAC包含蛋白质靶向部分(PTM;深色阴影矩形)、泛素连接酶结合部分(ULM;浅色阴影三角形)和任选的将PTM偶联或束缚至ULM的接头部分(L;黑线)。图1B示出了如本文所述的PROTAC的功能用途。简而言之,ULM识别并结合特定的E3泛素连接酶,并且PTM结合并募集靶蛋白,使其紧密接近E3泛素连接酶。典型地,E3泛素连接酶与E2泛素结合的蛋白质复合,且单独或通过E2蛋白质催化泛素(深色圆圈)连接到靶蛋白上的赖氨酸,此连接是通过异肽键达成。然后靶向聚泛素化蛋白质(最右侧)以便细胞的蛋白酶体机器使其降解。
具体实施方式
下述是提供以帮助本领域技术人员实践本公开的详细描述。本领域普通技术人员可以在本文描述的实施方案中进行修改和变化,而不脱离本公开的实质或范围。本文提及的所有出版物、专利申请、专利、附图和其他参考文献均全文以引用方式明确并入。
本发明描述了组合物和方法,所述组合物和方法涉及E3泛素连接酶蛋白(例如人小脑蛋白)一旦与靶蛋白通过结合E3泛素连接酶蛋白和靶蛋白的双官能或嵌合构建体邻近定位就会使靶蛋白泛素化的惊人和意外发现。因此,本公开提供了此类化合物和组合物,其包含偶联至蛋白质靶标结合部分(“PTM”)的E3泛素连接酶靶向部分(“ULM”),这将引起所选靶蛋白的泛素化,从而导致靶蛋白被蛋白酶体降解(参见图1A和图1B)。本公开还提供了组合物文库及其用途。
在某些方面,本公开提供了包含配体诸如小分子配体(即,分子量低于2,000、1,000、500或200道尔顿)的化合物,所述配体能够与泛素连接酶诸如IAP、VHL、MDM2或人小脑蛋白结合。该化合物还包含能够与靶蛋白结合的部分,以这样的方式使得靶蛋白置于泛素连接酶附近,以实现该蛋白的降解(和/或抑制)。除上述之外,小分子还可以意指该分子是非肽基的,即,它一般不视为肽,例如包含少于4、3或2个氨基酸。根据本说明书,PTM、ULM或PROTAC分子可以是小分子。
除非另外定义,否则本文所用的所有技术和科学术语具有与本发明所属领域的普通技术人员通常所理解相同的含义。本说明书中所用的术语仅用于描述特定实施方案,并非旨在限制本发明。
在提供值范围时,应当理解,在本发明内涵盖了每个中间值,至下限单位的十分之一,除非上下文另有明确规定(例如在含有多个碳原子的基团的情况下,在这种情况下,提供了落入该范围内的每个碳原子数),在该范围的上限和下限之间,以及在该所述范围内的任何其他所述值或中间值。这些较小范围的上限和下限可以独立地包含在更小范围内并且也涵盖于本发明内,从属于所述范围内的任何具体排除限值。在所述范围包含一个或两个限值的情况下,本发明中还包括排除所包括的那些限值中的任两个的范围。
下述术语用于描述本发明。在本文中未具体定义一个术语的情况下,那个术语是所属领域的一般技术人员在其用于描述本发明的上下文中应用那个术语时作为领域公认的含义给出。
如本文和所附权利要求中使用的冠词“一个”和“一种”在本文中用于指冠词的一个或多于一个(即,至少一个)语法对象,除非上下文另有明确说明。例如,“要素”意指一个要素或多于一个要素。
如本文在说明书和权利要求中所用,短语“和/或”应理解为意指如此结合的要素中的“任一者或两者”,即在某些情况下结合地存在且在其他情况下分离地存在的要素。用“和/或”列出的多个要素应该以相同的方式加以解释,即,如此结合的“一个或多个”要素。除了由“和/或”子句具体鉴定的要素之外,可以任选地存在其他要素,无论与具体鉴定的那些要素相关还是不相关。因此,作为非限制性实例,当与开放式语言例如“包含”结合使用时,对“A和/或B”的提及可以在一个实施方案中仅指A(任选地包括除了B之外的要素);在另一个实施方案中仅指B(任选地包括除了A之外的要素);在再一个实施方案中指A和B两者(任选地包括其他要素);等。
如本文在说明书和权利要求中使用的,“或”应该理解为具有与如上定义的“和/或”相同的含义。例如,当分开列表中的项目时,“或”或“和/或”应该解释为包含性的,即包含至少一个,但也包括许多要素或要素列表的多于一个,以及任选地,另外未列出的项目。只有明确指出相反的术语,例如“仅一个”或“恰好一个”,或者,当在权利要求中使用时,“由……组成”指许多要素或要素列表的恰好一个要素。一般而言,当之前为排他性术语例如“任一”、“之一”、“仅一个”或“恰好一个”时,如本文使用的,术语“或”应该仅解释为指示排他性替代物(即“一个或另一个但不是两者”)。
在权利要求以及上文说明书中,所有过渡短语、例如“包含”、“包括”、“携带”、“具有”、“含有”、“涉及”、“持有”、“组成”等等应理解为开放式的,即,意指包括但不限于。仅过渡短语“由……组成”和“基本上由……组成”应该分别为闭合式或半闭合式过渡短语,如美国专利局专利审查程序手册(United States Patent Office Manual of PatentExamining Procedures)第2111.03节中所述。
如本文在说明书和权利要求中使用的,提及一个或多个要素的列表,短语“至少一个”应该理解为意指选自要素列表中的任何一个或多个要素的至少一个要素,但不一定包括要素列表中具体列出的每个和每一个要素中的至少一个,并且不排除要素列表中的任何要素组合。该定义还允许除了在短语“至少一个”所指的要素列表内具体鉴定的要素之外,可以任选地存在无论与具体鉴定的那些要素相关还是不相关的要素。因此,作为非限制性实例,“A和B中的至少一个”(或等价地,“A或B中的至少一个”,或等价地“A和/或B中的至少一个”)在一个实施方案中,可以指至少一个,任选地包括多于一个A,而不存在B(并且任选地包括除了B之外的要素);在另一个实施方案中,指至少一个,任选地包括多于一个B,而不存在A(并且任选地包括除了A之外的要素);在再一个实施方案中,指至少一个,任选地包括多于一个A,以及至少一个,任选地包括多于一个B(以及任选地包括其他要素);等。
还应该理解,在本文描述的包括多于一个步骤或动作的某些方法中,该方法的步骤或动作的次序不一定限于其中叙述该方法的步骤或动作的次序,除非上下文另有说明。
术语“共同施用”或“联合疗法”指并行施用(同时施用两种或更多种治疗剂)和不同时间施用(一种或多种治疗剂的施用时间不同于其他治疗剂或药剂的施用时间),只要治疗剂在一定程度上,优选按照有效量同时存在于患者中。在某些优选方面,本文所述的一种或多种本发明化合物与至少一种其他生物活性剂(尤其包括抗癌剂)联合共同施用。在特别优选的方面,化合物的共同施用产生协同活性和/或治疗(包括抗癌)活性。
除非另外说明,否则如本文所用,术语“化合物”是指本文所公开的任何具体化合物,并且包括其互变异构体、区域异构体、几何异构体和适用的立体异构体,包括光学异构体(对映体)和其他立体异构体(非对映体),以及在上下文中,适用的可药用盐及其衍生物(包括前药形式)。设想的氘代小分子是其中药物分子中包含的一个或多个氢原子已被氘取代的那些。
在其在上下文中的使用内,术语化合物一般指单一化合物,但也可包括其他化合物,例如立体异构体、区域异构体和/或光学异构体(包括外消旋混合物)以及所公开化合物的特定对映体或富含对映体的混合物。在上下文中,该术语还指已进行修饰以促进化合物对活性部位的施用和递送的化合物的前药形式。应注意,在描述本文化合物中,尤其描述了许多取代基以及与其相关的变量。普通技术人员应理解,本文所述的分子是如下文一般描述的稳定化合物。示出键时,双键和单键均在所示化合物和众所周知的价态相互作用规则的背景内表示或理解。
术语“泛素连接酶”是指促进泛素向特定底物蛋白质的转移以靶向底物蛋白质从而进行降解的蛋白质家族。例如,人小脑蛋白是E3泛素连接酶蛋白,其单独或与E2泛素缀合酶组合引起泛素与靶蛋白上的赖氨酸的附接,并且随后靶向特定蛋白质底物以被蛋白酶体降解。因此,单独或与E2泛素缀合酶复合的E3泛素连接酶负责泛素至靶蛋白的转移。一般而言,泛素连接酶涉及多泛素化,使得第二泛素附接于第一泛素;第三泛素附接于第二泛素,如此等等。多泛素化将蛋白质标记用于被蛋白酶体降解。然而,存在一些泛素化事件,其限于单泛素化,其中仅单个泛素通过泛素连接酶加入底物分子。单泛素化蛋白质不被靶向蛋白酶体进行降解,而是可以在其细胞位置或功能方面改变,例如,经由结合具有能够结合泛素的结构域的其他蛋白质。更复杂的是,泛素的不同赖氨酸可被E3靶向以制备链。最常见的赖氨酸是泛素链上的Lys48。这是用于制备被蛋白酶体识别的聚泛素的赖氨酸。
术语“患者”或“受试者”在说明书自始至终用于描述用根据本公开的组合物对其提供治疗,包括预防性治疗的动物,优选人或驯养动物。对于特定动物(例如人患者)特异性的那些感染、状况或疾病状态的治疗,术语患者指该特定动物,包括驯养动物如犬或猫或者农场动物如马、牛、绵羊等。一般而言,在本公开中,除非另有说明或由该术语使用的上下文暗示,否则术语患者指人类患者。
术语“有效”用于描述化合物、组合物或组分的量,当在其预期用途的上下文内使用时,所述量实现预期结果。术语有效包含在本申请中另外描述或使用的所有其他有效量或有效浓度项。
化合物和组合物
在一个方面,本说明书提供了包含E3泛素连接酶结合部分(“ULM”)的化合物,所述ULM是人小脑蛋白E3泛素连接酶结合部分(“CLM”)。在一个实施方案中,根据以下结构,CLM偶联至化学接头(L):
(I)L-CLM
其中L是化学接头基团,并且CLM是人小脑蛋白E3泛素连接酶结合部分。本文示出的化合物中的部分的数目和/或相对位置仅为了举例而提供。如技术人员将理解的那样,如本文所述的化合物可以按照各个官能部分的任何所需数目和/或相对位置来合成。
除非上下文另外说明,否则术语ULM和CLM以其包含性意义使用。例如,术语ULM包含所有ULM,包括结合人小脑蛋白的那些(即,CLM)。另外,术语CLM包含所有可能的人小脑蛋白E3泛素连接酶结合部分。
在另一方面,本公开提供了可用于通过诱导靶蛋白的降解来调节蛋白质活性的双官能或多官能PROTAC化合物。在某些实施方案中,该化合物包含偶联(例如,共价、直接或间接连接)至结合靶蛋白的部分(即,蛋白质靶向部分或“PTM”)的CLM。在某些实施方案中,CLM和PTM经由化学接头(L)接合或偶联。CLM识别人小脑蛋白E3泛素连接酶,PTM识别靶蛋白,各个部分与其靶标的相互作用有利于通过将靶蛋白置于泛素连接酶蛋白附近来降解靶蛋白。示例性双官能化合物可以描绘为:
(II)PTM-CLM
在某些实施方案中,双官能化合物还包含化学接头(“L”)。例如,双官能化合物可以描绘为:
(III)PTM-L-CLM
其中PTM是蛋白质/多肽靶向部分,L是接头,并且CLM是人小脑蛋白E3连接酶结合部分。
在某些实施方案中,如本文所述的化合物包含多个PTM(靶向相同或不同的蛋白质靶标)、多个ULM、一个或多个ULM(即,特异性地结合至另一E3泛素连接酶的部分,例如VHL)或它们的组合。在本文所述的任何方面或实施方案中,PTM、CLM和ULM可以直接偶联或经由一个或多个化学接头偶联或它们的组合。在其中化合物具有多个ULM的另外的实施方案中,ULM可以用于相同的E3泛素连接酶,或者每个相应的ULM可以特异性地结合至不同的E3泛素连接酶。在其中化合物具有多个PTM的另外的实施方案中,PTM可以结合相同靶蛋白,或者每个相应的PTM可以特异性地结合至不同靶蛋白。
在另一个实施方案中,本说明书提供了一种化合物,其包含直接偶联或经由化学接头部分(L)偶联的多个CLM。例如,具有两个CLM的化合物可以描绘为:
(IV)CLM-CLM或
(V)CLM-L-CLM
在其中化合物包含多个CLM的某些实施方案中,CLM是相同的。在另外的实施方案中,包含多个CLM的化合物还包含至少一个与CLM直接偶联或经由化学接头(L)偶联或以这两种方式偶联的PTM。在某些另外的实施方案中,包含多个CLM的化合物还包含多个PTM。在另外的实施方案中,PTM是相同的或任选地不同的。在其中PTM不同的另外的实施方案中,各个PTM可以结合相同的蛋白质靶标或特异性地结合至不同的蛋白质靶标。
在另外的实施方案中,本说明书提供了一种化合物,其包含直接偶联或经由化学接头(L)偶联或以这两种方式偶联的至少两个不同CLM。例如,具有两个不同CLM的这样的化合物可以描绘为:
(VI)CLM-CLM’或
(VII)CLM-L-CLM’
其中CLM'表示结构上不同于CLM的人小脑蛋白E3泛素连接酶结合部分。在某些实施方案中,该化合物可包含多个CLM和/或多个CLM'。在其他实施方案中,包含至少两个不同CLM、多个CLM和/或多个CLM’的化合物还包含与CLM或CLM’直接偶联或经由化学接头偶联或以这两种方式偶联的至少一个PTM。在本文所述的任何实施方案中,包含至少两个不同CLM的化合物还可包含多个PTM。在另外的实施方案中,PTM是相同的或任选地不同的。在其中PTM不同的另外的实施方案中,各个PTM可以结合相同的蛋白质靶标或特异性地结合至不同的蛋白质靶标。在另外的实施方案中,PTM本身是ULM或CLM(或ULM'或CLM')。
在一个优选的实施方案中,CLM包含作为人小脑蛋白E3泛素连接酶(CRBN)配体的部分。在某些实施方案中,CLM包含“酰亚胺”分子类别的化学型。在某些另外的实施方案中,CLM包含邻苯二酰亚胺基团或其类似物或衍生物。在另外的实施方案中,CLM包含邻苯二酰亚胺-戊二酰亚胺基团或其类似物或衍生物。在另外的实施方案中,CLM包含由沙立度胺、来那度胺、泊马度胺和其类似物或衍生物组成的组的成员。
在另外的实施方案中,本说明书提供了如本文所述的化合物,包括其对映体、非对映体、溶剂化物和多晶型物,包括其药学上可接受的盐形式,例如酸和碱盐形式。
示例性人小脑蛋白结合和/或抑制化合物
在一个方面,本说明书提供了可用于结合和/或抑制人小脑蛋白E3泛素连接酶结合部分的化合物。在某些实施方案中,该化合物具有包含以下中的至少一种的化学结构(例如,该化合物具有选自以下的化学结构):
/>
/>
/>
其中:
W独立地选自CH
Q
R
R
R
R
R
X是C、CH、C=O或N;
X
R’选自H、卤素、胺、烷基(例如,C1-C3烷基)、取代的烷基(例如,取代的C1-C3烷基)、烷氧基(例如,C1-C3烷氧基)、取代的烷氧基(例如,取代的C1-C3烷氧基)、NR
n是0-4;并且
是单键或双键。
示例性CLM
在本文所述的任何化合物中,CLM包含选自以下的化学结构:
/>
/>
/>
其中:
W独立地选自CH
Q
R
R
R
R
R
X是C、CH、C=O或N;
X
R’选自H、卤素、胺、烷基(例如,C1-C3烷基)、取代的烷基(例如,取代的C1-C3烷基)、烷氧基(例如,C1-C3烷氧基)、取代的烷氧基(例如,取代的C1-C3烷氧基)、NR
n是0-4;
是单键或双键;并且
CLM共价接合至PTM、化学接头基团(L)、ULM、CLM(或CLM’)或它们的组合。
在本文所述的任何方面或实施方案中,CLM或CLM’经由R基团(例如,R、R
在本文所述的任何实施方案中,CLM或CLM’经由W、X、R、R
在本文所述的任何实施方案中,W、X、R
术语“独立地”在本文中用于指示独立应用的变量因应用不同而独立地改变。
术语“烷基”在其上下文中应该意指直链、支链或环状完全饱和的烃原子团或烷基,优选C
术语“烷氧基”是指单独结合至氧的烷基。
术语“烯基”是指含有至少一个C=C键的直链、支链或环状C
术语“炔基”是指含有至少一个C≡C键的直链、支链或环状C
术语“亚烷基”在使用时是指可以任选地被取代的-(CH
术语“未取代的”应该意指仅被氢原子取代。包括C
术语“取代的”或“任选被取代的”应该独立地(即,当存在多于一个取代基时,每个取代基独立于另一个取代基)意指在上下文内的分子上的任何地方的碳(或氮)位置处的一个或多个取代基(根据本公开的化合物中的部分上的独立地至多五个取代基,优选至多三个取代基,经常为1或2个取代基,并且可以包括其自身可以进一步取代的取代基),并且包括羟基、硫醇、羧基、氰基(C≡N)、硝基(NO
术语“取代的”(每个取代基独立于任何其他取代基)在其使用的上下文中还应该意指C
在上下文中,术语“芳基”或“芳香族”是指具有单环(例如苯、苯基、苯甲基)或稠环(例如萘基、蒽基、菲基等)的被取代(如本文另外所述)或未被取代的单价芳香族基团,并且能够在环上的任何可用稳定位置或如所呈现的化学结构中另外指示结合到根据本公开的化合物。在上下文中,芳基的其他实例可包括杂环芳香族环系,在环(单环)中具有一个或多个氮、氧或硫原子的“杂芳基”,例如咪唑、呋喃基、吡咯、呋喃基、噻吩、噻唑、吡啶、嘧啶、吡嗪、三唑、噁唑或稠环系统,例如吲哚、喹啉、吲哚嗪、氮杂吲哚嗪、苯并呋呫等,其可以任选地如上文所述被取代。可以提及的杂芳基包括含氮杂芳基,例如吡咯、吡啶、吡啶酮、哒嗪、嘧啶、吡嗪、吡唑、咪唑、三唑、三嗪、四唑、吲哚、异吲哚、吲哚嗪、氮杂吲哚嗪、嘌呤、吲唑、喹啉、二氢喹啉、四氢喹啉、异喹啉、二氢异喹啉、四氢异喹啉、喹嗪、酞嗪、萘啶、喹喏啉、喹唑啉、噌啉、喋啶、咪唑并吡啶、咪唑并三嗪、吡嗪并哒嗪、吖啶、啡啶、咔唑、咔唑啉、嘧啶、啡啉、菲烯、噁二唑、苯并咪唑、吡咯并吡啶、吡咯并嘧啶和吡啶并嘧啶;含硫芳香族杂环,例如噻吩和苯并噻吩;含氧芳香族杂环,例如呋喃、吡喃、环戊二烯并吡喃、苯并呋喃和异苯并呋喃;和包含2个或更多个选自氮、硫和氧的杂原子的芳香族杂环,例如噻唑、噻二唑、异噻唑、苯并噁唑、苯并噻唑、苯并噻二唑、吩噻嗪、异噁唑、呋呫、啡噁嗪、吡唑并噁唑、咪唑并噻唑、噻吩并呋喃、呋喃并吡咯、吡啶并噁嗪、呋喃并吡啶、呋喃并嘧啶、噻吩并嘧啶和噁唑等,所有这些可以任选地被取代。
术语“被取代的芳基”是指包含至少一个芳环或多个稠环的芳香族碳环基,所述多个稠环中的至少一个是芳香族,其中所述环被一个或多个取代基取代。例如,芳基可包含选自以下的取代基:-(CH
“羧基”表示基团--C(O)OR,其中R是氢、烷基、被取代的烷基、芳基、被取代的芳基、杂芳基或被取代的杂芳基,然而这些通用取代基的含义与本文所定义的相应基团的定义相同。
术语“杂芳基(heteroaryl)”或“杂芳基(hetaryl)”可以指(但决不限于)任选地被取代的喹啉(其可以连接到药效团或在喹啉环内的任何碳原子上被取代)、任选地被取代的吲哚(包括二氢吲哚)、任选地被取代的吲哚嗪、任选地被取代的氮杂吲哚嗪(2-氮杂吲哚嗪、3-氮杂吲哚嗪或4-氮杂吲哚嗪)、任选地被取代的苯并咪唑、苯并二唑、苯并呋喃、任选地被取代的咪唑、任选地被取代的异噁唑、任选地被取代的噁唑(优选被甲基取代)、任选地被取代的二唑、任选地被取代的三唑、四唑、任选地被取代的苯并呋喃、任选地被取代的噻吩、任选地被取代的噻唑(优选被甲基和/或硫醇取代)、任选地被取代的异噻唑、任选地被取代的三唑(优选被以下取代的1,2,3-三唑:甲基、三异丙基硅烷基、任选地被取代的-(CH
其中
S
R
R
R
Y
术语“杂环”是指含有至少一个杂原子(例如N、O或S)的环基,且可以是芳香族(杂芳基)或非芳香族。因此,取决于其使用的上下文,杂芳基部分包含在杂环的定义下。示例性的杂芳基如上所述。
示范性杂环包括:氮杂环丁烷基、苯并咪唑基、1,4-苯并二噁烷基、1,3-苯并间二氧杂环戊烯基、苯并噁唑基、苯并噻唑基、苯并噻吩基、二氢咪唑基、二氢吡喃基、二氢呋喃基、二噁烷基、二氧杂环戊烷基、亚乙基脲、1,3-二氧杂环戊烷、1,3-二噁烷、1,4-二噁烷、呋喃基、高哌啶基、咪唑基、咪唑啉基、咪唑啶基、吲哚啉基、吲哚基、异喹啉基、异噻唑啶基、异噻唑基、异噁唑啶基、异噁唑基、吗啉基、萘啶基、噁唑啶基、噁唑基、吡啶酮、2-吡咯烷酮、吡啶、哌嗪基、N-甲基哌嗪基、哌啶基、邻苯二甲酰亚胺、丁二酰亚胺、吡嗪基、吡唑啉基、吡啶基、嘧啶基、吡咯啶基、吡咯啉基、吡咯基、喹啉基、四氢呋喃基、四氢吡喃基、四氢喹啉、噻唑啶基、噻唑基、噻吩基、四氢噻吩、噁烷、氧杂环丁烷基、氧硫杂环戊烷基、噻烷等等。
杂环基团可以任选地被选自由以下组成的组的成员取代:烷氧基、被取代的烷氧基、环烷基、被取代的环烷基、环烯基、被取代的环烯基、酰基、酰基氨基、酰氧基、氨基、被取代的氨基、氨基酰基、氨基酰氧基、氧基氨基酰基、叠氮基、氰基、卤素、羟基、酮基、硫酮基、羧基、羧基烷基、硫代芳氧基、硫代杂芳氧基、硫代杂环氧基、硫醇、硫代烷氧基、被取代的硫代烷氧基、芳基、芳氧基、杂芳基、杂芳氧基、杂环、杂环氧基、羟胺基、烷氧基胺基、硝基、-SO-烷基、被-SO-取代的烷基、-SO芳基、-SO-杂芳基、-SO2-烷基、被-SO2-取代的烷基、-SO2-芳基、氧代(═O)和-SO2-杂芳基。此类杂环基可以具有单个环或多个稠环。氮杂环和杂芳基的实例包括(但不限于)吡咯、咪唑、吡唑、吡啶、吡嗪、嘧啶、哒嗪、吲哚嗪、异吲哚、吲哚、吲唑、嘌呤、喹嗪、异喹啉、喹啉、酞嗪、萘基吡啶、喹喏啉、喹唑啉、噌啉、喋啶、咔唑、咔啉、啡啶、吖啶、啡啉、异噻唑、吩嗪、异噁唑、啡噁嗪、吩噻嗪、咪唑啶、咪唑啉、哌啶、哌嗪、吲哚啉、N-吗啉基、哌啶基、四氢呋喃基和其类似基团,以及N-烷氧基-含氮杂环。术语“杂环”还包括双环基团,其中杂环中的任一个与苯环或环己烷环或另一杂环(例如吲哚基、喹啉基、异喹啉基、四氢喹啉基等)稠合。
术语“环烷基”可以指(但决不限于)衍生自如本文中所定义的单环或多环烷基或环烷烃的单价基团,例如环中具有三个至二十个碳原子的饱和单环烃基,包括(但不限于)环丙基、环丁基、环戊基、环己基、环庚基等。术语“被取代的环烷基”可以指(但决不限于)单环或多环烷基,且被一个或多个取代基取代,例如氨基、卤素、烷基、被取代的烷基、碳氧基、碳基巯基、芳基、硝基、巯基或磺酸基,而这些通用取代基具有与如此图例中所定义的相应基团的定义相同的含义。
术语“烃基”应该意指含有碳和氢且可以是完全饱和的、部分不饱和的或芳族的化合物,并且包括芳基、烷基、烯基和炔基。
术语“低级烷基”是指甲基、乙基或丙基
术语“低级烷氧基”是指甲氧基、乙氧基或丙氧基。
更具体地讲,CLM的非限制性实例包括下文所示的那些以及起于组合下述化合物的1种或多种不同特征的“杂合”分子或化合物:
/>
/>
其中:
W独立地选自CH
R
R
R
R
R
R
CLM的R是H;
R’是H或者PTM、PTM’、化学接头基团(L)、ULM、CLM、CLM’的附接点,
Q1和Q2各自独立地是被独立地选自H或C1-C3烷基的基团取代的C或N;
是单键或双键;并且
Rn包含官能团或原子。
在本文所述的任何实施方案中,W、R
在本文所述的任何实施方案中,R
在本文所述的任何实施方案中,Q
在本文所述的任何方面或实施方案中,R
在本文描述的任何方面或实施方案中,CLM选自:
/>
其中R’是卤素,并且R
在某些情况下,“CLM”可以是结合至人小脑蛋白E3连接酶的酰亚胺。这些酰亚胺和接头附接点可以是但不限于下述结构:
示例性接头
在某些实施方案中,如本文所述的化合物包含经由化学接头(L)化学连接或偶联至一个或多个PTM(例如,PTM和/或PTM’)、ULM(例如,ULM、ULM’和/或CLM’)的一个或多个CLM。在某些实施方案中,接头基团L是包含一个或多个共价连接的结构单元(例如,-A
在某些实施方案中,接头基团是–(A
(A
接头的q是大于或等于1的整数;
每个A
R
在某些实施方案中,接头的q是大于或等于0的整数。在某些实施方案中,q是大于或等于1的整数。
在某些实施方案(例如其中q大于2)中,A
在某些实施方案(例如其中接头的q是2)中,A
在某些实施方案(例如其中接头的q是1)中,接头基团L的结构是–A
在某些实施方案中,接头(L)包含由选自以下的通式结构表示的基团:
-NR(CH
接头的n可以是0至10;
接头的R可以是H、低级烷基;
接头的R1和R2可通过连接的N形成环。
在某些实施方案中,接头(L)包含由选自以下的通式结构表示的基团:
-N(R)-(CH2)
-O-(CH2)
-O-(CH2)
-N(R)-(CH2)
-(CH2)
-(CH2)
/>
/>
其中
接头的m、n、o、p、q和r独立地是0、1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20;
当数目为零时,不存在N-O或O-O键
所述接头的R是H、甲基和乙基;
所述接头的X是H和F
其中所述接头的m可以是2、3、4、5
/>
/>
/>
/>
/>
/>
/>
其中所述接头的每个n和m可以独立地是0、1、2、3、4、5、6。在本文所述的任何方面或实施方案中,接头(L)选自:
/>
/>
其中每个n和m独立地选自0、1、2、3、4、5或6。
在本文所述的任何方面或实施方案中,接头(L)选自:
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
其中每个m、n、o、p、q和r独立地是0、1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20。
在本文所述的任何方面或实施方案中,L选自:
/>
/>
/>
/>
/>
/>
/>
/>
在另外的实施方案中,接头(L)包含选自但不限于下文所示结构的结构,其中虚线指示与PTM或ULM部分的附接点:
/>
其中:
W
Y
n是0-10;并且
虚线指示与所述PTM或ULM部分的附接点。
在另外的实施方案中,接头(L)包含选自但不限于下文所示结构的结构,其中虚线指示与PTM或ULM部分的附接点:
其中:
W
Y
Q
R
n是0-10;并且
虚线指示与所述PTM或ULM部分的附接点。
在其他实施方案中,接头基团是任选被取代的(聚)乙二醇,其具有1到约100个乙二醇单元,约1到约50个乙二醇单元,1到约25个乙二醇单元,约1到10个乙二醇单元,1到约8个乙二醇单元和1到6个乙二醇单元,2到4个乙二醇单元;或任选被取代的烷基,其内散杂有任选被取代的O、N、S、P或Si原子。在某些实施方案中,接头被芳基、苯基、苯甲基、烷基、亚烷基或杂环基取代。在某些实施方案中,接头可以是不对称或对称的。
在本文所述化合物的任何实施方案中,接头基团可以是如本文所述的任何合适部分。在一个实施方案中,接头是取代或未取代的聚乙二醇基团,其大小范围是约1到约12个乙二醇单元、1到约10个乙二醇单元、约2到约6个乙二醇单元、约2到5个乙二醇单元、约2到4个乙二醇单元。
在另一个实施方案中,本公开涉及一种化合物,其包含结合到靶蛋白或多肽的PTM基团,所述PTM基团通过泛素连接酶发生泛素化并且直接化学连接到或通过接头部分L连接到ULM基团(例如CLM),或者PTM替代地是ULM’基团(例如CLM’),所述ULM’基团也是泛素连接酶结合部分,可以与如上文所述的ULM基团相同或不同并且直接连接到ULM基团或通过接头部分连接到ULM基团;并且L是如上文所述的接头部分,可以存在或不存在并且使ULM化学(共价)连接至PTM;或其药学上可接受的盐、对映异构体、立体异构体、溶剂化物或多晶型物。
在某些实施方案中,接头基团L是包含一个或多个共价连接的结构单元的基团,所述结构单元独立地选自:
。X选自O、N、S、S(O)和SO
尽管ULM基团和PTM基团可通过对于接头化学性质来说适当且稳定的任何基团共价连接至接头基团,但在本公开的优选方面,接头独立地共价结合至ULM基团和PTM基团,优选地通过酰胺、酯、硫酯、酮基、氨基甲酸酯(氨甲酸酯)、碳或醚共价结合,所述基团中的每一个可插入到ULM基团和PTM基团上的任何地方,以使得泛素连接酶上的ULM基团与待降解的靶蛋白上的PTM基团达成最大的结合。(需注意,在其中PTM基团是ULM基团的某些方面,用于降解的靶蛋白可以是泛素连接酶本身)。在某些优选方面,接头可连接到ULM和/或PTM基团上的任选被取代的烷基、亚烷基、烯基或炔基、芳基或杂环基。
在另外的实施方案中,q是1至100、1至90、1至80、1至70、1至60、1至50、1至40、1至30、1至20或1至10的整数。
在某些实施方案中,接头(L)选自:
/>
在其他实施方案中,接头基团是任选被取代的(聚)乙二醇,其具有1到约100个乙二醇单元,约1到约50个乙二醇单元,1到约25个乙二醇单元,约1到10个乙二醇单元,1到约8个乙二醇单元和1到6个乙二醇单元,2到4个乙二醇单元;或任选被取代的烷基,其内散杂有任选被取代的O、N、S、P或Si原子。在某些实施方案中,接头被芳基、苯基、苯甲基、烷基、亚烷基或杂环基取代。在某些实施方案中,接头可以是不对称或对称的。
在本文所述化合物的任何实施方案中,接头基团可以是如本文所述的任何合适部分。在一个实施方案中,接头是取代或未取代的聚乙二醇基团,其大小范围是约1到约12个乙二醇单元、1到约10个乙二醇单元、约2到约6个乙二醇单元、约2到5个乙二醇单元、约2到4个乙二醇单元。
尽管CLM(或ULM)基团和PTM基团可通过对于接头化学性质来说适当且稳定的任何基团共价连接至接头基团,但在本公开的优选方面,接头独立地共价结合至CLM基团和PTM基团,优选地通过酰胺、酯、硫酯、酮基、氨基甲酸酯(氨甲酸酯)、碳或醚共价结合,所述基团中的每一个可插入到CLM基团和PTM基团上的任何地方,以使得泛素连接酶上的CLM基团与待降解的靶蛋白上的PTM基团达成最大的结合。(需注意,在其中PTM基团是ULM基团的某些方面,用于降解的靶蛋白可以是泛素连接酶本身)。在某些优选方面,接头可连接到CLM和/或PTM基团上的任选被取代的烷基、亚烷基、烯基或炔基、芳基或杂环基。
在某些实施方案中,“L”可以是具有4-24个直链原子的直链,直链中的碳原子可以被氧、氮、酰胺、氟化碳等取代,例如以下:
/>
在某些实施方案中,“L”可以是非直链,并且可以是脂族或芳族或杂芳族环状部分,“L”的一些实例包括但不限于以下:
/>
其中:
上述结构中的“X”可以是具有2至14个原子的直链,并且所述链可含有杂原子,例如氧;并且
上述结构中的“Y”可以是O、N、S(O)
示例性PTM
在本公开的优选方面,PTM基团是结合至靶蛋白的基团。PTM基团的靶标具有多种类型,并且选自在细胞中表达的蛋白质,使得序列的至少一部分存在于细胞中并且可结合至PTM基团。术语“蛋白质”包括足够长度的寡肽和多肽序列,使得它们可结合至根据本公开的PTM基团。如本文其他地方所述,真核系统或微生物系统中的任何蛋白质(包括病毒、细菌或真菌)是由根据本公开的化合物介导的泛素化的靶标。优选地,靶蛋白是真核蛋白质。在某些方面,蛋白质结合部分是卤代烷(优选C
根据本公开的PTM基团包含例如特异性结合蛋白质(结合靶蛋白)并包括小分子靶蛋白部分的以下非限制性实例的任何部分:Hsp90抑制剂、激酶抑制剂、雄激素受体抑制剂、HDM2和MDM2抑制剂、靶向人BET含溴结构域蛋白质的化合物、HDAC抑制剂、人赖氨酸甲基转移酶抑制剂、血管生成抑制剂、核激素受体化合物、免疫抑制化合物和靶向芳烃受体(AHR)的化合物等等。下文所述的组合物举例说明了这九种类型的小分子靶蛋白结合部分的一些成员。此类小分子靶蛋白结合部分还包括这些组合物的药学上可接受的盐、对映体、溶剂化物和多晶型物,以及可靶向目的蛋白质的其他小分子。这些结合部分优选通过接头连接至泛素连接酶结合部分,以便在泛素连接酶附近呈递靶蛋白(蛋白质靶标部分与之结合)以进行泛素化和降解。
根据本公开的靶蛋白是可结合至蛋白质靶标部分或PTM基团并作用于泛素连接酶或被其降解的任何蛋白质。通常,靶蛋白可包括例如结构蛋白、受体、酶、细胞表面蛋白、与细胞整合功能相关的蛋白质(包括参与催化活性、芳香酶活性、运动活性、解旋酶活性、代谢过程(合成代谢和分解代谢)、抗氧化活性、蛋白水解、生物合成的蛋白质)、具有激酶活性、氧化还原酶活性、转移酶活性、水解酶活性、裂解酶活性、异构酶活性、连接酶活性、酶调节剂活性、信号转导活性、结构分子活性、结合活性(蛋白质、脂质碳水化合物)、受体活性、细胞运动性、膜融合、细胞通讯、生物过程调节、发育、细胞分化、对刺激的反应的蛋白质、行为蛋白、细胞粘附蛋白、参与细胞死亡的蛋白质、参与运输的蛋白质(包括蛋白质转运活性、核转运、离子转运蛋白活性、通道转运蛋白活性、载体活性、通透酶活性、分泌活性、电子转运蛋白活性、发病、分子伴侣调节蛋白活性、核酸结合活性、转录调节蛋白活性、细胞外组织和生物发生活性、翻译调节蛋白活性。目的蛋白质可包括来自真核生物和原核生物的蛋白质,包括作为药物治疗靶标的人类,其他动物,包括驯养的动物,用于确定抗生素和其他抗微生物剂的靶标的微生物,以及植物,甚至病毒等等。
在其他实施方案中,PTM基团是卤代烷基,其中所述烷基的长度大小通常在约1或2个碳至约12个碳的范围内,通常长度为约2至10个碳,通常长度为约3个碳至约8个碳,更通常长度为约4个碳至约6个碳。卤代烷基通常是直链烷基(但也可以使用支链烷基),并且用至少一个卤基(优选单个卤基,通常单个氯基)封端。用于本公开的卤代烷基PT基团优选由化学结构–(CH
在另一个实施方案中,本公开提供了化合物文库。该文库包含超过一种化合物,其中每种组合物具有式A-B,其中A是泛素路径蛋白质结合部分(优选如本文另外公开的E3泛素连接酶部分),并且B是分子文库的蛋白质结合成员,其中A偶联(优选通过接头部分)至B,并且其中泛素路径蛋白质结合部分识别泛素路径蛋白质,具体地讲E3泛素连接酶,例如人小脑蛋白。在一个具体实施方案中,该文库包含结合至随机靶蛋白结合元件(例如化学化合物文库)的特定人小脑蛋白E3泛素连接酶结合部分。因此,靶蛋白不是预先确定的,并且该方法可用于确定推定的蛋白质结合元件的活性及其作为靶标在泛素连接酶降解后的药理学价值。
本公开可用于治疗多种疾病状态和/或状况,包括蛋白质失调并且患者将从蛋白质降解中受益的任何疾病状态和/或状况。
在另一方面,本说明书提供了治疗组合物,其包含有效量的如本文所述化合物或其盐形式,以及药学上可接受的载体、添加剂或赋形剂,和任选地另外的生物活性剂。治疗组合物调节患者或受试者(例如动物,例如人)中的蛋白质降解,并且可以用于治疗或改善通过降解的蛋白质调节的疾病状态或状况。在某些实施方案中,如本文所述的治疗组合物可用于实现目的蛋白质的降解,以治疗或改善疾病,例如癌症(例如前列腺癌)和肯尼迪病。在某些另外的实施方案中,疾病是前列腺癌。
在替代的方面,本公开涉及一种通过降解用以调节疾病状态或状况的蛋白质或多肽来治疗有需要的受试者的疾病状态或者改善疾病或病症的症状的方法,该方法包括向所述患者或受试者施用有效量例如治疗有效量的至少一种如上所述的化合物,任选地与药学上可接受的载体、添加剂或赋形剂和任选地另外的生物活性剂组合,其中所述组合物能够有效治疗或改善受试者的疾病或病症或其症状。根据本公开的方法可用于通过施用有效量的至少一种本文所述化合物治疗多种疾病状态或状况,包括癌症。疾病状态或状况可以是由微生物剂或其他外源性剂(例如病毒、细菌、真菌、原生动物或其他微生物)引起的疾病,或者可以是由蛋白质的过表达引起的疾病状态,其导致疾病状态和/或状况。
在另一方面,本说明书提供了使用根据本公开的化合物鉴别生物系统中的所关注蛋白质降解的影响的方法。
术语“靶蛋白”用于描述蛋白质或多肽,其是用于结合根据本公开的化合物和通过下文的泛素连接酶降解的靶标。此类小分子靶蛋白结合部分还包括这些组合物的药学上可接受的盐、对映体、溶剂化物和多晶型物,以及可靶向目的蛋白质的其他小分子。这些结合部分通过接头基团L连接至CLM或ULM基团。
可结合至蛋白质靶标部分并被泛素连接酶结合部分所结合的连接酶降解的靶蛋白包括任何蛋白质或肽,包括其片段、其类似物和/或其同源物。靶蛋白包括具有任何生物功能或活性(包括结构、调节、激素、酶、遗传、免疫、收缩、储存、运输和信号转导)的蛋白质和肽。在某些实施方案中,靶蛋白包括结构蛋白、受体、酶、细胞表面蛋白、与细胞整合功能相关的蛋白质(包括参与催化活性、芳香酶活性、运动活性、解旋酶活性、代谢过程(合成代谢和分解代谢)、抗氧化活性、蛋白水解、生物合成的蛋白质)、具有激酶活性、氧化还原酶活性、转移酶活性、水解酶活性、裂解酶活性、异构酶活性、连接酶活性、酶调节剂活性、信号转导活性、结构分子活性、结合活性(蛋白质、脂质碳水化合物)、受体活性、细胞运动性、膜融合、细胞通讯、生物过程调节、发育、细胞分化、对刺激的反应的蛋白质、行为蛋白、细胞粘附蛋白、参与细胞死亡的蛋白质、参与运输的蛋白质(包括蛋白质转运活性、核转运、离子转运蛋白活性、通道转运蛋白活性、载体活性、通透酶活性、分泌活性、电子转运蛋白活性、发病、分子伴侣调节蛋白活性、核酸结合活性、转录调节蛋白活性、细胞外组织和生物发生活性、翻译调节蛋白活性。目的蛋白质可包括来自真核生物和原核生物的蛋白质,包括细菌、病毒、真菌和寄生虫,包括作为药物治疗靶标的人、细菌、病毒、真菌和寄生虫,其他动物,包括驯养动物、用于确定抗生素和其他抗微生物剂的靶标的微生物,以及植物,甚至病毒等等。
更具体地讲,多种用于人类治疗的药物靶标代表蛋白质靶标部分可结合到其上并掺入根据本公开的化合物中的蛋白质靶标。这些包括可用于在多种多基因疾病中恢复功能的蛋白质,包括例如B7.1和B7、TINFRlm、TNFR2、NADPH氧化酶、BclIBax和凋亡途径中的其他配偶体、C5a受体、HMG-CoA还原酶、PDE V磷酸二酯酶类型、PDE IV磷酸二酯酶4型、PDE I、PDEII、PDEIII、角鲨烯环化酶抑制剂、CXCR1、CXCR2、一氧化氮(NO)合酶、环加氧酶1、环加氧酶2、5HT受体、多巴胺受体、G蛋白(即Gq)、组胺受体、5-脂氧合酶、类胰蛋白酶丝氨酸蛋白酶、胸苷酸合成酶、嘌呤核苷磷酸化酶、锥虫GAPDH、糖原磷酸化酶、碳酸酐酶、趋化因子受体、JAW STAT、RXR及类似物、HIV 1蛋白酶、HIV 1整合酶、流感神经氨酸酶、乙型肝炎逆转录酶、钠通道、多重耐药性(MDR)、蛋白质P-糖蛋白(和MRP)、酪氨酸激酶、CD23、CD124、酪氨酸激酶p56lck、CD4、CD5、IL-2受体、IL-1受体、TNF-αR、ICAM1、Cat+通道、VCAM、VLA-4整合素、选择素、CD40/CD40L、newokinin和受体、肌苷一磷酸脱氢酶、p38 MAP激酶、RaslRaflMEWERK通路、白介素-1转换酶、半胱天冬酶、HCV、NS3蛋白酶、HCV NS3 RNA解旋酶、甘氨酰胺核糖核苷酸甲酰转移酶、鼻病毒3C蛋白酶、单纯疱疹病毒-1(HSV-I)、蛋白酶、巨细胞病毒(CMV)蛋白酶、聚(ADP-核糖)聚合酶、周期蛋白依赖性激酶、血管内皮生长因子、催产素受体、微粒体转运蛋白抑制剂、胆汁酸转运抑制剂、5α还原酶抑制剂、血管紧张素11、甘氨酸受体、去甲肾上腺素再摄取受体、内皮素受体、神经肽Y和受体、雌激素受体、雄激素受体(AR)、腺苷受体、腺苷激酶和AMP脱氨酶、嘌呤能受体(P2Y1、P2Y2、P2Y4、P2Y6、P2X1-7)、法尼基转移酶、香叶酰转移酶、NGF的TrkA受体、β-淀粉样蛋白、酪氨酸激酶Flk-IIKDR、玻连蛋白受体、整合素受体、Her-21neu、端粒酶抑制、细胞溶质磷脂酶A2和EGF受体酪氨酸激酶。另外的蛋白质靶标包括例如蜕皮激素20-单加氧酶、GABA门控氯离子通道、乙酰胆碱酯酶、电压敏感性钠通道蛋白质、钙释放通道和氯离子通道。另外的靶蛋白包括乙酰辅酶A羧化酶、腺苷酸琥珀酸合成酶、原卟啉原氧化酶和烯醇丙酮莽草酸磷酸合酶。
卤代烷烃脱卤素酶是根据本公开的特定化合物的另一个靶标。含有氯代烷烃肽结合部分(C
这些蛋白质靶标可用于鉴定与蛋白质结合的化合物部分的筛查中,并且通过将该部分掺入根据本公开的化合物内,可以改变蛋白质的活性水平以实现治疗最终结果。
术语“蛋白质靶标部分”或PTM用于描述这样的小分子,其与靶蛋白或者其他目的蛋白质或多肽结合,并且将该蛋白质或多肽置于/呈递于泛素连接酶附近,使得可发生通过泛素连接酶的蛋白质或多肽的降解。小分子靶蛋白结合部分的非限制性实例包括Hsp90抑制剂、激酶抑制剂、MDM2抑制剂、靶向人BET含溴结构域蛋白的化合物、HDAC抑制剂、人赖氨酸甲基转移酶抑制剂、血管生成抑制剂、免疫抑制化合物和靶向芳烃受体(AHR)的化合物等等。下文所述的组合物举例说明了这九种类型的小分子靶蛋白的一些成员。
根据本公开的示例性蛋白质靶标部分包括卤代烷卤化酶抑制剂、Hsp90抑制剂、激酶抑制剂、MDM2抑制剂、靶向人BET含溴结构域蛋白的化合物、HDAC抑制剂、人赖氨酸甲基转移酶抑制剂、血管生成抑制剂、免疫抑制化合物和靶向芳烃受体(AHR)的化合物。
下文所述的组合物举例说明了这些类型的小分子靶蛋白结合部分的一些成员。此类小分子靶蛋白结合部分还包括这些组合物的药学上可接受的盐、对映体、溶剂化物和多晶型物,以及可靶向目的蛋白质的其他小分子。下文引用的参考文献以引用的方式整体并入本文。
I.热休克蛋白90(HSP90)抑制剂:
如本文所用的HSP90抑制剂包括但不限于:
1.Vallee等人,"Tricyclic Series of Heat Shock Protein 90(HSP90)Inhibitors Part I:Discovery of Tricyclic Imidazo[4,5-C]Pyridines as PotentInhibitors of the HSP90 Molecular Chaperone(2011)J.Med.Chem.54:7206中鉴定的HSP90抑制剂,包括YKB(N-[4-(3H-咪唑并[4,5-C]吡啶-2-基)-9H-芴-9-基]-琥珀酰胺):
衍生化,其中接头基团L或–(L-CLM)基团例如经由末端酰胺基附接;
2.HSP90抑制剂p54(改性)(8-[(2,4-二甲基苯基)磺酰基]-3]戊-4-炔-1-基-3H-嘌呤-6-胺):
衍生化,其中接头基团L或–(L-CLM)基团例如经由末端乙炔基团附接;
3.Brough等人,"4,5-Diarylisoxazole HSP90 Chaperone Inhibitors:Potential Therapeutic Agents for the Treatment of Cancer”,J.MED.CHEM.,第51卷,第196页(2008)中鉴定的HSP90抑制剂(改性),包括化合物2GJ(5-[2,4-二羟基-5-(1-甲基乙基)苯基]-n-乙基-4-[4-(吗啉-4-基甲基)苯基]异噁唑-3-甲酰胺),其具有以下结构:
衍生化,其中接头基团L或–(L-CLM)基团例如经由酰胺基(在胺处或在胺上的烷基处)附接;
4.Wright等人,Structure-Activity Relationships in Purine-BasedInhibitor Binding to HSP90 Isoforms,Chem Biol.2004年6月;11(6):775-85中鉴定的HSP90抑制剂(改性),包括具有以下结构的HSP90抑制剂PU3:
衍生化,其中接头基团L或–(L-CLM)例如经由丁基附接;以及
5.HSP90抑制剂格尔德霉素((4E,6Z,8S,9S,10E,12S,13R,14S,16R)-13-羟基-8,14,19-三甲氧基-4,10,12,16-四甲基-3,20,22-三氧代-2-氮杂双环[16.3.1](衍生化)或其任何衍生物(如17-烷基氨基-17-去甲氧基格尔德霉素(“17-AAG”)或17-(2-二甲基氨基乙基)氨基-17-去甲氧基格尔德霉素(“17-DMAG”))(衍生化,其中接头基团L或–(L-CLM)基团例如经由酰胺基附接)。
II.激酶和磷酸酶抑制剂:
如本文所用的激酶抑制剂包括但不限于:
1.埃罗替尼衍生物酪氨酸激酶抑制剂:
其中R是例如经由醚基附接的接头基团L或–(L-CLM)基团;
2.激酶抑制剂舒尼替尼(衍生化):
衍生化,其中R是例如附接至吡咯部分的接头基团L或–(L-CLM)基团;
3.激酶抑制剂索拉非尼(衍生化):
衍生化,其中R是例如附接至酰胺部分的接头基团L或–(L-CLM)基团;
4.激酶抑制剂达沙替尼(衍生化):
衍生化,其中R是例如附接至嘧啶的接头基团L或–(L-CLM);
5.激酶抑制剂拉帕替尼(衍生化):
衍生化,其中接头基团L或–(L-CLM)基团例如经由磺酰基甲基的末端甲基附接;
6.激酶抑制剂U09-CX-5279(衍生化):
衍生化,其中接头基团L或–(L-CLM)基团例如经由胺(苯胺)、羧酸或胺α附接至环丙基或经由环丙基附接;
7.Millan等人,Design and Synthesis of Inhaled P38 Inhibitors for theTreatment of Chronic Obstructive Pulmonary Disease,J.MED.CHEM第54卷,第7797页(2011)中鉴定的激酶抑制剂,包括具有以下结构的激酶抑制剂Y1W和Y1X(衍生化):
/>
YIX(1-乙基-3-(2-{[3-(1-甲基乙基)[1,2,4]三唑并[4,3-a]吡啶-6-基]磺酰基}苄基)脲,衍生化,其中接头基团L或–(L-CLM)基团例如经由异丙基附接;
YIW
1-(3-叔丁基-1-苯基-1H-吡唑-5-基)-3-(2-{[3-(1-甲基乙基)-[1,2,4]三唑并[4,3-a]吡啶-6-基]磺酰基}苄基)脲
衍生化,其中接头基团L或–(L-CLM)基团例如优选经由异丙基或叔丁基附接;
8.Schenkel等人,Discovery of Potent and Highly SelectiveThienopyridine Janus Kinase 2Inhibitors J.Med.Chem.,2011,54(24),第8440-8450页中鉴定的激酶抑制剂,包括具有以下结构的化合物6TP和0TP(衍生化):
6TP
4-氨基-2-[4-(叔丁基磺酰胺基)苯基]-N-甲基噻吩并[3,2-c]吡啶-7-甲酰胺噻吩并吡啶19
衍生化,其中接头基团L或–(L-CLM)基团例如经由结合至酰胺部分的末端甲基附接;
0TP
4-氨基-N-甲基-2-[4-(吗啉-4-基)苯基]噻吩并[3,2-c]吡啶-7-甲酰胺
噻吩并吡啶8
衍生化,其中接头基团L或–(L-CLM)基团例如经由结合至酰胺部分的末端甲基附接;
9.Van Eis等人,"2,6-Naphthyridines as potent and selective inhibitorsof the novel protein kinase C isozymes”,Biorg.Med.Chem.Lett.2011年12月15日;21(24):7367-72中鉴定的激酶抑制剂,包括具有以下结构的激酶抑制剂07U:
07U
2-甲基-N~1~-[3-(吡啶-4-基)-2,6-二氮杂萘-1-基]丙烷-1,2-二胺
衍生化,其中接头基团L或–(L-CLM)基团例如经由仲胺或末端氨基附接;
10.Lountos等人,”Structural Characterization of Inhibitor Complexeswith Checkpoint Kinase 2(Chk2),a Drug Target for Cancer Therapy”,J.STRUCT.BIOL第176卷,第292页(2011)中鉴定的激酶抑制剂,包括具有以下结构的激酶抑制剂YCF:
衍生化,其中接头基团L或–(L-CLM)基团例如经由任一末端羟基附接;
11.Lountos等人,”Structural Characterization of Inhibitor Complexeswith Checkpoint Kinase 2(Chk2),a Drug Target for Cancer Therapy”,J.STRUCT.BIOL.第176卷,第292页(2011)中鉴定的激酶抑制剂,包括具有以下结构的激酶抑制剂XK9和NXP(衍生化):
XK9
N-{4-[(1E)-N-(N-羟基脒基)乙烷腙基]苯基}-7-硝基-1H-吲哚-2-甲酰胺;
NXP
N-{4-[(1E)-N-脒基乙烷腙基]苯基}-1H-吲哚-3-甲酰胺
衍生化,其中接头基团L或–(L-CLM)基团例如经由末端羟基(XK9)或腙基(NXP)附接;
12.激酶抑制剂阿法替尼(衍生化)(N-[4-[(3-氯-4-氟苯基)氨基]-7-[[(3S)-四氢-3-呋喃基]氧基]-6-喹唑啉基]-4(二甲基氨基)-2-丁酰胺)(衍生化,其中接头基团L或–(L-CLM)基团例如经由脂族胺基团附接);
13.激酶抑制剂福坦替尼(衍生化)([6-({5-氟-2-[(3,4,5-三甲氧基苯基)氨基]嘧啶-4-基}氨基)-2,2-二甲基-3-氧代-2,3-二氢-4H-吡啶并[3,2-b]-1,4-噁嗪-4-基]甲基磷酸二钠六水合物)(衍生化,其中接头基团L或–(L-CLM)基团例如经由甲氧基附接);
14.激酶抑制剂吉非替尼(衍生化)(N-(3-氯-4-氟-苯基)-7-甲氧基-6-(3-吗啉-4-基丙氧基)喹唑啉-4-胺):
衍生化,其中接头基团L或–(L-CLM)基团例如经由甲氧基或醚基附接;
15.激酶抑制剂乐伐替尼(衍生化)(4-[3-氯-4-(环丙基氨基甲酰基氨基)苯氧基]-7-甲氧基-喹啉-6-甲酰胺)(衍生化,其中接头基团L或–(L-CLM)基团例如经由环丙基附接);
16.激酶抑制剂凡德他尼(衍生化)(N-(4-溴-2-氟苯基)-6-甲氧基-7-[(1-甲基哌啶-4-基)甲氧基]喹唑啉-4-胺)(衍生化,其中接头基团L或–(L-CLM)基团例如经由甲氧基或羟基附接);
17.激酶抑制剂维莫非尼(衍生化)(丙烷-1-磺酸{3-[5-(4-氯苯基)-1H-吡咯并[2,3-b]吡啶-3-羰基]-2,4-二氟-苯基}-酰胺),衍生化,其中接头基团L或–(L-CLM)基团例如经由磺酰基丙基附接;
18.激酶抑制剂格列卫(衍生化):
(衍生化,其中作为接头基团L或–(L-CLM)基团的R例如经由酰胺基或经由苯胺基附接);
19.激酶抑制剂帕唑帕尼(衍生化)(VEGFR3抑制剂):
衍生化,其中R是例如附接至苯基部分或经由苯胺基附接的接头基团L或–(L-CLM)基团;
20.激酶抑制剂AT-9283(衍生化)Aurora激酶抑制剂
其中R是例如附接至苯基部分的接头基团L或–(L-CLM)基团;
21.激酶抑制剂TAE684(衍生化)ALK抑制剂
其中R是例如附接至苯基部分的接头基团L或–(L-CLM)基团;
22.激酶抑制剂尼罗替尼(衍生化)Abl抑制剂:
衍生化,其中R是例如附接至苯基部分或苯胺基的接头基团L或–(L-CLM)基团;
23.激酶抑制剂NVP-BSK805(衍生化)JAK2抑制剂
衍生化,其中R是例如附接至苯基部分或二唑基的接头基团L或–(L-CLM)基团;
24.激酶抑制剂克唑替尼衍生化Alk抑制剂
衍生化,其中R是例如附接至苯基部分或二唑基的接头基团L或–(L-CLM)基团;
25.激酶抑制剂JNJ FMS(衍生化)抑制剂
衍生化,其中R是例如附接至苯基部分的接头基团L或–(L-CLM)基团;/>
26.激酶抑制剂佛瑞替尼(衍生化)Met抑制剂
衍生化,其中R是例如附接至苯基部分或者喹啉部分上的羟基或醚基的接头基团L或–(L-CLM)基团;
27.变构蛋白酪氨酸磷酸酶抑制剂PTP1B(衍生化):
衍生化,其中接头基团L或–(L-CLM)基团例如附接在R处,如图所示;
28.酪氨酸磷酸酶SHP-2结构域的抑制剂(衍生化):
衍生化,其中接头基团L或–(L-CLM)基团例如附接在R处;
29.BRAF(BRAF
衍生化,其中接头基团L或–(L-CLM)基团例如附接在R处;
30.酪氨酸激酶ABL的抑制剂(衍生化)
衍生化,其中接头基团L或–(L-CLM)基团例如附接在R处;
31.激酶抑制剂OSI-027(衍生化)mTORC1/2抑制剂
衍生化,其中接头基团L或–(L-CLM)基团例如附接在R处;
32.激酶抑制剂OSI-930(衍生化)c-Kit/KDR抑制剂
衍生化,其中接头基团L或–(L-CLM)基团例如附接在R处;以及
33.激酶抑制剂OSI-906(衍生化)IGF1R/IR抑制剂
衍生化,其中接头基团L或–(L-CLM)基团例如附接在R处。
其中,在I-XVII部分描述的任何实施方案中,“R”表示哌嗪部分上接头基团L或–(L-CLM)基团的附接位点。
III.HDM2/MDM2抑制剂:
如本文所用的HDM2/MDM2抑制剂包括但不限于:
1.Vassilev等人,In vivo activation of the p53 pathway by small-molecule antagonists of MDM2,SCIENCE第303卷,第844-848页(2004)和Schneekloth等人,Targeted intracellular protein degradation induced by a small molecule:Enroute to chemical proteomics,Bioorg.Med.Chem.Lett.18(2008)5904–5908中鉴定的HDM2/MDM2抑制剂,包括(或附加)如下所述的化合物nutlin-3、nutlin-2和nutlin-1(衍生化),以及其所有衍生物和类似物:
(衍生化,其中接头基团L或–(L-CLM)基团附接在例如甲氧基处或作为羟基);
/>
(衍生化,其中接头基团L或–(L-CLM)基团附接在例如甲氧基或羟基处);
(衍生化,其中接头基团L或–(L-CLM)基团例如经由甲氧基或作为羟基附接);以及
2.反式-4-碘-4'-硼烷基-查耳酮
(衍生化,其中接头基团L或–(L-CLM)基团例如经由羟基附接)。
IV.靶向人BET含溴结构域蛋白的化合物:
在某些实施方案中,“PTM”可以是结合至溴结构域和额外末端结构域(BET)蛋白BRD2、BRD3和BRD4的配体。靶向人BET含溴结构域蛋白的化合物包括但不限于与如下所述的靶标相关的化合物,其中“R”或“接头”表示用于接头基团L或–(L-CLM)基团附接的位点,例如:
1.JQ1,Filippakopoulos等人,Selective inhibition of BETbromodomains.Nature(2010):
2.I-BET,Nicodeme等人,Supression of Inflammation by a SyntheticHistone Mimic.Nature(2010),Chung等人,Discovery and Characterization of SmallMolecule Inhibitors of the BET Family Bromodomains.J.Med Chem.(2011):
3.Hewings等人,3,5-Dimethylisoxazoles Act as Acetyl-lysine BromodomainLigands.J.Med.Chem.(2011)54 6761-6770中所述的化合物。
4.I-BET151,Dawson等人,Inhibition of BET Recruitment to Chromatin asan Efective Treatment for MLL-fusion Leukemia.Nature(2011):
5.咔唑型(US 2015/0256700)
6.吡咯并吡啶酮型(US 2015/0148342)
7.四氢喹啉型(WO 2015/074064)
8.三唑并吡嗪型(WO 2015/067770)
9.吡啶酮型(WO 2015/022332)
10.喹唑啉酮型(WO 2015/015318)
11.二氢吡啶并吡嗪酮型(WO 2015/011084)
(其中在每种情况下,R或L或接头表示例如接头基团L或–(L-CLM)基团的附接位点)。
在本文所述的任何方面或实施方案中,要求保护的结构PTM可以由作为BET/BRD4配体的三环二氮杂环庚三烯或三环氮杂环庚三烯组成(PTM-a),其中虚线指示接头连接轨迹,并且三个位点被定义为可附接的接头:
其中:
A和B独立地是芳环、杂芳环、5元碳环、6元碳环、5元杂环、6元杂环、噻吩、吡咯、吡唑、吡啶、嘧啶、吡嗪,任选地被烷基、烷氧基、卤素、腈或另一芳环或杂芳环取代,其中A稠合至中心氮杂环庚三烯(Y1=C)或二氮杂环庚三烯(Y1=N)部分;
Y1、Y2和Y3和Y4可以是碳、氮或氧,以形成作为三唑或异噁唑的稠合5元芳环;并且
Z1是甲基或低级烷基。
作为BET/BRD4配体的PTM-a的片段在文献(WO 2016/069578;WO2014/001356;WO2016/050821;WO 2015/195863;WO 2014/128111)中有所描述。
在本文所述的包含结构CLM-L-PTM-a的任何方面或实施方案中,PTM-a可由以下通式结构表示,其中虚线指示可能的接头连接点。在结构PTM-aa至PTM-ai中,X和Y的取代模式可以是单取代或双取代。
/>
在本文所述的任何方面或实施方案中,作为BET/BRD4配体的PTM-a的结构包括以下结构,其中虚线指示BET/BRD4配体与接头之间的连接点:
/>
/>
在某些实施方案中,本说明书提供了但不限于以下示例性BET PROTAC(化合物1或2),包括其盐、前药、多晶型物、类似物、衍生物和氘代形式:
V.HDAC抑制剂:
HDAC抑制剂(衍生化)包括但不限于:
1.Finnin,M.S等人,Structures of Histone Deacetylase Homologue Bound tothe TSA and SAHA Inhibitors.Nature 40,188-193(1999)。
(衍生化,其中“R”表示例如接头基团L或–(L-CLM)基团的附接位点);以及
2.PCT WO0222577(“脱乙酰酶抑制剂”)的式(I)所定义的化合物(衍生化,其中接头基团L或–(L-CLM)基团例如经由羟基附接);
VI.人赖氨酸甲基转移酶抑制剂:
人赖氨酸甲基转移酶抑制剂包括但不限于:
1.Chang等人,Structural Basis for G9a-Like protein LysineMethyltransferase Inhibition by BIX-1294.Nat.Struct.Biol.(2009)16(3)312。
(衍生化,其中“R”表示例如接头基团L或–(L-CLM)基团的附接位点);
2.Liu,F等人,Discovery of a 2,4-Diamino-7-aminoalkoxyquinazoline asaPotent and Selective Inhibitor of Histone Methyltransferase G9a.J.Med.Chem.(2009)52(24)7950。
(衍生化,其中“R”表示例如接头基团L或–(L-CLM)基团的潜在附接位点);
3.阿扎胞苷(衍生化)(4-氨基-1-β-D-呋喃核糖基-1,3,5-三嗪-2(1H)-酮)(衍生化,其中接头基团L或–(L-CLM)基团例如经由羟基或氨基附接);以及
4.地西他滨(衍生化)(4-氨基-1-(2-脱氧-b-D-赤式-呋喃戊糖基)-1,3,5-三嗪-2(1H)-酮)(衍生化,其中接头基团L或–(L-CLM)基团例如经由任一羟基或在氨基处附接)。
VII.血管生成抑制剂:
血管生成抑制剂包括但不限于:
1.GA-1(衍生化)及其衍生物和类似物,具有如Sakamoto等人,Development ofProtacs to target cancer-promoting proteins for ubiquitination anddegradation,Mol Cell Proteomics 2003年12月;2(12):1350-8中所述的结构并结合至其中所述的接头;
2.雌二醇(衍生化),其可以结合至接头基团L或–(L-CLM)基团,如Rodriguez-Gonzalez等人,Targeting steroid hormone receptors for ubiquitination anddegradation in breast and prostate cancer,Oncogene(2008)27,7201–7211中整体所述;
3.雌二醇、睾酮(衍生化)和相关衍生物,包括但不限于DHT及其衍生物和类似物,具有如Sakamoto等人,Development of Protacs to target cancer-promoting proteinsfor ubiquitination and degradation,Mol Cell Proteomics 2003年12月;2(12):1350-8中整体所述的结构并结合至其中所述的接头基团L或–(L-CLM)基团;以及
4.卵假散囊菌素、烟曲霉素(衍生化)及其衍生物和类似物,具有如Sakamoto等人,Protacs:chimeric molecules that target proteins to the Skp1-Cullin-F boxcomplex for ubiquitination and degradation Proc Natl Acad Sci USA.2001年7月17日;98(15):8554-9和美国专利号7,208,157中整体所述的结构并结合至其中所述的接头基团L或–(L-CLM)基团。
VIII.免疫抑制化合物:
免疫抑制化合物包括但不限于:
1.AP21998(衍生化),具有如Schneekloth等人,Chemical Genetic Control ofProtein Levels:Selective in Vivo Targeted Degradation,J.AM.CHEM.SOC.2004,126,3748-3754中整体所述的结构并结合至其中所述的接头基团L或–(L-CLM)基团;
2.糖皮质激素(例如,氢化可的松、泼尼松、泼尼松龙和甲基泼尼松龙)(衍生化,其中接头基团L或–(L-CLM)基团将例如结合至任何羟基)和二丙酸倍氯米松(衍生化,其中接头基团或–(L-CLM)例如结合至丙酸酯);
3.甲氨喋呤(衍生化,其中接头基团或–(L-CLM)基团可以例如结合至任一末端羟基);
4.环孢素(衍生化,其中接头基团或–(L-CLM)基团可以例如结合在任何丁基处);
5.他克莫司(FK-506)和雷帕霉素(衍生化,其中接头基团L或–(L-CLM)基团可以例如结合在甲氧基之一处);以及
6.放线菌素(衍生化,其中接头基团L或–(L-CLM)基团可以例如结合在异丙基之一处)。
IX.靶向芳烃受体(AHR)的化合物:
靶向芳烃受体(AHR)的化合物包括但不限于:
1.芹菜素(以结合至接头基团L或–(L-CLM)基团的方式衍生化,如Lee等人,Targeted Degradation of the Aryl Hydrocarbon Receptor by the PROTAC Approach:A Useful Chemical Genetic Tool,ChemBioChem第8卷,第17期,第2058–2062页,2007年11月23日中整体所述);以及
2.SR1和LGC006(衍生化,使得结合接头基团L或–(L-CLM)),如Boitano等人,ArylHydrocarbon Receptor Antagonists Promote the Expansion of Human HematopoieticStem Cells,Science,2010年9月10日:第329卷第5997期,第1345-1348页中所述。
X.靶向RAF受体(激酶)的化合物:
PLX4032
(衍生化,其中“R”表示例如用于接头基团L或–(L-CLM)基团附接的位点)。
XI.靶向FKBP的化合物:
(衍生化,其中“R”表示例如用于接头基团L或–(L-CLM)基团附接的位点)。
XII.靶向雄激素受体(AR)的化合物
1.雄激素受体的RU59063配体(衍生化)
(衍生化,其中“R”表示例如用于接头基团L或–(L-CLM)基团附接的位点)。
2.雄激素受体的SARM配体(衍生化)
/>
(衍生化,其中“R”表示例如用于接头基团L或–(L-CLM)基团附接的位点)。
3.雄激素受体配体DHT(衍生化)
(衍生化,其中“R”表示例如用于接头基团L或–(L-CLM)基团附接的位点)。
4.MDV3100配体(衍生化)
5.ARN-509配体(衍生化)
6.六氢苯并异噁唑
7.四甲基环丁烷
8.在本文所述的任何方面或实施方案中,PTM是结合至雄激素受体(AR)的化学部分(ABM)。各种雄激素受体结合化合物已在文献中有所描述,包括各种雄激素衍生物,例如睾酮、二氢睾酮和美曲勃龙(也称为甲雌三烯醇酮或R1881),以及非类固醇化合物,例如比卡鲁胺、恩杂鲁胺,其中一些如上所述。所属领域的一般技术人员将了解,这些雄激素受体结合化合物可以潜在地用作PROTAC化合物中的ABM部分。此类文献包括但不限于G.F.Allan等人,Nuclear Receptor Signaling,2003,1,e009;R.H.Bradbury等人,Bioorganic&Medicinal Chemistry Letters,2011 5442–5445;C.Guo等人,Bioorganic&MedicinalChemistry Letters,2012 2572-2578;P.K.Poutiainen等人,J.Med.Chem.2012,55,6316-6327;A.Pepe等人,J.Med.Chem.2013,56,8280–8297;M.E.Jung等人,J.Med.Chem.2010,53,2779–2796,这些文献以引用方式并入本文
在本文所述的任何方面或实施方案中,ABM包含但不限于选自下文所示结构的结构,其中虚线表示接头部分或ULM如CLM的附接点:
其中:
W
Y
Y
Q是具有0-4个杂原子的3-6元环,其任选地被0-6个R
R
W
每个R
R
在本文所述的任何方面或实施方案中,W
在本文所述的任何方面或实施方案中,W
在本文所述的任何方面或实施方案中,W
在本文所述的任何方面或实施方案中,ABM包含选自下文所示的以下结构的结构,其中
其中:
R
R
Y
R
R
在本文所述的任何方面或实施方案中,每个R
在本文所述的任何方面或实施方案中,ABM包含选自下文所示的以下结构的结构,其中
其中:
R
R
Y
R
X是N或C。
在本文所述的任何方面或实施方案中,R
在本文所述的任何方面或实施方案中,ABM包含下文所示的结构,其中虚线表示接头部分或ULM或CLM的附接点:
其中:
W
每个R
每个R
Y
Y
Y
R
W
每个R
表示可以是立体特异性((R)或(S))或非立体特异性的键。
在本文所述的任何方面或实施方案中,W
在本文所述的任何方面或实施方案中,W
在本文所述的任何方面或实施方案中,W
在本文所述的任何方面或实施方案中,ABM包含但不限于选自下文所示结构的结构,其中虚线表示接头部分或ULM的附接点:
其中:
W
每个R
每个R
Y
R
W
每个R
在本文所述的任何实施方案中,W
在某些另外的实施方案中,W
和/>
在本文所述的任何方面或实施方案中,W2选自:
在本文所述的任何方面或实施方案中,ABM选自:
/>
/>
/>
/>
/>
在本文所述的任何方面或实施方案中,ABM包含以下结构:
其中:
其中W
Y
Q是具有0-2个杂原子的4元脂环,其任选地被0-6个R
R
W
每个R
R
在本文所述的任何方面或实施方案中,本说明书提供了一种雄激素受体结合化合物,其包含以下结构:
其中:
W
Y
Y
Q是具有0-4个杂原子的3-6元脂环或芳环,其任选地被0-6个R
R
W
每个R
R
在本文所述的任何方面或实施方案中,雄激素受体结合部分具有以下结构:
其中:
W
每个R
每个R
Y
Q是任选地被0-4个R
Y4是键或NH;
Y5是键、C=O或C=S;并且
每个W
在本文所述的任何方面或实施方案中,W
在本文所述的任何方面或实施方案中,W
在本文所述的任何方面或实施方案中,W
在本文所述的任何方面或实施方案中,雄激素结合部分具有以下结构:
其中:
W
Y
Q是具有1或2个杂原子的5元芳环;
R
W
每个R
在本文所述的任何方面或实施方案中,W
在本文所述的任何方面或实施方案中,W
其中每个R
每个R
在本文所述的任何方面或实施方案中,W
在本文所述的任何方面或实施方案中,Q是
在本文所述的任何方面或实施方案中,W
在本文所述的任何方面或实施方案中,(Y
在本文所述的任何方面或实施方案中,ABM包含但不限于选自下文所示结构的结构,其中虚线表示接头部分或ULM如CLM的附接点:
其中:
W
每个R
每个R
Y
Y
R
W
每个R
在本文所述的任何方面或实施方案中,W
在本文所述的任何方面或实施方案中,W
在本文所述的任何方面或实施方案中,W2选自:
在本文所述的任何方面或实施方案中,ABM包含下文所示的结构,其中虚线表示接头部分或ULM或CLM的附接点:
其中:
W
每个R
每个R
Y
Y
Y
R
W
每个R
表示可以是立体特异性((R)或(S))或非立体特异性的键。
在本文所述的任何实施方案中,W
在某些另外的实施方案中,W
在某些另外的实施方案中,W
在某些实施方案中,ABM的雄激素受体结合化合物选自:
反-2-氯-4-[3-氨基-2,2,4,4-四甲基环丁氧基]苯甲腈;
顺-2-氯-4-[3-氨基-2,2,4,4-四甲基环丁氧基]苯甲腈;
反6-氨基-N-[3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基]哒嗪-3-甲酰胺;
反N-[3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基]氨基甲酸叔丁酯;
反4-氨基-N-[3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基]苯甲酰胺;
反5-氨基-N-[3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基]吡嗪-2-甲酰胺;
反2-氨基-N-[3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基]嘧啶-5-甲酰胺;
4-甲氧基-N-[(1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基]苯甲酰胺;
反1-(2-羟基乙基)-N-[3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基]-1H-吡唑-4-甲酰胺;
反6-氨基-N-[3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基]吡啶-3-甲酰胺;
反4-[(5-羟戊基)氨基]-N-[3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基]苯甲酰胺;和
反2-({5-[(4-{[3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基]氨甲酰基}苯基)氨基戊基}氧基)乙酸叔丁酯;和
N-((1r,3r)-3-(4-氰基苯氧基)-2,2,4,4-四甲基环丁基)-4-甲基苯甲酰胺。
在某些实施方案中,本说明书提供了但不限于以下示例性雄激素受体PROTAC分子(PROTAC 3至PROTAC-30),包括其盐、前药、多晶型物、类似物、衍生物和氘代形式:
/>
/>
/>
/>
/>
/>
/>
XIII.靶向雌激素受体(ER)的化合物ICI-182780
1.雌激素受体配体
(衍生化,其中“R”表示用于接头基团L或–(L-CLM)基团附接的位点)。
在本文所述的任何实施方案或方面,PTM可由式PTM-I表示:
PTM-I
其中:
X
X
R
至少一个R
至少一个R
虚线指示至少一个接头、CLM、CLM’、PTM、PTM’或它们的组合的附接位点。
在本文所述的任何实施方案或方面,PTM可由式PTM-I表示:
其中:
X
X
R
每个R
每个R
PTM-I在相应的环上包含至少一个R
虚线指示至少一个接头、CLM、CLM’、PTM、PTM’或它们的组合的附接位点。
在本文所述的任何实施方案或方面,PTM-I具有以下中的至少一者:两个R
在本文所述的任何实施方案或方面,PTM可由式PTM-II表示:
PTM-II
其中:
X
X
R
R
R
虚线指示至少一个接头、CLM、CLM’、PTM、PTM’或它们的组合的附接位点。
在本文所述的方面或实施方案中,O(CO)R
在本文所述的任何实施方案或方面,PTM-I或PTM-II的O-低级烷基具有碳数为1至3的烷基链。
在本文所述的方面或实施方案中,本公开提供了式(I
式(I
其中:
每个X
指示至少一个接头、CLM、CLM’、PTM、PTM’或它们的组合的附接位点;
每个R
每个R
每个R
在本文描述的任何方面或实施方案中,PTM由式(II
式(II
其中:
X
指示至少一个接头、CLM、CLM’、PTM、PTM’或它们的组合的附接位点;
每个R
每个R
每个R
在某些实施方案中,以下中的至少一者:
式(II
式(II
式(II
式(II
XIV.靶向甲状腺激素受体(TR)的化合物
1.甲状腺激素受体配体(衍生化)
(衍生化,其中“R”表示用于接头基团L或–(L-CLM)基团附接的位点,并且MOMO表示甲氧基)。
XV.靶向HIV蛋白酶的化合物
1.HIV蛋白酶的抑制剂(衍生化)
(衍生化,其中“R”表示用于接头基团L或–(L-CLM)基团附接的位点)。参见J.Med.Chem.2010,53,521-538。
2.HIV蛋白酶的抑制剂
(衍生化,其中“R”表示用于接头基团L或–(L-CLM)基团附接的潜在位点)。参见J.Med.Chem.2010,53,521-538。
XVI.靶向HIV整合酶的化合物
1.HIV整合酶的抑制剂(衍生化)
(衍生化,其中“R”表示用于接头基团L或–(L-CLM)基团附接的位点)。参见J.Med.Chem.2010,53,6466。
2.HIV整合酶的抑制剂(衍生化)
3.HIV整合酶的抑制剂Isetntress(衍生化)
(衍生化,其中“R”表示用于接头基团L或–(L-CLM)基团附接的位点)。参见J.Med.Chem.2010,53,6466。
XVII.靶向HCV蛋白酶的化合物
1.HCV蛋白酶的抑制剂(衍生化)
(衍生化,其中“R”表示用于接头基团L或–(L-CLM)基团附接的位点)。
XVIII.靶向酰基蛋白硫酯酶1和2的化合物(APT1和APT2)
1.APT1和APT2的抑制剂(衍生化)
(衍生化,其中“R”表示用于接头基团L或–(L-CLM)基团附接的位点)。参见Angew.Chem.Int.Ed.2011,50,9838–9842,其中L是如本文其他地方所述的接头基团,并且所述CLM基团如本文其他地方所述,使得–(L-CLM)将CLM基团结合至PTM基团,如本文其他地方所述。
VIV.靶向Tau蛋白的化合物
在本文所述的任何方面或实施方案中,PTM可包含Tau蛋白结合部分。例如,PTM可以由式I、式II、式III、式IV、式V、式VI、式VII、式VIII、式IX、式X或式XI表示:
,其中:
A、B、C、D、E和F独立地选自任选被取代的5或6元芳基或杂芳基环、任选被取代的4至7元环烷基或杂环烷基,其中圆圈之间的接触指示环稠合;并且
L
在某些实施方案中,PTM的A、B、C、D、E和F的芳基和杂芳基环任选被1至3个取代基取代,所述取代基各自独立地选自烷基、烯基、卤代烷基、卤素、羟基、烷氧基、氟代烷氧基、氨基、烷基氨基、二烷基氨基、酰基氨基、三氟甲基和氰基,其中所述烷基和烯基进一步任选被取代。
在本文所述的任何方面或实施方案中,A、B、C、F或它们的组合中的至少一者的环选自任选被取代的5或6元芳基或杂芳基环;
在本文所述的任何方面或实施方案中,PTM具有式I的化学结构,其中:
A、B和C环独立地是5或6元稠合芳基或杂芳基环;
L
D选自6元芳基、杂芳基或杂环烷基,
其中A、B、C和D任选被烷基、卤代烷基、卤素、羟基、烷氧基、氨基、烷基氨基、二烷基氨基或氰基取代。
在本文所述的任何方面或实施方案中,PTM具有式I的化学结构,其中:
A和C是苯基或6元杂芳基环;
B是5元杂芳基环;
L
D是6元杂芳基或6元杂环烷基环;
其中每个A、B、C和D任选独立地被烷基、卤代烷基、卤素、羟基、烷氧基、氨基、二烷基氨基或氰基取代,并且其中A、B、C和D环中任何一个的氮原子并非与杂原子或碳原子直接连接,另一个杂原子与所述杂原子或碳原子直接附接。
在其他实施方案中,PTM具有式III或IV的化学结构,其中A、B和C是5或6元稠合芳基或杂芳基环,L
在本文所述的任何方面或实施方案中,PTM由以下化学结构表示:
/>
其中:
R
R
R
其中PTM经由L与ULM偶联。
在本文所述的任何方面或实施方案中,PTM共价偶联至一个或多个ULM(VLM或CLM)基团,或者与如本文所述的一个或多个ULM(VLM或CLM)基团附接的接头。
在本文所述的任何方面或实施方案中,PTM由以下化学结构表示:
/>
/>
其中:
R
R
在本文所述的任何方面或实施方案中,PTM由以下化学结构表示:
/>
在本文所述的任何方面或实施方案中,PTM的接头附接点如虚线所示:
治疗组合物
药物组合物代表本公开的另外方面,所述药物组合物包含与药学有效量的载体、添加剂或赋形剂组合的、有效量的如本文所述的至少一种双官能化合物和本文其他地方所述的一种或多种化合物的组合。
适用时,本公开包括包含药学上可接受的盐,特别是如本文所述的化合物的酸或碱加成盐的组合物。用于制备可根据此方面使用的上述基础化合物的药学上可接受的酸加成盐的酸是形成无毒酸加成盐的那些酸,即含有药学上可接受的阴离子的盐,例如盐酸盐、氢溴酸盐、氢碘酸盐、硝酸盐、硫酸盐、硫酸氢盐、磷酸盐、酸式磷酸盐、乙酸盐、乳酸盐、柠檬酸盐、酸式柠檬酸盐、酒石酸盐、酒石酸氢盐、琥珀酸盐、马来酸盐、富马酸盐、葡糖酸盐、蔗糖酸盐、苯甲酸盐、甲磺酸盐、乙磺酸盐、苯磺酸盐、对甲苯磺酸盐和双羟萘酸盐[即1,1'-亚甲基-双-(2-羟基-3萘甲酸)]盐等等。
药学上可接受的碱加成盐也可以用于制备根据本公开的化合物或衍生物的药学上可接受的盐形式。在性质中为酸性的、可以用作制备本文化合物的药学可接受的碱性盐的试剂的化学碱是与此类化合物形成无毒碱性盐的那些化学碱。此类无毒碱性盐包括但不限于源自此类药学上可接受的阳离子的那些,所述阳离子例如碱金属阳离子(例如钾和钠)和碱土金属阳离子(例如钙、锌和镁)、铵或水溶性胺加成盐如N-甲基葡糖胺-(葡甲胺),以及低级烷醇铵和药学上可接受的有机胺的其他碱性盐等等。
如本文所述的化合物可根据本公开通过经口、肠胃外或局部途径以单一剂量或分份剂量施用。活性化合物的施用范围可以从连续(静脉内滴注)到每天几次经口施用(例如,Q.I.D.),并且可以包括经口、局部、肠胃外、肌内、静脉内、皮下、透皮(其可以包括渗透增强剂)、经颊、舌下和栓剂施用,以及其他施用途径。肠溶衣经口片剂也可以用于增强来自经口施用途径的化合物的生物利用度。最有效的剂型取决于所选择的特定药剂的药代动力学以及患者中的疾病的严重程度。也可以使用作为用于鼻内、气管内或肺部施用的喷雾剂、薄雾或气溶胶的根据本公开的化合物施用。因此,本公开还涉及药物组合物,其包含有效量的如本文所述的化合物,任选地与药学上可接受的载体、添加剂或赋形剂组合。根据本公开的化合物可以立即释放、中间释放或持续或控制释放形式施用。持续或控制释放形式优选经口施用,也可以栓剂和透皮或其他局部形式施用。以脂质体形式的肌内注射也可以用于控制或维持化合物在注射部位处的释放。
如本文所述的组合物可使用一种或多种药学上可接受的载体以常规方式配制,并且还可以在控制释放制剂中施用。可以用于这些药物组合物中的药学可接受的载体包括但不限于离子交换剂、氧化铝、硬脂酸铝、卵磷脂、血清蛋白例如人血清白蛋白、缓冲物质例如磷酸盐、甘氨酸、山梨酸、山梨酸钾、饱和植物脂肪酸的偏甘油酯混合物、水、盐或电解质例如硫酸醇溶蛋白、磷酸氢二钠、磷酸氢钾、氯化钠、锌盐、胶体二氧化硅、三硅酸镁、聚乙烯吡咯烷酮、纤维素基物质、聚乙二醇、羧甲基纤维素钠、聚丙烯酸酯、蜡、聚乙烯-聚氧丙烯嵌段聚合物、聚乙二醇和羊毛脂。
如本文所述的组合物可以经口、肠胃外、吸入喷雾、局部、直肠、鼻腔、口腔、阴道或经由植入的储库施用。如本文所用的术语“肠胃外”包括皮下、静脉内、肌内、关节内、滑膜内、胸骨内、鞘内、肝内、病灶内和颅内注射或输注技术。优选地,组合物经口、腹膜内或静脉内施用。
如本文所述的组合物的无菌可注射形式可以是水性或油性悬浮液。可以根据本领域已知的技术,使用合适的分散剂或润湿剂和悬浮剂配制这些悬浮液。无菌可注射制剂还可以是在无毒的肠胃外可接受的稀释剂或溶剂中的无菌可注射溶液或悬浮液,例如作为在1,3-丁二醇中的溶液。在可以采用的可接受的媒介物和溶剂有水、林格氏溶液和等渗氯化钠溶液。另外,无菌的不挥发性油照常规用作溶剂或悬浮介质。为此目的,可以采用任何温和的不挥发性油,包括合成的甘油单酯或甘油二酯。脂肪酸,例如油酸及其甘油酯衍生物可用于制备注射剂,如天然的药学可接受的油,例如橄榄油或蓖麻油一样,尤其以其聚氧乙基化形式。这些油溶液或悬浮液也可以含有长链醇稀释剂或分散剂,例如Ph.Helv或类似的醇。
如本文所述的药物组合物可以以任何经口可接受的剂型经口施用,包括但不限于胶囊、片剂、水性悬浮液或溶液。在用于经口使用的片剂的情况下,常用的载体包括乳糖和玉米淀粉。通常还添加润滑剂,如硬脂酸镁。对于以胶囊形式的经口施用,有用的稀释剂包括乳糖和干玉米淀粉。当需要水性悬浮液用于经口使用时,将活性成分与乳化剂和悬浮剂混合。需要时,还可以加入某些甜味剂、调味剂或着色剂。
可替代地,如本文所述的药物组合物可以以栓剂的形式施用,用于直肠施用。这些可以通过将试剂与合适的非刺激性赋形剂混合来制备,所述赋形剂在室温下为固体但在直肠温度下为液体,并且因此将在直肠中融化以释放药物。此类材料包括可可脂、蜂蜡和聚乙二醇。
如本文所述的药物组合物也可以局部施用。对于这些区域或器官中的每一个,容易制备合适的局部制剂。对于下肠道的局部施用可以以直肠栓剂制剂(参见上文)或以合适的灌肠剂制剂实现。也可以使用局部可接受的透皮贴剂。
对于局部应用,药物组合物可以配制成合适的软膏,所述软膏含有悬浮或溶解于一种或多种载体中的活性组分。用于局部施用本发明的化合物的载体包括但不限于矿物油、液体凡士林、白凡士林、丙二醇、聚氧乙烯、聚氧丙烯化合物、乳化蜡和水。在本发明的某些优选方面,可以将化合物涂布到支架上,所述支架将要手术植入患者体内,以便抑制或减少支架在患者中发生阻塞的可能性。
可替代地,可以将药物组合物配制成合适的洗剂或乳膏,所述洗剂或乳膏含有悬浮或溶解于一种或多种药学可接受的载体中的活性组分。合适的载体包括但不限于矿物油、脱水山梨糖醇单硬脂酸酯、聚山梨醇酯60、十六烷基酯蜡、鲸蜡硬脂醇、2-辛基十二烷醇、苯甲醇和水。
对于眼部使用,药物组合物可以配制为在等渗、pH调节的无菌盐水中的微粉化悬浮液,或者优选地,配制为在等渗、pH调节的无菌盐水中的溶液,使用或不使用防腐剂如苯扎氯铵。可替代地,对于眼部使用,可以将药物组合物配制成软膏例如凡士林。
如本文所述的药物组合物还可通过鼻气溶胶或吸入施用。此类组合物根据药物制剂领域众所周知的技术制备,并且可以制备为在盐水中的溶液,采用苯甲醇或其他合适的防腐剂、增强生物利用度的吸收促进剂、碳氟化合物和/或其他常规增溶剂或分散剂。
可以与载体材料组合以制备单一剂型的如本文所述的药物组合物中化合物的量将根据所治疗的宿主和疾病、特定的施用模式而变。优选地,组合物应配制成包含约0.05毫克至约750毫克或更多、更优选地约1毫克至约600毫克、甚至更优选地约10毫克至约500毫克的活性成分,该活性成分单独存在或与至少一种根据本公开的其他化合物组合。
还应该理解,用于任何特定患者的具体剂量和治疗方案将取决于各种因素,包括所采用的特定化合物的活性、年龄、体重、一般健康、性别、饮食、施用时间、排泄率、药物组合、以及治疗医师的判断和待治疗的特定疾病或状况的严重程度。
需要根据本文所述的方法使用化合物治疗的患者或受试者可通过向患者(受试者)施用单独或与如本文其他地方鉴定的其他已知红血球生成刺激剂组合的有效量的根据本公开的化合物来治疗,所述化合物包括其药学上可接受的盐、溶剂化物或多晶型物,任选地在药学上可接受的载体或稀释剂中。
这些化合物可通过任何适当的途径施用,例如经口、肠胃外、静脉内、皮内、皮下或局部,包括透皮、在液体中、乳膏、凝胶或固体形式或通过气溶胶形式。
活性化合物包括在药学可接受的载体或稀释剂中,其量足以向患者递送对于所需适应症的治疗有效量,而不在所治疗的患者中引起严重的毒性效应。用于所有本文提及状况的活性化合物的优选剂量在约10ng/kg至300mg/kg的范围内,优选地0.1至100mg/kg/天,更一般地0.5至约25mg/千克接受者/患者的体重/天。典型的局部剂量范围为在合适的载体中0.01-5重量%。
该化合物方便地以任何合适的单位剂型施用,包括但不限于每单位剂型含有小于1mg、1mg至3000mg,优选5至500mg的活性成分的单位剂型。约25-250mg的经口剂量通常是方便的。
优选施用活性成分以达到约0.00001-30mM,优选约0.1-30μM的活性化合物的峰值血浆浓度。这可以例如通过任选地在盐水或含水介质中的活性成分溶液或制剂的静脉内注射来实现,或作为活性成分的推注来施用。经口施用也适合于生成活性剂的有效血浆浓度。
药物组合物中活性化合物的浓度将取决于药物的吸收、分布、失活和排泄速率,以及本领域技术人员已知的其他因素。应注意,剂量值也将随着待缓解的状况的严重程度而变。还应理解,对于任何特定受试者,应该根据个体需要和施用或监督组合物施用的个人的专业判断,随着时间过去调整特定剂量方案,并且本文所述的浓度范围仅为示例性的,并且不预期限制请求保护的组合物的范围或实践。活性成分可以一次施用,或者可以分成许多较小的剂量,以不同的时间间隔施用。
经口组合物通常将包含惰性稀释剂或可食用载体。它们可以包封在明胶胶囊中或压制成片剂。为了经口治疗施用的目的,可以将活性化合物或其前药衍生物与赋形剂混合,并且以片剂、锭剂或胶囊的形式使用。可包含药学上相容的粘合剂和/或佐剂材料作为组合物的一部分。
片剂、丸剂、胶囊、锭剂等等可以含有下述成分或类似性质的化合物中的任一种:粘合剂如微晶纤维素、黄蓍胶或明胶;赋形剂如淀粉或乳糖,分散剂如海藻酸、Primogel或玉米淀粉;润滑剂如硬脂酸镁或Sterotes;助流剂如胶体二氧化硅;甜味剂如蔗糖或糖精;或调味剂如薄荷、水杨酸甲酯或橙子调味料。当剂量单位形式是胶囊时,除上述类型的材料之外,它还可以含有液体载体如脂肪油。另外,剂量单位形式可以含有修改剂量单位的物理形式的各种其他物质,例如糖包衣、虫胶或肠溶剂。
活性化合物或其药学可接受的盐可以作为酏剂、悬浮液、糖浆剂、晶片、口香糖等等的组分施用。除活性化合物之外,糖浆剂还可以含有蔗糖作为甜味剂与某些防腐剂、染料以及着色剂和调味料。
活性化合物或其药学上可接受的盐也可以与不损害所需作用的其他活性物质混合,或与补充所需作用的物质混合,例如红细胞生成刺激剂,包括EPO和达依泊汀α等。在本发明的某些优选方面,根据本公开的一种或多种化合物与另一种生物活性剂共同施用,例如红细胞生成刺激剂或伤口愈合剂,包括抗生素,如本文其他地方所述。
用于肠胃外、皮内、皮下或局部施用的溶液或悬浮液可包含下列组分:无菌稀释剂如注射用水、盐水溶液、不挥发性油、聚乙二醇、丙三醇、丙二醇或其他合成溶剂;抗菌剂如苯甲醇或对羟基苯甲酸甲酯;抗氧化剂如抗坏血酸或亚硫酸氢钠;螯合剂如乙二胺四乙酸;缓冲剂如乙酸盐、柠檬酸盐或磷酸盐和用于调节张力的试剂如氯化钠或葡萄糖。肠胃外制剂可以包封在由玻璃或塑料制成的安瓿、一次性注射器或多剂量小瓶中。
如果静脉内施用,则优选的载体是生理盐水或磷酸盐缓冲盐水(PBS)。
在一个实施方案中,活性化合物与载体一起制备,所述载体将保护化合物免于从体内快速消除,例如控制释放制剂,包括植入物和微囊化递送系统。可以使用可生物降解的、生物相容的聚合物,例如乙烯乙酸乙烯酯、聚酐、聚乙醇酸、胶原、聚原酸酯和聚乳酸。用于制备此类制剂的方法对于本领域技术人员是显而易见的。
脂质体悬浮液也可以是药学可接受的载体。这些可以根据本领域技术人员已知的方法制备,例如,如美国专利号4,522,811中所述(其以引用的方式整体并入本文)。例如,脂质体制剂可以通过将适当的脂质(例如硬脂酰磷脂酰乙醇胺、硬脂酰磷脂酰胆碱、arachadoyl磷脂酰胆碱和胆固醇)溶解于有机溶剂中来制备,然后蒸发所述有机溶剂,在容器的表面上留下干燥脂质的薄膜。然后将活性化合物的水溶液引入容器中。然后用手旋转容器,以从容器的侧面释放脂质材料且分散脂质聚集体,从而形成脂质体悬浮液。
治疗方法
在另一方面,本说明书提供了治疗组合物,其包含有效量的如本文所述化合物或其盐形式和药学上可接受的载体。治疗组合物调节患者或受试者(例如动物,例如人)中的蛋白质降解,并且可以用于治疗或改善通过降解的蛋白质调节的疾病状态或状况。
如本文使用的,术语“治疗(treat)”、“治疗(treating)”和“治疗(treatment)”等指对患者提供益处的任何动作,对于所述患者可以施用本文化合物,所述益处包括通过本文化合物与之结合的蛋白质调节的任何疾病状态或状况的治疗。上文阐述了可以使用根据本公开的化合物治疗的疾病状态或病状,包括癌症。
本说明书提供了如本文所述的治疗组合物,用于实现目的蛋白质的降解,以治疗或改善疾病,例如癌症。在某些另外的实施方案中,疾病是多发性骨髓瘤。因此,在另一方面,本说明书提供了泛素化/降解细胞中的靶蛋白的方法。在某些实施方案中,该方法包括施用如本文所述的双官能化合物,该双官能化合物包含例如优选通过如本文另外所述的接头部分连接的CLM和PTM,其中CLM与PTM偶联且其中CLM识别泛素路径蛋白质(例如,泛素连接酶,优选E3泛素连接酶,如人小脑蛋白)并且PTM识别靶蛋白,使得当靶蛋白被置于泛素连接酶附近时,将发生靶蛋白的降解,从而实现靶蛋白的降解/靶蛋白效应的抑制及蛋白质水平的控制。通过本公开提供的蛋白质水平的控制提供了疾病状态或状况的治疗,其通过降低细胞(例如,患者的细胞)中该蛋白质的水平而通过靶蛋白调节。在某些实施方案中,该方法包括施用有效量的如本文所述的化合物,任选地包含药学上可接受的赋形剂、载体、佐剂、另一种生物活性剂或它们的组合。
在另外的实施方案中,本说明书提供了用于治疗或改善受试者或患者(例如动物,例如人类)的疾病、病症或其症状的方法,该方法包括向有需要的受试者施用组合物,该组合物包含有效量(例如治疗有效量)的本文所述化合物或其盐形式,和药学上可接受的赋形剂、载体、助剂、另一种生物活性剂或它们的组合,其中所述组合物能够有效治疗或改善受试者的疾病或病症或其症状。
在另一方面,本说明书提供了使用根据本公开的化合物鉴别生物系统中的所关注蛋白质降解的影响的方法。
在另一个实施方案中,本公开涉及治疗需要治疗通过蛋白质调节的疾病状态或状况治疗的人类患者的方法,其中该蛋白质的降解在该患者中产生治疗效果,所述方法包括向有需要的患者施用有效量的根据本公开的化合物,任选地与另一种生物活性剂组合。疾病状态或状况可以是由微生物剂或其他外源性剂(例如病毒、细菌、真菌、原生动物或其他微生物)引起的疾病,或者可以是由蛋白质的过表达引起的疾病状态,其导致疾病状态和/或状况。
术语“疾病状态或状况”用于描述任何疾病状态或状况,其中发生蛋白质失调(即,患者中表达的蛋白质的量升高),并且其中患者中一种或多种蛋白质的降解可以为对此有需要的患者提供有益疗法或症状的缓解。在某些情况下,疾病状态或状况可以被治愈。
可使用根据本公开的化合物治疗的疾病状态或状况包括例如哮喘、自体免疫疾病如多发性硬化症、各种癌症、纤毛病、腭裂、糖尿病、心脏病、高血压、发炎性肠病、智力迟钝、情绪障碍、肥胖症、屈光不正、不育症、Angelman综合征、Canavan病、乳糜泻、夏-马-图三氏病(Charcot-Marie-Tooth disease)、囊肿性纤维化、杜氏肌营养不良、血色病、血友病、克氏综合征、神经纤维瘤病、苯丙酮尿症、多囊性肾病(PKD1)或4(PKD2)、Prader-Willi综合征、镰状细胞病、Tay-Sachs病、Turner综合征。
可使用根据本公开的化合物治疗的其他疾病状态或状况包括阿尔茨海默病、肌萎缩性脊髓侧索硬化症(Lou Gehrig病)、神经性厌食症、焦虑症、动脉粥样硬化、注意力不足过动症、自闭症,双相情感障碍、慢性疲劳综合症、慢性阻塞性肺病、克罗恩病、冠心病、痴呆、抑郁症、1型糖尿病、2型糖尿病、癫痫、格林-巴利综合征、肠易激综合征、狼疮、代谢综合征、多发性硬化、心肌梗死、肥胖症、强迫症、恐慌症、帕金森病、牛皮癣、类风湿性关节炎、结节病、精神分裂症、中风、血栓闭塞性脉管炎、妥瑞症、血管炎。
可使用根据本公开的化合物治疗的其他疾病状态或状况包括铜蓝蛋白缺乏症、软骨成长不全II型、软骨发育不全、尖头、2型戈谢病、急性间歇性卟啉症、卡纳万病、腺瘤性结肠息肉病、ALA脱水酶缺乏症、腺苷酸琥珀酸裂解酶缺乏症、肾上腺生殖综合征、肾上腺脑白质营养不良、ALA-D卟啉症、ALA脱水酶缺乏症、黑尿症、亚历山大病、黑尿性褐黄病、α1-抗胰蛋白酶缺乏症、α-1蛋白酶抑制剂、肺气肿、肌萎缩性脊髓侧索硬化症、
在整个说明书中使用术语“瘤形成”或“癌症”是指导致癌性或恶性肿瘤(即,通过细胞增殖而生长的异常组织)形成和生长的病理过程,所述形成和生长通常比正常组织更快并且在引发新增长的刺激停止后继续生长。恶性肿瘤显示出部分或完全缺乏与正常组织的结构组织和功能协调,并且大多数侵入周围组织,转移到若干个部位,并且在尝试移除后可能复发并且除非充分治疗,否则导致患者死亡。如本文所用,术语瘤形成用于描述所有癌性疾病状态,并且包括或涵盖与恶性血源性、腹水和实体肿瘤相关的病理过程。可用本发明的化合物单独地或与至少一种另外的抗癌剂组合地治疗的示例性癌症包括鳞状细胞癌、基底细胞癌、腺癌、肝细胞癌和肾细胞癌;膀胱、肠道、乳房、子宫颈、结肠、食道、头、肾、肝、肺、颈、卵巢、胰腺、前列腺和胃的癌症;白血病;良性和恶性淋巴瘤,尤其是伯基特氏淋巴瘤(Burkitt’s lymphoma)和非霍奇金淋巴瘤(Non-Hodgkin’slymphoma);良性和恶性黑素瘤;骨髓增生性疾病;肉瘤,包括尤文氏肉瘤(Ewing’s sarcoma)、血管内皮瘤、卡波西肉瘤(Kaposi’s sarcoma)、脂肪肉瘤、肌肉瘤、外周神经上皮瘤、滑膜肉瘤、神经胶质瘤、星形细胞瘤、少突神经胶质瘤、室管膜瘤、胶质母细胞瘤、神经母细胞瘤、节细胞神经瘤、神经节细胞胶质瘤、髓母细胞瘤、松果体细胞肿瘤、脑膜瘤、脑膜肉瘤、神经纤维瘤和许旺细胞瘤(Schwannomas);肠道癌、乳癌、前列腺癌、子宫颈癌、子宫癌、肺癌、卵巢癌、睾丸癌、甲状腺癌、星形细胞瘤、食道癌、胰脏癌、胃癌、肝癌、结肠癌、黑素瘤;癌肉瘤、霍奇金病、维尔姆斯瘤和畸胎癌。可使用根据本公开的化合物治疗的另外的癌症包括例如T-谱系急性淋巴母细胞白血病(T-ALL)、T谱系淋巴母细胞淋巴瘤(T-LL)、外周T细胞淋巴瘤、成人T细胞白血病、Pre-B ALL、Pre-B淋巴瘤、大B细胞淋巴瘤、Burkitts淋巴瘤、B细胞ALL、费城染色体阳性ALL和费城染色体阳性CML。
术语“生物活性剂”用于描述除了根据本公开的化合物之外的试剂,其与本发明化合物组合用作具有生物活性的试剂,以帮助实现对于其使用本发明化合物的预期疗法、抑制和/或防止/预防。本文使用的优选生物活性剂包括药理学活性与所用或所施用的本发明化合物类似的那些试剂,并且包括例如抗癌剂、抗病毒剂,尤其包括抗HIV剂和抗HCV剂、抗微生物剂、抗真菌剂等。
术语“另外的抗癌剂”用于描述可以与根据本公开的化合物组合以治疗癌症的抗癌剂。这些剂包括例如依维莫司、曲贝替定、阿布拉生、TLK286、AV-299、DN-101、帕唑帕尼、GSK690693、RTA 744、ON0910.Na、AZD 6244(ARRY-142886)、AMN-107、TKI-258、GSK461364、AZD 1152、恩扎妥林、凡德他尼、ARQ-197、MK-0457、MLN8054、PHA-739358、R-763、AT-9263、FLT-3抑制剂、VEGFR抑制剂、EGFR TK抑制剂、极光激酶抑制剂、PIK-1调节剂、Bcl-2抑制剂、HDAC抑制剂、c-MET抑制剂、PARP抑制剂、Cdk抑制剂、EGFR TK抑制剂、IGFR-TK抑制剂、抗HGF抗体、PI3激酶抑制剂、AKT抑制剂、mTORC1/2抑制剂、JAK/STAT抑制剂、检查点-1或2抑制剂、病灶粘附激酶抑制剂、Map激酶(mek)抑制剂、VEGF截获抗体、培美曲塞、埃罗替尼、达沙替尼、尼罗替尼、德卡坦尼、帕尼单抗、氨柔比星、奥戈伏单抗、Lep-etu、诺拉曲特、azd2171、巴他布林、奥法木单抗、扎木单抗、艾特咔林(edotecarin)、汉防己碱、鲁比替康、替米利芬、奥利默森、替西木单抗、伊匹单抗、棉子酚、Bio 111、131-I-TM-601、ALT-110、BIO 140、CC8490、西仑吉肽、吉马替康、IL13-PE38QQR、INO 1001、IPdR
术语“抗HIV剂”或“另外的抗HIV剂”包括例如核苷逆转录酶抑制剂(NRTI)、其他非核苷类逆转录酶抑制剂(即不能代表本公开的那些)、蛋白酶抑制剂、融合抑制剂等,其示例性化合物可包括例如3TC(拉米夫定)、AZT(齐多夫定),(-)-FTC、ddI(地达诺新)、ddC(扎西他滨)、阿巴卡韦(ABC)、替诺福韦(PMPA)、D-D4FC(Reverset)、D4T(司他夫定)、Racivir、L-FddC、L-FD4C、NVP(奈韦拉平)、DLV(地拉夫定)、EFV(依法韦仑)、SQVM(甲磺酸沙奎那韦)、RTV(利托那韦)、IDV(茚地那韦)、SQV(沙奎那韦)、NFV(奈非那韦)、APV(安普那韦)、LPV(洛匹那韦)、融合抑制剂如T20等、其融合物和混合物,包括目前正处于临床试验或开发阶段的抗HIV化合物。
可与根据本公开的化合物共同施用的其他抗HIV剂包括例如其他NNRTI(即,除了根据本公开的NNRTI之外),可选自奈韦拉平(BI-R6-587)、地拉夫定(U-90152S/T)、依法韦仑(DMP-266)、UC-781(N-[4-氯-3-(3-甲基-2-丁烯基氧基)苯基]-2-甲基-3-呋喃甲酰胺、依曲韦林(TMC125)、曲韦定(Ly300046.HCl)、MKC-442(依米韦林,coactinon)、HI-236、HI-240、HI-280、HI-281、利匹韦林(TMC-278)、MSC-127、HBY 097、DMP266、黄芩苷(TJN-151)ADAM-II(3’,3’-二氯-4’,4”-二甲氧基-5’,5”-双(甲氧基羰基)-6,6-二苯基己烯酸甲酯)、3-溴-5-(1-5-溴-4-甲氧基-3-(甲氧基羰基)苯基)庚-1-烯基)-2-甲氧基苯甲酸甲酯(Alkenyldiarylmethane类似物,Adam类似物)、(5-氯-3-(苯亚磺酰基)-2’-吲哚甲酰胺)、AAP-BHAP(U-104489或PNU-104489)、卡普韦林(AG-1549、S-1153)、阿替韦啶(U-87201E)、金精三羧酸(SD-095345)、1-[(6-氰基-2-吲哚基)羰基]-4-[3-(异丙基氨基)-2-吡啶基]哌嗪、1-[5-[[N]-(甲基)甲基磺酰基氨基]-2-吲哚基羰基-4-[3-(异丙基氨基)-2-吡啶基]哌嗪、1-[3-(乙基氨基)-2-[吡啶基]-4-[(5-羟基-2-吲哚基)羰基]哌嗪、1-[(6-甲酰基-2-吲哚基)羰基]-4-[3-(异丙基氨基)-2-吡啶基]哌嗪、1-[[5-(甲基磺酰基氧基)-2-吲哚基)羰基]-4-[3-(异丙基氨基)-2-吡啶基]哌嗪、U88204E、双(2-硝基苯基)砜(NSC 633001)、卡拉内酯A(NSC675451)、卡拉内酯B、6-苄基-5-甲基-2-(环己基氧基)嘧啶-4-酮(DABO-546)、DPC961、E-EBU、E-EBU-dm、E-EPSeU、E-EPU、膦甲酸(Foscavir)、HEPT(1-[(2-羟乙氧基)甲基]-6-(苯硫基)胸腺嘧啶)、HEPT-M(1-[(2-羟乙氧基)甲基]-6-(3-甲基苯基)硫代)胸腺嘧啶)、HEPT-S(1-[(2-羟乙氧基)甲基]-6-(苯硫基)-2-硫代胸腺嘧啶)、海棠果素P、L-737,126、米歇尔胺A(NSC650898)、米歇尔胺B(NSC649324)、米歇尔胺F、6-(3,5-二甲基苄基)-1-[(2-羟乙氧基)甲基]-5-异丙基尿嘧啶、6-(3,5-二甲基苄基)-1-(乙氧基甲基)-5-异丙基尿嘧啶、NPPS、E-BPTU(NSC 648400)、奥替普拉(4-甲基-5-(吡嗪基)-3H-1,2-二硫杂环戊烯-3-硫酮)、N-{2-(2-氯-6-氟苯乙基)-N’-(2-噻唑基)硫脲(PETT Cl,F衍生物)、N-{2-(2,6-二氟苯乙基)-N’-[2-(5-溴吡啶基)]硫脲(PETT衍生物)、N-{2-(2,6-二氟苯乙基)-N’-[2-(5-甲基吡啶基)]硫脲{PETT吡啶基衍生物]、N-[2-(3-氟呋喃基)乙基]-N’-[2-(5-氯吡啶基)]硫脲、N-[2-(2-氟-6-乙氧基苯乙基)]-N’-[2-(5-溴吡啶基)]硫脲、N-(2-苯乙基)-N’-(2-噻唑基)硫脲(LY-73497)、L-697,639、L-697,593、L-697,661、3-[2-(4,7-二氟苯并噁唑-2-基)乙基}-5-乙基-6-甲基(吡啶-2(1H)-硫酮(2-吡啶酮衍生物)、3-[[(2-甲氧基-5,6-二甲基-3-吡啶基)甲基]胺]-5-乙基-6-甲基(吡啶-2(1H)-硫酮、R82150、R82913、R87232、R88703、R89439(洛韦胺)、R90385、S-2720、舒拉明钠、TBZ(噻唑并苯并咪唑,NSC625487)、噻唑并异吲哚-5-酮、(+)(R)-9b-(3,5-二甲基苯基-2,3-二氢噻唑并[2,3-a]异吲哚-5(9bH)-酮、替韦拉平(R86183)、UC-38和UC-84等。
适用时,术语“药学上可接受的盐”在本说明书中通篇用于描述本文所述的一种或多种化合物的盐形式,其用于增加化合物在患者的胃肠道的胃液中的溶解度,以便促进化合物的溶出度和生物利用度。适用时,药学上可接受的盐包括源自药学上可接受的无机或有机碱和酸的盐。合适的盐包括源自碱金属如钾和钠的盐,碱土金属如钙、镁和铵盐,以及制药领域中众所周知的众多其他酸和碱。钠盐和钾盐作为根据本公开的磷酸盐的中和盐是特别优选的。
术语“药学上可接受的衍生物”在本说明书中通篇用于描述任何药学上可接受的前药形式(例如酯、酰胺、其他前药基团),在施用于患者时,其直接或间接提供本发明化合物或本发明化合物的活性代谢产物。
一般合成方法
如本文所述的双官能分子的合成实现和优化可以逐步或模块化的方式进行。例如,如果不能立即获得合适的配体,则鉴定与靶分子结合的化合物可涉及高通量或中通量的筛查活动。初始配体需要迭代设计和优化循环来改善次优方面并不罕见,如通过合适的体外和药理学和/或ADMET测定所确定的那样。优化/SAR活动的一部分将是探测配体的耐受取代的位置,并且可能是附接本文先前提到的化学接头的合适位置。在可获得晶体学或NMR结构数据的情况下,这些可用于集中这样的合成任务。
以非常类似的方式,可以鉴定和优化E3连接酶的配体,即ULM/CLM。
利用PTM和ULM(例如CLM),本领域技术人员可以使用已知的合成方法将它们在有或没有接头部分的情况下进行组合。接头部分可以合成为具有一系列组成、长度和柔韧性,并且被官能化,使得PTM和ULM基团可以顺序地附接至接头的远端。因此,可以在体外和体内药理学和ADMET/PK研究中实现和分析双官能分子文库。与PTM和ULM基团一样,最终的双官能分子可以进行迭代设计和优化循环,以鉴定具有期望特性的分子。
本申请中描述的示例性化合物可通过连接根据方案2-30、2-31、2-40、2-41、2-45和2-46制备的右手关键片段来合成。本申请中要求保护的代表性化合物的详细制备在方案3-10、3-56、3-58和3-72中进一步描述。
A.示例性人小脑蛋白配体通用合成方案
合成方案2-30、2-31、2-40、2-41、2-45和2-46描述了用于制备CRBN配体的途径,以及连接有局部接头部分的CRBN配体。
制备中间产物的一般合成方案2-30。
制备中间产物的一般合成方案2-31。
制备中间产物的一般合成方案2-40。
制备中间产物的一般合成方案2-41。
制备中间产物的一般合成方案2-45。
/>
制备中间产物的一般合成方案2-46。
B.示例性PROTAC通用合成方案
合成方案3-10、3-56、3-58和3-72描述了用于制备本申请中要求保护的代表性嵌合化合物的途径。
制备请求保护的化合物的一般合成方案3-10。
制备请求保护的化合物的一般合成方案3-56。
制备请求保护的化合物的一般合成方案3-58。
制备请求保护的化合物的一般合成方案3-72。
示例性PROTAC
2-(4-(4-氯苯基)-2,3,9-三甲基-6H-噻吩并[3,2-f][1,2,4]三唑并[4,3-a][1,4]二氮杂环庚三烯-6-基)-N-(3-(3-((3-(2,4-二氧代-3,4-二氢嘧啶-1(2H)-基)喹啉-6-基)氧基)丙氧基)丙基)乙酰胺
合成方案:
向(S)-2-(4-(4-氯苯基)-2,3,9-三甲基-6H-噻吩并[3,2-f][1,2,4]三唑并[4,3-a][1,4]二氮杂环庚三烯-6-基)乙酸(20.6mg,0.051mmol)和1-(5-(3-(3-氨基丙氧基)丙氧基)喹啉-3-基)嘧啶-2,4(1H,3H)-二酮盐酸盐(21.6mg,0.053mmol)的DCM(1mL)溶液中添加二异丙基乙胺(0.022mL,0.128mmol)、HATU(20.1mg,0.053mmol),并将其在室温下搅拌2小时。用NaHCO3溶液洗涤反应物,分离有机层并干燥。通过二氧化硅柱色谱(10%MeOH/DCM)纯化产物,得到2-(4-(4-氯苯基)-2,3,9-三甲基-6H-噻吩并[3,2-f][1,2,4]三唑并[4,3-a][1,4]二氮杂环庚三烯-6-基)-N-(3-(3-((3-(2,4-二氧代-3,4-二氢嘧啶-1(2H)-基)喹啉-6-基)氧基)丙氧基)丙基)乙酰胺(25mg,65%)
LCMS(m/e+)=753.35[M+H]
示例性PROTAC
N-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基)-6-(4-(6-((1-(2,6-二氧代哌啶-3-基)-6-氧代-1,6-二氢哒嗪-4-基)氧基)己基)哌嗪-1-基)烟酰胺合成方案第1部分—N-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基)-6-(哌嗪-1-基)烟酰胺的合成
步骤1:6-(4-(叔丁氧基羰基)哌嗪-1-基)烟酸的合成
将6-氯烟酸(1.6g,10.0mmol)溶解于N,N-二甲基乙酰胺(15mL)中,向其中添加哌嗪-1-羧酸叔丁酯(1.9g,10.0mmol)和乙基二异丙胺(2.6g,20mmol),然后在130℃下搅拌过夜。减压浓缩反应混合物,向所得的残余物中添加1M NaOH水溶液(10mL),然后用CHCl
LC-MS(Agilent LCMS 1200-6120,色谱柱:Waters X-Bridge C18(50mm*4.6mm*3.5μm);柱温:40℃;流速:2.0mL/min;流动相:在1.6分钟内从95%[水+10mM NH
化学式:C
步骤2:4-(5-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基氨基甲酰基)吡啶-2-基)哌嗪-1-羧酸叔丁酯的合成
将6-(4-(叔丁氧基羰基)哌嗪-1-基)烟酸(614mg,2.0mmol)、4-((1r,3r)-3-氨基-2,2,4,4-四甲基环丁氧基)-2-氯苯甲腈盐酸盐(630mg,2.0mmol)、2-(7-氮杂-1H-苯并三唑-1-基)-1,1,3,3-四甲基脲六氟磷酸酯(1.1g,3.0mmol)和乙基二异丙胺(516mg,4.0mmol)在二氯甲烷(20mL)中的混合物在室温下搅拌过夜。添加水(50mL),并用二氯甲烷(50mL x 3)萃取。将合并的有机层用盐水(50mL x 2)洗涤,经无水硫酸钠干燥。浓缩溶剂,得到残余物,将其通过硅胶柱色谱(石油醚/乙酸乙酯=1/1)进行纯化,得到4-(5-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基氨基甲酰基)吡啶-2-基)哌嗪-1-羧酸叔丁酯(977mg,86%收率),呈白色固体。
LC-MS(Agilent LCMS 1200-6120,色谱柱:Waters X-Bridge C18(50mm*4.6mm*3.5μm);柱温:40℃;流速:2.0mL/min;流动相:在1.6分钟内从95%[水+10mM NH
1
化学式:C
得自HNMR数据的总H计数:38。
步骤3:N-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基)-6-(哌嗪-1-基)烟酰胺盐酸盐的合成
将4-(5-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基氨基甲酰基)吡啶-2-基)哌嗪-1-羧酸叔丁酯(405mg,0.7mmol)在HCl/1,4-二氧杂环己烷(10mL)中的混合物在室温下搅拌4小时。真空去除溶剂,得到N-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基)-6-(哌嗪-1-基)烟酰胺盐酸盐(353mg,100%收率),呈白色固体。
LC-MS(Agilent LCMS 1200-6120,色谱柱:Waters X-Bridge C18(50mm*4.6mm*3.5μm);柱温:40℃;流速:2.0mL/min;流动相:在1.6分钟内从95%[水+10mM NH
化学式:C
合成方案第2部分:
步骤4:4,5-二氯-2-(4-甲氧基苄基)哒嗪-3(2H)-酮的合成
将4,5-二氯哒嗪-3(2H)-酮(5.0g,30.5mmol)、1-(氯甲基)-4-甲氧基苯(7.1g,45.7mmol)和碳酸钾(12.6g,91.5mmol)在N’,N’-二甲基甲酰胺(100mL)中的混合物在室温下搅拌12小时。将该混合物倒入水中,并用乙酸乙酯(100mL x 3)萃取。将合并的有机相真空浓缩,并通过硅胶柱色谱(石油醚/乙酸乙酯=3/1)纯化残余物,得到4,5-二氯-2-(4-甲氧基苄基)哒嗪-3(2H)-酮(6.3g,73%收率),呈白色固体。
LC-MS(Agilent LCMS 1200-6110,色谱柱:Waters X-Bridge C18(50mm x 4.6mmx 3.5μm);柱温:40℃;流速:1.5mL/min;流动相:在1.5分钟内从95%[水+0.05%TFA]和5%[CH
化学式:C
步骤5:5-(6-(苄氧基)己氧基)-4-氯-2-(4-甲氧基苄基)哒嗪-3(2H)-酮的合成
在0℃下,向6-(苄氧基)己-1-醇(1.04g,50mmol)的无水THF(100mL)溶液中添加60%NaH(240mg,60mmol),然后将其搅拌30分钟,添加4,5-二氯-2-(4-甲氧基苄基)哒嗪-3(2H)-酮(1.42g,50mmol),将所得混合物回流过夜。冷却至室温后,将该混合物用NH
1
得自HNMR数据的总H计数:29。
步骤6:5-(6-(苄氧基)己氧基)-4-氯哒嗪-3(2H)-酮的合成
在0°下,向5-(6-(苄氧基)己氧基)-4-氯-2-(4-甲氧基苄基)哒嗪-3(2H)-酮(450mg,1mmol)的CH
LC-MS(Agilent LCMS 1200-6110,色谱柱:Waters X-Bridge C18(50mm x 4.6mmx 3.5μm);柱温:40℃;流速:1.5mL/min;流动相:在1.5分钟内从95%[水+0.05%TFA]和5%[CH
化学式:C
步骤7:3-(4-(6-(苄氧基)己氧基)-5-氯-6-氧代哒嗪-1(6H)-基)哌啶-2,6-二酮的合成
将5-(6-(苄氧基)己氧基)-4-氯哒嗪-3(2H)-酮(250mg,0.74mmol)、3-溴哌啶-2,6-二酮(143mg,0.74mmol)和碳酸钾(205mg,1.48mmol)在乙腈(40mL)中的混合物在室温下搅拌3天,然后过滤。将滤液浓缩并通过硅胶柱色谱(石油醚/乙酸乙酯=3/2)进行纯化,得到3-(4-(6-(苄氧基)己氧基)-5-氯-6-氧代哒嗪-1(6H)-基)哌啶-2,6-二酮(180mg,54%收率),呈浅黄色凝胶。
1
得自HNMR数据的总H计数:26。
步骤8:3-(4-(6-羟基己氧基)-6-氧代哒嗪-1(6H)-基)哌啶-2,6-二酮的合成
将3-(4-(6-(苄氧基)己氧基)-5-氯-6-氧代哒嗪-1(6H)-基)哌啶-2,6-二酮(180mg,0.4mmol)和10%活性炭载钯(100mg)在MeOH(20mL)中的混合物在1atm氢气气氛和室温下搅拌2小时。过滤去除固体,浓缩滤液,得到3-(4-(6-羟基己氧基)-6-氧代哒嗪-1(6H)-基)哌啶-2,6-二酮(118mg,90%收率),呈浅黄色固体。
1
得自HNMR数据的总H计数:21。
步骤9:6-(1-(2,6-二氧代哌啶-3-基)-6-氧代-1,6-二氢哒嗪-4-基氧基)己醛的合成
向3-(4-(6-羟基己氧基)-6-氧代哒嗪-1(6H)-基)哌啶-2,6-二酮(64mg,0.2mmol)的CH
LC-MS(Agilent LCMS 1200-6110,色谱柱:Waters X-Bridge C18(50mm x4.6mmx3.5μm);柱温:40℃;流速:1.5mL/min;流动相:在1.5分钟内从95%[水+0.05%TFA]和5%[CH
化学式:C
步骤10:N-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基)-6-(4-(6-(1-(2,6-二氧代哌啶-3-基)-6-氧代-1,6-二氢哒嗪-4-基氧基)己基)哌嗪-1-基)烟酰胺的合成
/>
向N-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基)-6-(哌嗪-1-基)烟酰胺盐酸盐(100mg,0.2mmol)的MeOH(5mL)溶液中添加6-(1-(2,6-二氧代哌啶-3-基)-6-氧代-1,6-二氢哒嗪-4-基氧基)己醛(64mg,0.2mmol)的CH
LC-MS(Agilent LCMS 1200-6120;流动相:在3.0分钟内从95%[水+10mM NH
[CH
HPLC(Agilent HPLC 1200,色谱柱:Waters X-Bridge C18(150mm x4.6mm x 3.5μm);柱温:40℃;流速:1.0mL/min;流动相:在10分钟内从95%[水+10mM NH
1
化学式:C
得自HNMR数据的总H计数:49。
示例性PROTAC
N-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基)-6-(4-(5-((3-(2,6-二氧代哌啶-3-基)-2-甲基-4-氧代-3,4-二氢喹唑啉-8-基)氧基)戊基)哌嗪-1-基)烟酰胺
合成方案
步骤1:3-(8-羟基-2-甲基-4-氧代喹唑啉-3(4H)-基)哌啶-2,6-二酮的合成
在室温下,向2-氨基-3-羟基苯甲酸(2.0g,13.1mmol)和咪唑(2.0g,29.4mmol)在乙腈(30mL)中的搅拌混合物中添加乙酰氯(2.0mL,28.7mmol)。将该混合物在室温下搅拌2天。向该混合物中添加3-氨基-哌啶-2,6-二酮氯化氢(2.2g,13.1mmol)、咪唑(2.0g,29.4mmol)和亚磷酸三苯酯(4.11mL,15.7mmol),并加热至回流3天。向该混合物中添加水(60mL)和浓HCl,直至pH=1。真空去除溶剂。向残余物中添加水(50mL)。用乙酸乙酯(2x50mL)萃取水层。向水层中添加碳酸氢钠(1.8g)至pH=7-8,并将该混合物在室温下搅拌,得到悬浮液。将该悬浮液过滤并干燥,得到3-(8-羟基-2-甲基-4-氧代-4H-喹唑啉-3-基)-哌啶-2,6-二酮(230mg,6%收率),呈灰色固体。
1
得自HNMR数据的总H计数:13。
步骤2:3-(8-(5-氯戊氧基)-2-甲基-4-氧代喹唑啉-3(4H)-基)哌啶-2,6-二酮的合成
在室温下,向3-(8-羟基-2-甲基-4-氧代-4H-喹唑啉-3-基)-哌啶-2,6-二酮(91mg,0.32mmol)和5-氯戊基-4-甲基苯磺酸酯(88mg,0.32mmol)的DMF(10mL)溶液中添加K
1
得自HNMR数据的总H计数:21。
步骤3:N-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基)-6-(4-(5-(3-(2,6-二氧代哌啶-3-基)-2-甲基-4-氧代-3,4-二氢喹唑啉-8-基氧基)戊基)哌嗪-1-基)烟酰胺的合成
将3-(8-(5-氯戊氧基)-2-甲基-4-氧代喹唑啉-3(4H)-基)哌啶-2,6-二酮(15mg,0.038mmol)、DIEA(25mg,0.19mmol)、KI(6mg,0.038mmol)和N-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基)-6-(哌嗪-1-基)烟酰胺(20mg,0.038mmol)在CH
LC-MS(Agilent LCMS 1200-6120;流动相:在3.0分钟内从95%[水+10mM NH
HPLC(Agilent HPLC 1200,色谱柱:Waters X-Bridge C18(150mm x4.6mm x 3.5μm);柱温:40℃;流速:1.0mL/min;流动相:在10分钟内从95%[水+10mM NH
1
化学式:C
得自HNMR数据的总H计数:51。
示例性PROTAC
N-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基)-6-(4-(5-((2-(2,6-二氧代哌啶-3-基)-1,1-二氧代-3-氧代-2,3-二氢苯并[d]异噻唑-6-基)氧基)戊基)哌嗪-1-基)烟酰胺
合成方案
步骤1:6-((5-羟基戊基)氧基)苯并[d]异噻唑-3(2H)-酮1,1-二氧化物的合成
在氮气气氛下,向戊烷-1,5-二醇(1.73g,16.7mmol)的N,N-二甲基甲酰胺(15.0mL)溶液中添加氢化钠(266mg,6.66mmol)。将反应混合物在室温下搅拌1小时。然后添加6-硝基苯并[d]异噻唑-3(2H)-酮1,1-二氧化物(760mg,3.33mmol),并在70℃下搅拌12小时。冷却至室温后,真空去除溶剂。用乙酸乙酯(30mL x3)和水(30mL)萃取残余物。用盐水(5mL)洗涤有机层。将合并的有机相经无水硫酸钠干燥,过滤并真空浓缩。用甲醇(3mL)洗涤残余物,得到6-((5-羟基戊基)氧基)苯并[d]异噻唑-3(2H)-酮1,1-二氧化物(560mg,59%),呈浅黄色固体。
Agilent LC-MS(Agilent LCMS 1200-6120,色谱柱:Waters X-Bridge C18(50mm*4.6mm*3.5μm);柱温:40℃;流速:2.0mL/min;流动相:在1.6分钟内从95%[水+10mMNH
步骤2:5-((1,1-二氧代-3-氧代-2,3-二氢苯并[d]异噻唑-6-基)氧基)戊基甲磺酸酯的合成
在氮气下,向6-((5-羟基戊基)氧基)苯并[d]异噻唑-3(2H)-酮(120mg,0.421mmol)的四氢呋喃(10.0mL)溶液中添加三乙胺(85.1mg,0.841mmol)和甲磺酰氯(38.5mg,0.336mmol)。将所得反应混合物在室温下搅拌0.5小时。将溶剂真空浓缩。用二氯甲烷(10mL x 3)和水(20mL)萃取残余物。将有机相干燥并真空浓缩,得到呈黄色油状物的粗制物5-((1,1-二氧代-3-氧代-2,3-二氢苯并[d]异噻唑-6-基)氧基)戊基甲磺酸酯,将其用于下一步骤,无需进一步纯化。
Agilent LC-MS(Agilent LCMS 1200-6120,色谱柱:Waters X-Bridge C18(30mm*4.6mm*3.5μm);柱温:40℃;流速:2.0mL/min;流动相:在0.5分钟内从90%[水+10mMNH
步骤3:N-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基)-6-(4-(5-((1,1-二氧代-3-氧代-2,3-二氢苯并[d]异噻唑-6-基)氧基)戊基)哌嗪-1-基)烟酰胺
向5-((1,1-二氧代-3-氧代-2,3-二氢苯并[d]异噻唑-6-基)氧基)戊基甲磺酸酯(0.421mmol)的乙腈(5mL)溶液中添加碳酸钾(291mg,2.11mmol)和N-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基)-6-(哌嗪-1-基)烟酰胺盐酸盐(212mg,0.421mmol)。将所得反应混合物在90℃下搅拌16小时。将溶剂真空浓缩。用乙酸乙酯(20mL x 3)和水(20mL)萃取残余物。将有机相干燥并真空浓缩。通过制备型HPLC纯化残余物,得到N-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基)-6-(4-(5-((1,1-二氧代-3-氧代-2,3-二氢苯并[d]异噻唑-6-基)氧基)戊基)哌嗪-1-基)烟酰胺(34mg,11%,两步),呈浅黄色固体。
Agilent LC-MS(Agilent LCMS 1200-6120,色谱柱:Waters X-Bridge C18(30mm*4.6mm*3.5μm);柱温:40℃;流速:2.0mL/min;流动相:在0.5分钟内从90%[水+10mMNH
步骤4:N-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基)-6-(4-(5-((2-(2,6-二氧代哌啶-3-基)-1,1-二氧代-3-氧代-2,3-二氢苯并[d]异噻唑-6-基)氧基)戊基)哌嗪-1-基)烟酰胺的合成
向N-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基)-6-(4-(5-((1,1-二氧代-3-氧代-2,3-二氢苯并[d]异噻唑-6-基)氧基)戊基)哌嗪-1-基)烟酰胺(30mg,0.0408mmol)的1,4-二氧杂环己烷/N,N-二甲基甲酰胺向(5mL/0.5mL)溶液中添加3-溴哌啶-2,6-二酮(11.8mg,0.0612mmol)和叔丁醇钾(9.16mg,0.0816mmol)。将该反应混合物在100℃下搅拌过夜。冷却至室温后,添加冰水(2.0mL)并用盐酸(1N)调节至PH=2至3,然后用乙酸乙酯(20.0mL x 3)萃取。将合并的有机相用盐水(5.0mL)洗涤,经无水硫酸钠干燥,过滤并真空浓缩。通过制备型HPLC和制备型TLC(二氯甲烷/甲醇=10:1)纯化残余物,得到N-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基)-6-(4-(5-((2-(2,6-二氧代哌啶-3-基)-1,1-二氧代-3-氧代-2,3-二氢苯并[d]异噻唑-6-基)氧基)戊基)哌嗪-1-基)烟酰胺(6.8mg,20%),呈白色固体。
LC-MS(Agilent LCMS 1200-6120,色谱柱:Waters X-Bridge C18(50mm*4.6mm*3.5μm);柱温:40℃;流速:2.0mL/min;流动相:在3.0分钟内从95%[水+10mM NH
HPLC(Agilent HPLC 1200,色谱柱:Waters X-Bridge C18(150mm*4.6mm*3.5μm);柱温:40℃;流速:1.0mL/min;流动相:在10分钟内从95%[水+10mM NH
1
化学式:C
得自HNMR数据的总H计数:48。
示例性PROTAC
N-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基)-6-(4-(2-((2-(2,4-二氟苯基)-1-氧代异吲哚啉-4-基)氧基)乙基)哌嗪-1-基)烟酰胺
合成方案:
步骤1:N-(2,4-二氟苯基)-3-甲氧基-2-甲基苯甲酰胺的合成
将3-甲氧基-2-甲基苯甲酸(5g,30mmol)、草酰氯(5.6g,150mmol)和N,N-二甲基甲酰胺(0.1ml)在二氯甲烷(20ml)中的混合物在室温下搅拌2小时。TLC显示反应完成。减压蒸发挥发物,得到呈黄色油状物的3-甲氧基-2-甲基苯甲酰氯(粗制物),将其用于下一步骤,无需进一步纯化。将3-甲氧基-2-甲基苯甲酰氯(粗制物)、2,4-二氟苯胺(3.8g,30mmol)和三乙胺(12g,120mmol)在二氯甲烷(20ml)中的混合物在室温下搅拌1小时。TLC显示反应完成。将反应混合物用二氯甲烷(20ml)稀释,用盐水(20ml)洗涤,经无水硫酸钠干燥并减压浓缩,得到粗残余物,将其通过硅胶快速色谱进行纯化,得到N-(2,4-二氟苯基)-3-甲氧基-2-甲基苯甲酰胺(5.8g,收率69%),呈黄色油状物。
步骤2:2-(溴甲基)-N-(2,4-二氟苯基)-3-甲氧基苯甲酰胺的合成
将N-(2,4-二氟苯基)-3-甲氧基-2-甲基苯甲酰胺(5.8g,20.9mmol)、N-溴代琥珀酰亚胺(3.9g,31.4mmol)和AIBN(2,2’-偶氮二(2-甲基丙腈))(342mg,2.09mmol)在四氯化碳(30mL)中的混合物在70℃下搅拌过夜。减压蒸发挥发物,并通过硅胶快速柱色谱(用10-20%乙酸乙酯的己烷溶液洗脱)进行纯化,得到2-(溴甲基)-N-(2,4-二氟苯基)-3-甲氧基苯甲酰胺(5.9g,收率80%),呈白色固体。
LC_MS:(ES
步骤3:2-(2,4-二氟苯基)-4-甲氧基异吲哚-1-酮的合成
在0℃下,向2-(溴甲基)-N-(2,4-二氟苯基)-3-甲氧基苯甲酰胺(2.0g,5.6mmol)的无水四氢呋喃(20mL)溶液中添加叔丁醇钾(1M的四氢呋喃溶液,8.4ml,8.4mmol),并将所得混合物在0℃下搅拌2小时。TLC显示反应完成。将反应混合物在水(50ml)和乙酸乙酯(50ml)之间分配。将有机层收集,用盐水(20ml x 2)洗涤,经无水硫酸钠干燥并减压浓缩,得到粗残余物,将其通过硅胶快速色谱(用20%乙酸乙酯的己烷溶液洗脱)进行纯化,得到2-(2,4-二氟苯基)-4-甲氧基异吲哚-1-酮(500mg,收率33%),呈黄色固体。
步骤4:2-(2,4-二氟苯基)-4-羟基异吲哚啉-1-酮的合成
将2-(2,4-二氟苯基)-4-甲氧基异吲哚-1-酮(200mg,0.727mmol)在溴化氢的乙酸溶液(33%,3ml)中的混合物在100℃下搅拌2天。TLC显示反应完成。减压蒸发挥发物,得到粗残余物,将其通过硅胶快速色谱(用30-50%乙酸乙酯的己烷溶液洗脱)进行纯化,得到2-(2,4-二氟苯基)-4-羟基异吲哚-1-酮(180mg,收率95%),呈黄色油状物。
LC_MS:(ES+):m/z 262.1[M+H]
步骤5:4-(烯丙氧基)-2-(2,4-二氟苯基)异吲哚啉-1-酮的合成
在0℃下,向2-(2,4-二氟苯基)-4-羟基异吲哚-1-酮(180mg,0.68mmol)、三苯基膦(539mg,2.06mmol)和丙-2-烯-1-醇(119mg,2.06mmol)在四氢呋喃(5ml)中的搅拌溶液中添加偶氮二羧酸二异丙酯(416mg,2.06mmol)的四氢呋喃(2ml)溶液,并将反应混合物在0℃下搅拌30分钟。TLC显示反应完成。减压蒸发挥发物,得到粗残余物,将其通过硅胶快速色谱(用10-20%乙酸乙酯的己烷溶液洗脱)进行纯化,得到4-(烯丙氧基)-2-(2,4-二氟苯基)异吲哚啉-1-酮(180mg,收率87%),呈无色油状物。
LC_MS:(ES+):m/z 302.2[M+H]
步骤6:2-((2-(2,4-二氟苯基)-1-氧代异吲哚啉-4-基)氧基)乙醛的合成
在-78℃下,将富含臭氧的氧气蒸汽鼓泡通过4-(烯丙氧基)-2-(2,4-二氟苯基)异吲哚啉-1-酮(180mg,0.59mmol)的二氯甲烷(20mL)溶液,直到反应混合物变成深蓝色。在-78℃下,用氧气吹扫溶液20分钟,以去除过量的臭氧。然后,在-78℃下,向反应混合物中添加二甲基硫醚(1.5ml,20.4mmol);使该混合物加温至室温并搅拌过夜。TLC显示反应完成。减压浓缩反应混合物,得到2-((2-(2,4-二氟苯基)-1-氧代异吲哚啉-4-基)氧基)乙醛(180mg,100%),将其用于下一步骤,无需进一步纯化。
1
化学式:C
得自HNMR数据的总H计数:11;
步骤7:N-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基)-6-(4-(2-((2-(2,4-二氟苯基)-1-氧代异吲哚啉-4-基)氧基)乙基)哌嗪-1-基)烟酰胺的合成
在室温下,向2-((2-(2,4-二氟苯基)-1-氧代异吲哚啉-4-基)氧基)乙醛(160mg,0.53mmol)、N-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基)-6-(哌嗪-1-基)烟酰胺(300mg,0.6mmol,示例性PROTAC 29的合成的中间产物)和乙酸(2滴)在甲醇(3mL)中的搅拌溶液中添加氰基硼氢化钠(150mg,2.4mmol)。将反应混合物在室温下搅拌过夜。TLC显示反应完成。将反应混合物在乙酸乙酯(40ml)和水(20ml)之间分配。将有机层收集,用盐水(20ml)洗涤,经无水硫酸钠干燥并减压浓缩,得到粗残余物,将其通过制备型TLC(用10%甲醇的二氯甲烷溶液洗脱)进行纯化,得到N-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基)-6-(4-(2-((2-(2,4-二氟苯基)-1-氧代异吲哚啉-4-基)氧基)乙基)哌嗪-1-基)烟酰胺(50mg,收率12%,3步),呈浅黄色固体。
1
化学式:C
得自HNMR数据的总H计数:40;
LC_MS:(ES+):m/z 755.6[M+H]
示例性PROTAC
N-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基)-6-(4-(5-((2-(2,4-二氟苯基)-1,3-二氧代异吲哚啉-5-基)氧基)戊基)哌嗪-1-基)烟酰胺
合成方案:
步骤1:2-(2,4-二氟苯基)-5-羟基异吲哚啉-1,3-二酮的合成
在室温下,向4-羟基邻苯二甲酸(2g,10.98mmol)的乙腈(50ml)溶液中分批添加1,1'-羰基二咪唑(3.9g,24.16mmol)。搅拌30分钟后,添加2,4-二氟苯胺(1.6g,12.08mmol),并将所得混合物在70℃下搅拌3小时。TLC显示反应完成。将反应混合物在乙酸乙酯(50ml)和水(50ml)之间分配,将有机层用盐水(50ml×2)洗涤,经无水硫酸钠干燥并减压浓缩,得到粗残余物,将其通过硅胶快速柱色谱(用25-35%乙酸乙酯的己烷溶液洗脱)进行纯化,得到2-(2,4-二氟苯基)-5-羟基异吲哚啉-1,3-二酮(2.1g,收率70%),呈黄色固体。
LC_MS:(ES
1
化学式:C
得自HNMR数据的总H计数:7。
步骤2:2-(2,4-二氟苯基)-5-((5-羟基戊基)氧基)异吲哚啉-1,3-二酮的合成
将2-(2,4-二氟苯基)-5-羟基异吲哚啉-1,3-二酮(300mg,1.09mmol)、5-羟基戊基4-甲基苯磺酸酯(282mg,1.09mmol)和碳酸钾(301mg,2.18mmol)在N,N-二甲基甲酰胺(5ml)中的混合物在50℃下搅拌过夜。TLC显示反应完成。将反应混合物在乙酸乙酯(30ml)和水(30ml)之间分配,将有机层用盐水(30ml×2)洗涤,经无水硫酸钠干燥并减压浓缩,得到粗残余物,将其通过硅胶快速柱色谱(用40-50%乙酸乙酯的己烷溶液洗脱)进行纯化,得到2-(2,4-二氟苯基)-5-((5-羟基戊基)氧基)异吲哚啉-1,3-二酮(217mg,收率55%),呈白色固体。
LC_MS:(ES
1
化学式:C
得自HNMR数据的总H计数:16。
步骤3:5-((2-(2,4-二氟苯基)-1,3-二氧代异吲哚啉-5-基)氧基)戊基4-甲基苯磺酸酯的合成
在0℃下,向2-(2,4-二氟苯基)-5-((5-羟基戊基)氧基)异吲哚啉-1,3-二酮(217mg,0.60mmol)、三乙胺(122mg,1.20mmol)和N,N-二甲基吡啶-4-胺(7.3mg,0.06mmol)的二氯甲烷(20ml)溶液中添加4-甲苯磺酰氯(171mg,0.90mmol),使反应混合物加温至室温并搅拌过夜。TLC显示反应完成。将反应混合物用二氯甲烷(30ml)稀释,用水(50ml)洗涤,然后用盐水(50ml)洗涤,经无水硫酸钠干燥并减压浓缩,得到粗残余物,将其通过硅胶快速色谱(用30-50%乙酸乙酯的己烷溶液洗脱)进行纯化,得到5-((2-(2,4-二氟苯基)-1,3-二氧代异吲哚啉-5-基)氧基)戊基4-甲基苯磺酸酯(208mg,收率67%),呈白色固体。
LC_MS:(ES
1
化学式:C
得自HNMR数据的总H计数:23。
步骤4:N-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基)-6-(4-(5-((2-(2,4-二氟苯基)-1,3-二氧代异吲哚啉-5-基)氧基)戊基)哌嗪-1-基)烟酰胺的合成
向5-((2-(2,4-二氟苯基)-1,3-二氧代异吲哚啉-5-基)氧基)戊基4-甲基苯磺酸酯(110mg,0.21mmol)、N-乙基-N-异丙基丙烷-2-胺(55mg,0.43mmol)和碘化钾(3mg,0.02mmol)在N,N-二甲基甲酰胺(2ml)中的搅拌溶液中添加N-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基)-6-(哌嗪-1-基)烟酰胺(100mg,0.21mmol,示例性PROTAC29的合成的中间产物),并且在氮气下,将该混合物在50℃下搅拌过夜。TLC显示反应完成。将反应混合物在乙酸乙酯(50ml)和水(30ml)之间分配,将有机层收集并用盐水(20ml×2)洗涤,经无水硫酸钠干燥并减压浓缩,得到粗残余物,将其通过硅胶快速柱色谱(用2-5%甲醇的二氯甲烷溶液洗脱)进行纯化,得到N-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基)-6-(4-(5-((2-(2,4-二氟苯基)-1,3-二氧代异吲哚啉-5-基)氧基)戊基)哌嗪-1-基)烟酰胺(98.4mg,收率57%),呈白色固体。
LC_MS:(ES
1
化学式:C
得自HNMR数据的总H计数:45。
示例性PROTAC
N-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基)-6-(4-(5-((2-(6-氰基-2-氧代-1,2-二氢吡啶-3-基)-1,3-二氧代异吲哚啉-5-基)氧基)戊基)哌嗪-1-基)烟酰胺
合成方案
步骤1:5-(5-羟基-1,3-二氧代异吲哚啉-2-基)-6-甲氧基氰基吡啶的合成
将5-氨基-6-甲氧基氰基吡啶(600mg,4.02mmol)和5-羟基异苯并呋喃-1,3-二酮(660mg,4.02mmol)在冰醋酸(4mL)中的混合物在100℃下搅拌过夜,然后冷却至室温。添加水(40mL)。将该混合物用饱和碳酸氢钠中和至pH>7。用乙酸乙酯(20mL x 3)萃取混合物。将合并的有机层用盐水(10mL x3)洗涤,经无水硫酸钠干燥,过滤并真空浓缩。用乙醚洗涤残余物,得到5-(5-羟基-1,3-二氧代异吲哚啉-2-基)-6-甲氧基氰基吡啶(650mg,55%),呈黄色固体。
LC-MS(Agilent LCMS 1200-6120,色谱柱:Waters X-Bridge C18(30mm*4.6mm*3.5μm);柱温:40℃;流速:2.0mL/min;流动相:在0.5分钟内从90%[水+10mM NH
步骤2:5-(5-(5-氯戊氧基)-1,3-二氧代异吲哚啉-2-基)-6-甲氧基氰基吡啶的合成
将5-(5-羟基-1,3-二氧代异吲哚啉-2-基)-6-甲氧基氰基吡啶(200mg,0.68mmol)、碳酸钾(188mg,1.36mmol)和5-氯戊基4-甲基苯磺酸酯(187mg,0.68mmol)在二甲基亚砜(5mL)中的混合物在40℃下搅拌2小时。使所得混合物冷却至室温。添加水(20mL)和乙酸乙酯(20mL)。将有机层分离,用盐水(10mL x 2)洗涤,经无水硫酸钠干燥,过滤并真空浓缩,得到粗产物,将其通过制备型TLC(乙酸乙酯/石油醚=1:1)进行纯化,得到5-(5-(5-氯戊氧基)-1,3-二氧代异吲哚啉-2-基)-6-甲氧基氰基吡啶(100mg,37%),呈黄色固体
步骤3:N-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基)-6-(4-(5-(2-(6-氰基-2-甲氧基吡啶-3-基)-1,3-二氧代异吲哚啉-5-基氧基)戊基)哌嗪-1-基)烟酰胺的合成
将5-(5-(5-氯戊氧基)-1,3-二氧代异吲哚啉-2-基)-6-甲氧基氰基吡啶(100mg,025mmol)、乙基二异丙胺(96.8mg,0.75mmol)、碘化钾(41.5mg,0.25mmol)和N-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基)-6-(哌嗪-1-基)烟酰胺(117mg,0.25mmol)在二甲基亚砜(3mL)中的混合物在70℃下搅拌过夜。使所得混合物冷却至室温。添加水(20mL)和乙酸乙酯(20mL)。将有机层分离,用盐水(50mL x 2)洗涤,经无水硫酸钠干燥,过滤并真空浓缩,得到粗产物,将其通过制备型TLC(乙酸乙酯)进行纯化,得到N-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基)-6-(4-(5-(2-(6-氰基-2-甲氧基吡啶-3-基)-1,3-二氧代异吲哚啉-5-基氧基)戊基)哌嗪-1-基)烟酰胺(53mg,34%),呈黄色固体。
步骤4:5-(5-(5-(4-(5-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基氨基甲酰基)吡啶-2-基)哌嗪-1-基)戊氧基)-1,3-二氧代异吲哚啉-2-基)-6-羟基吡啶酰胺
将N-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基)-6-(4-(5-(2-(6-氰基-2-甲氧基吡啶-3-基)-1,3-二氧代异吲哚啉-5-基氧基)戊基)哌嗪-1-基)烟酰胺(70mg,0.084mmol)在溴化氢/冰醋酸(48重量%,0.5mL)中的混合物在45℃下搅拌5小时。使所得混合物冷却至室温。添加水(20mL)。将该混合物用饱和碳酸氢钠中和至pH>7,并用乙酸乙酯(10mL x2)萃取。将合并的有机层用盐水(10mL x2)洗涤,经无水硫酸钠干燥,过滤并真空浓缩,得到5-(5-(5-(4-(5-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基氨基甲酰基)吡啶-2-基)哌嗪-1-基)戊氧基)-1,3-二氧代异吲哚啉-2-基)-6-羟基吡啶酰胺(50mg,71%),呈白色固体。
步骤5:N-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基)-6-(4-(5-(2-(6-氰基-2-羟基吡啶-3-基)-1,3-二氧代异吲哚啉-5-基氧基)戊基)哌嗪-1-基)烟酰胺的合成
向5-(5-(5-(4-(5-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基氨基甲酰基)吡啶-2-基)哌嗪-1-基)戊氧基)-1,3-二氧代异吲哚啉-2-基)-6-羟基吡啶酰胺(45mg,0.053mmol)和三乙胺(21.2mg,0.21mmol)的二氯甲烷(4mL)溶液中添加三氟乙酸酐(44.1mg,0.21mmol)。将该混合物搅拌2小时。将该混合物倒入冰水(40mL)中。添加二氯甲烷(40mL)。将有机层分离,用盐水(10mL x2)洗涤,经无水硫酸钠干燥,过滤并真空浓缩。将残余物溶解于四氢呋喃(5mL)和水(5mL)中并搅拌过夜。添加乙酸乙酯(10mL)。将有机层分离,用盐水(10mL x2)洗涤,经无水硫酸钠干燥,过滤并真空浓缩,得到粗产物,将其通过制备型HPLC进行纯化,得到N-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基)-6-(4-(5-(2-(6-氰基-2-羟基吡啶-3-基)-1,3-二氧代异吲哚啉-5-基氧基)戊基)哌嗪-1-基)烟酰胺(6.8mg,16%),呈白色固体
LC-MS(Agilent LCMS 1200-6110,色谱柱:Waters X-Bridge C18(50mm*4.6mm*3.5μm);柱温:40℃;流速:2.0mL/min;流动相:在1.6分钟内从95%[水+0.05%TFA]和5%[CH
HPLC(Agilent HPLC 1200,色谱柱:L-column2 ODS(150mm*4.6mm*5.0μm);柱温:40℃;流速:1.0mL/min;流动相:在10分钟内从95%[水+0.1%TFA]和5%[CH
1
化学式:C
得自HNMR数据的总H计数:45
示例性PROTAC 43的合成
N-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基)-4-(4-((4-(1,3-二氧代-2-(6-氧代-1,6-二氢吡啶-3-基)异吲哚啉-5-基)哌嗪-1-基)甲基)哌啶-1-基)苯甲酰胺
合成方案
步骤1:4-[4-(羟甲基)-1-哌啶基]苯甲酸的合成
向4-[4-(羟甲基)-1-哌啶基]苯甲酸乙酯(52g,197.47mmol,1当量)在四氢呋喃(250mL)、甲醇(250mL)和水(250mL)中的溶液中添加氢氧化钠(31.6g,0.79mmol,4当量)。将该混合物在30℃下搅拌12小时。薄层色谱(石油醚:乙酸乙酯=1:1)显示反应完成。用盐酸(2M)将该混合物调节至pH3~4并过滤。将滤饼真空干燥。用乙酸乙酯(500mL)研磨残余物,得到4-[4-(羟甲基)-1-哌啶基]苯甲酸(35g,148.76mmol,75%收率),呈白色固体。
1
化学式:C
得自HNMR数据的总H计数:17。
步骤2:N-[3-(3-氯-4-氰基-苯氧基)-2,2,4,4-四甲基-环丁基]-4-[4-(羟甲基)-1-哌啶基]苯甲酰胺的合成
向4-[4-(羟甲基)-1-哌啶基]苯甲酸(38g,161.51mmol,1当量)和4-(3-氨基-2,2,4,4-四甲基-环丁氧基)-2-氯-苯甲腈(50.9g,161.51mmol,1当量,盐酸盐)的二甲基甲酰胺(800mL)溶液中添加二异丙基乙胺(83.5g,646.04mmol,112mL,4当量)。将该混合物在30℃下搅拌10分钟,然后添加邻-(7-氮杂苯并三唑-1-基)-n,n,n’,n’-四甲基脲六氟磷酸酯(64.48g,169.59mmol,1.05当量)。将该混合物在30℃下搅拌1小时。LCMS显示反应完成,并且可以检测到所需MS。将该混合物倒入水(4L)中并过滤。将滤饼浓缩并用甲醇(500mL x 2)研磨,得到N-[3-(3-氯-4-氰基-苯氧基)-2,2,4,4-四甲基-环丁基]-4-[4-(羟甲基)-1-哌啶基]苯甲酰胺(72g,137.89mmol,85%收率,95%纯度),呈白色固体。
LCMS:MS(ESI)m/z:496.1[M+1]
1
得自HNMR数据的总H计数:34。
步骤3:N-[3-(3-氯-4-氰基-苯氧基)-2,2,4,4-四甲基-环丁基]-4-(4-甲酰基-1-哌啶基)苯甲酰胺的合成
向N-[3-(3-氯-4-氰基-苯氧基)-2,2,4,4-四甲基-环丁基]-4-[4-(羟甲基)-1-哌啶基]苯甲酰胺(65g,131.04mmol,1当量)的二氯甲烷(700mL)溶液中添加Dess-Martin试剂(76.70g,180.83mmol,1.38当量)。将该混合物在30℃下搅拌2小时。薄层色谱(二氯甲烷:甲醇=1:1)显示反应完成。用饱和碳酸氢钠将反应调节至pH8~9。将该混合物用水(3L)稀释,并用二氯甲烷(1.5L x3)萃取。将合并的有机相用饱和盐水(1.5L x 2)洗涤,用无水硫酸钠干燥,过滤并真空浓缩。通过硅胶色谱(二氯甲烷:甲醇=100:0至50:1)纯化残余物,得到N-[3-(3-氯-4-氰基-苯氧基)-2,2,4,4-四甲基-环丁基]-4-(4-甲酰基-1-哌啶基)苯甲酰胺(34.6g,67.94mmol,51%收率,97%纯度),呈白色固体。
1
化学式:C
得自HNMR数据的总H计数:32。
步骤4:5-氟-2-(6-甲氧基吡啶-3-基)异吲哚啉-1,3-二酮的合成
将5-氟-1,3-二氢-2-苯并呋喃-1,3-二酮(100.0mg,602μmol)、6-甲氧基吡啶-3-胺(82.1mg,662μmol)、乙酸钠(59.2mg,722μmol)和乙酸(499μL,8.74mmol)的混合物一边搅拌一边在118℃下加热2小时。通过LCMS(CF-820-1)监测反应,其显示具有与所需产物一致的质量的主峰。将反应冷却至90℃并用水(2mL)淬灭。使混合物冷却至室温。过滤所得沉淀并用水洗涤。将该物质干燥,得到呈浅紫色固体的所需产物5-氟-2-(6-甲氧基吡啶-3-基)-2,3-二氢-1H-异吲哚-1,3-二酮(149.1mg,547μmol,91.4%收率)。
1
LCMS m/e+=273.16[M+H]
步骤5:4-(2-(6-甲氧基吡啶-3-基)-1,3-二氧代异吲哚啉-5-基)哌嗪-1-羧酸叔丁酯的合成
向哌嗪-1-羧酸叔丁酯(34.0mg,183μmol)和5-氟-2-(6-甲氧基吡啶-3-基)-2,3-二氢-1H-异吲哚-1,3-二酮(50.0mg,183μmol)的甲基吡咯烷酮(1.0mL)溶液中添加N,N-二异丙基乙胺(95.5μL,549μmol)。将该反应混合物在120℃下加热2小时。通过LCMS监测反应,其显示具有与所需产物一致的质量的主峰和具有与起始材料一致的质量的小峰。将反应在120℃下再搅拌16小时。LCMS显示具有与所需产物一致的质量的主峰。将反应混合物用水(2mL)淬灭,并用EtOAc(2mL)萃取。将有机层用盐水(1mL)洗涤,经Na2SO4干燥,过滤并减压浓缩。在用DCM/MeOH(梯度100:0至95:5)洗脱的Teledyne Combiflash ISCO上,通过硅胶色谱纯化粗物质。将含有产物的级分减压浓缩,得到呈白色固体的所需产物4-[2-(6-甲氧基吡啶-3-基)-1,3-二氧代-2,3-二氢-1H-异吲哚-5-基]哌嗪-1-羧酸叔丁酯(39.6mg,90.3μmol,49.3%收率)。
LCMS m/e+=439.33[M+H]
1
步骤6:2-(6-氧代-1,6-二氢吡啶-3-基)-5-(哌嗪-1-基)异吲哚啉-1,3-二酮的合成
将4-[2-(6-甲氧基吡啶-3-基)-1,3-二氧代-2,3-二氢-1H-异吲哚-5-基]哌嗪-1-羧酸叔丁酯(39.6mg,90.3μmol)在4.0M盐酸的1,4-二氧杂环己烷(1.0mL,4.00mmol)溶液中的溶液在100℃下搅拌16小时。将反应混合物
减压浓缩,得到白色固体2-(6-氧代-1,6-二氢吡啶-3-基)-5-(哌嗪-1-基)-2,3-二氢-1H-异吲哚-1,3-二酮盐酸盐(32.5mg,90.0μmol,100%收率)。将该物质用于下一反应中,无需任何进一步纯化。
LCMS m/e+=425.22[M+H]
步骤7:N-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基)-4-(4-((4-(1,3-二氧代-2-(6-氧代-1,6-二氢吡啶-3-基)异吲哚啉-5-基)哌嗪-1-基)甲基)哌啶-1-基)苯甲酰胺的合成
向4-(4-甲酰基哌啶-1-基)-N-[(1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基]苯甲酰胺(44.4mg,90.0μmol)和2-(6-氧代-1,6-二氢吡啶-3-基)-5-(哌嗪-1-基)-2,3-二氢-1H-异吲哚-1,3-二酮盐酸盐(32.5mg,90.0μmol)的二氯乙烷(1.0mL)溶液中添加三乙胺(37.4μL,269μmol)和三乙酰氧基硼氢化钠(57.0mg,269μmol)。使反应混合物在室温下搅拌5小时。通过LCMS监测反应混合物,其显示具有与所需产物一致的质量的峰和具有与起始材料一致的质量的峰。使反应混合物在室温下再搅拌16小时。LMCS显示具有与所需产物一致的质量的主峰。将反应混合物用NaHCO3水溶液(1mL)淬灭,并用DCM(1mL)萃取。将有机层经Na2SO4干燥,过滤并减压浓缩。在用DCM/MeOH(梯度100:0至90:10)洗脱的Teledyne Combiflash ISCO上,通过硅胶色谱纯化粗物质。将含有产物的级分合并并减压浓缩,得到呈黄色固体的所需产物N-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基)-4-(4-((4-(1,3-二氧代-2-(6-氧代-1,6-二氢吡啶-3-基)异吲哚啉-5-基)哌嗪-1-基)甲基)哌啶-1-基)苯甲酰胺(30mg,37.3μmol,41.5%收率)。
1
LCMS m/e+=802.57[M+
示例性PROTAC
N-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基)-6-(4-(2-(2-((2-(2,6-二氧代哌啶-3-基)-1-氧代-1,2-二氢异喹啉-3-基)甲氧基)乙氧基)乙基)哌嗪-1-基)烟酰胺
合成方案:
步骤1:N-(2,6-二氧代哌啶-3-基)-2-碘苯甲酰胺的合成
在100mL圆底烧瓶中置入2-碘苯甲酸(5.0g,20.16mmol,1.00当量)、N,N-二甲基甲酰胺(40mL)、HATU(7.66g,20.15mmol,1.00当量)、DIEA(7.80g,60.35mmol,3.00当量),搅拌10分钟后,添加3-氨基哌啶-2,6-二酮(3.30g,25.76mmol,1.00当量)。将所得溶液在室温下搅拌2小时。然后通过添加500mL水/冰来淬灭反应。过滤收集固体。真空浓缩所得混合物。这得到6.48g(90%)的N-(2,6-二氧代哌啶-3-基)-2-碘苯甲酰胺,呈灰白色固体。
LC-MS(ES
步骤2:([2-[2-(丙-2-炔-1-基氧基)乙氧基]乙氧基]甲基)苯的合成
在用氮气惰性气氛吹扫并保持的250mL 3颈圆底烧瓶中置入2-[2-(苄氧基)乙氧基]乙-1-醇(10.0g,50.96mmol,1.00当量)、N,N-二甲基甲酰胺(100mL)。然后,在搅拌30分钟后,在0℃下,分数批添加氢化钠(2.4g,100.00mmol,1.20当量)。在0℃下,一边搅拌,一边向其中逐滴添加3-溴丙-1-炔(7.285g,61.24mmol,1.20当量)的N,N-二甲基甲酰胺(30mL)溶液。将所得溶液在室温下搅拌过夜。然后通过添加300mL水/冰来淬灭反应。用乙酸乙酯(300mL)萃取所得溶液,再合并有机层。用盐水(300mL)洗涤所得混合物。将该混合物经无水硫酸钠干燥。将残余物施加到使用乙酸乙酯/石油醚(1/4)的硅胶柱上。这得到9.5g(80%)的([2-[2-(丙-2-炔-1-基氧基)乙氧基]乙氧基]甲基)苯,呈浅黄色油状物。
LC-MS(ES
步骤3:2-(3-(2-(2-(苄氧基)乙氧基)乙氧基)丙-1-炔基)-N-(2,6-二氧代哌啶-3-基)苯甲酰胺的合成
在用氮气惰性气氛吹扫并保持的25mL圆底烧瓶中置入N-(2,6-二氧代哌啶-3-基)-2-碘苯甲酰胺(1.5g,4.1mmol,1.00当量)、N,N-二甲基甲酰胺(20mL)、(PPh
LC-MS(ES
步骤4:3-[3-([2-[2-(苄氧基)乙氧基]乙氧基]甲基)-1-氧代-1,2-二氢异喹啉-2-基]哌啶-2,6-二酮的合成
在用氮气惰性气氛吹扫并保持的25mL圆底烧瓶中置入2-(3-[2-[2-(苄氧基)乙氧基]乙氧基]丙-1-炔-1-基)-N-(2,6-二氧代哌啶-3-基)苯甲酰胺(1.0g,2.15mmol,1.00当量)的N,N-二甲基甲酰胺(10mL)溶液、Pd(OAc)
LC-MS(ES
步骤5:3-(3-[[2-(2-羟基乙氧基)乙氧基]甲基]-1-氧代-1,2-二氢异喹啉-2-基)哌啶-2,6-二酮的合成
在用氮气惰性气氛吹扫并保持的100mL 3颈圆底烧瓶中置入3-[3-([2-[2-(苄氧基)乙氧基]乙氧基]甲基)-1-氧代-1,2-二氢异喹啉-2-基]哌啶-2,6-二酮(420.0mg,0.90mmol,1.00当量)、二氯甲烷(10mL)。然后在-78℃下,一边搅拌一边逐滴添加BBr
LC-MS(ES
步骤6:2-(2-[[2-(2,6-二氧代哌啶-3-基)-1-氧代-1,2-二氢异喹啉-3-基]甲氧基]乙氧基)乙基4-甲基苯-1-磺酸酯的合成
在50mL圆底烧瓶中置入3-(3-[[2-(2-羟基乙氧基)乙氧基]甲基]-1-氧代-1,2-二氢异喹啉-2-基)哌啶-2,6-二酮(212.0mg,0.57mmol,1.00当量)、二氯甲烷(10.0mL)、TsCl(215.4mg,1.13mmol,2.00当量)、三乙胺(171.0mg,1.69mmol,3.00当量)、4-二甲基氨基吡啶(6.98mg,0.06mmol,0.10当量)。将所得溶液在室温下搅拌3小时。用二氯甲烷(100mL)萃取所得溶液,再合并有机层。用盐水(100mL)洗涤所得混合物。将该混合物经无水硫酸钠干燥。将残余物施加到使用乙酸乙酯/石油醚(4/1)的硅胶柱上。这得到238.0mg(80%)的2-(2-[[2-(2,6-二氧代哌啶-3-基)-1-氧代-1,2-二氢异喹啉-3-基]甲氧基]乙氧基)乙基4-甲基苯-1-磺酸酯,呈浅黄色油状物。
LC-MS(ES
步骤7:6-[4-[2-(2-[[2-(2,6-二氧代哌啶-3-基)-1-氧代-1,2-二氢异喹啉-3-基]甲氧基]乙氧基)乙基]哌嗪-1-基]-N-[(1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基]吡啶-3-甲酰胺的合成
在用氮气惰性气氛吹扫并保持的20mL微波管中置入6-(哌嗪-1-基)-N-[(1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基]吡啶-3-甲酰胺(65.0mg,0.14mmol,1.00当量)、乙腈(5.0mL)、碳酸钾(71.3mg,0.52mmol,4.00当量)、2-(2-[[2-(2,6-二氧代哌啶-3-基)-1-氧代-1,2-二氢异喹啉-3-基]甲氧基]乙氧基)乙基4-甲基苯-1-磺酸酯(68.0mg,0.13mmol,1.00当量)、NaI(19.38mg,0.13mmol,1.00当量)。将所得溶液在75℃下在油浴中搅拌24小时。滤出固体。真空浓缩所得混合物。然后通过制备型HPLC柱纯化:XBridgeShield RP18 OBD色谱柱,5um,19*150mm;流动相A:水(10mmol/L NH
1
LC-MS(ES
化学式:C
得自HNMR数据的总H计数:50
示例性PROTAC
N-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基)-6-(4-(5-((3-(2,4-二氧代-3,4-二氢嘧啶-1(2H)-基)喹啉-6-基)氧基)戊基)哌嗪-1-基)烟酰胺
合成方案
步骤1:甲基5-(3-溴喹啉-6-基氧基)戊-1-醇的合成
将3-溴喹啉-6-醇(700mg,3.1mmol)、5-溴戊-1-醇(518mg,3.1mmol)和碳酸钾(856mg,6.2mmol)在N,N-二甲基甲酰胺(5mL)中的混合物在80℃下加热6小时。将反应混合物冷却至室温。添加水(10mL),并用乙酸乙酯(20mL x 3)萃取。将合并的有机层用水(20mLx 2)和盐水(20mL)洗涤,经无水硫酸钠干燥。浓缩溶剂,得到残余物,将其通过硅胶柱色谱(石油醚/乙酸乙酯=1/1)进行纯化,得到5-(3-溴喹啉-6-基氧基)戊-1-醇(750mg,78%收率),呈黄色固体。
LC-MS(Agilent LCMS 1200-6120,色谱柱:Waters X-Bridge C18(50mm x 4.6mmx 3.5μm);柱温:40℃;流速:2.0mL/min;流动相:在1.6分钟内从95%[水+10mM NH
步骤2:1-(6-(5-羟基戊氧基)喹啉-3-基)嘧啶-2,4(1H,3H)-二酮的合成
在氩气气氛下,将5-(3-溴喹啉-6-基氧基)戊-1-醇(496mg,1.6mmol)、嘧啶-2,4(1H,3H)-二酮(538mg,4.8mmol)、磷酸钾(1.0g,4.8mmol)、碘化亚铜(304mg,1.6mmol)、N-(2-氰基苯基)吡啶酰胺(357mg,1.6mmol)的二甲基亚砜(10mL)溶液在120℃下加热5小时。将反应混合物冷却至室温。添加水(10mL),并用乙酸乙酯(20mL x 2)萃取。将合并的有机层用盐水(10mL x 2)洗涤,经无水硫酸钠干燥。去除溶剂,并通过硅胶柱色谱(甲醇/二氯甲烷=1/20)纯化残余物,得到1-(6-(5-羟基戊氧基)喹啉-3-基)嘧啶-2,4(1H,3H)-二酮(200mg,37%收率),呈灰白色固体。
LC-MS(Agilent LCMS 1200-6120,色谱柱:Waters X-Bridge C18(50mm x 4.6mmx 3.5μm);柱温:40℃;流速:2.0mL/min;流动相:在1.6分钟内从95%[水+10mM NH
步骤3:5-(3-(2,4-二氧代-3,4-二氢嘧啶-1(2H)-基)喹啉-6-基氧基)戊醛的合成
将1-(6-(5-羟基戊氧基)喹啉-3-基)嘧啶-2,4(1H,3H)-二酮(150mg,0.4mmol)和戴斯-马丁过碘烷(559mg,1.3mmol)在二氯甲烷(15mL)中的混合物在室温下搅拌过夜。过滤反应混合物,并用二氯甲烷(10mL x 2)洗涤滤饼。浓缩滤液,并通过制备型TLC(二氯甲烷/甲醇=5/1)纯化残余物,得到5-(3-(2,4-二氧代-3,4-二氢嘧啶-1(2H)-基)喹啉-6-基氧基)戊醛(100mg,67%收率),呈黄色固体。
LC-MS(Agilent LCMS 1200-6120,色谱柱:Waters X-Bridge C18(50mm x 4.6mmx 3.5μm);柱温:40℃;流速:2.0mL/min;流动相:在1.6分钟内从95%[水+10mM NH
步骤4:N-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基)-6-(4-(5-(3-(2,4-二氧代-3,4-二氢嘧啶-1(2H)-基)喹啉-6-基氧基)戊基)哌嗪-1-基)烟酰胺的合成
/>
将5-(3-(2,4-二氧代-3,4-二氢嘧啶-1(2H)-基)喹啉-6-基氧基)戊醛(100mg,0.29mmol)、N-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基)-6-(哌嗪-1-基)烟酰胺盐酸盐(149mg,0.29mmol)、氰基硼氢化钠(36mg,0.58mmol)在甲醇(5mL)溶液和冰醋酸(0.5mL)中的混合物在室温下搅拌过夜。添加水(10mL),并用二氯甲烷(20mL x 3)萃取。将合并的有机层用盐水(10mL x 2)洗涤,经无水硫酸钠干燥。浓缩溶剂,得到残余物,将其通过制备型HPLC纯化,得到N-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基)-6-(4-(5-(3-(2,4-二氧代-3,4-二氢嘧啶-1(2H)-基)喹啉-6-基氧基)戊基)哌嗪-1-基)烟酰胺(23mg,10%收率),呈白色固体。
LC-MS(Agilent LCMS 1200-6120,色谱柱:Waters X-Bridge C18(50mm x 4.6mmx 3.5μm);柱温:40℃;流速:2.0mL/min;流动相:在3.0分钟内从95%[水+10mM NH
HPLC(Agilent HPLC 1200,色谱柱:Waters X-Bridge C18(150mm x4.6mm x 3.5μm);柱温:40℃;流速:1.0mL/min;流动相:在10分钟内从95%[水+10mM NH
1
化学式:C
得自HNMR数据的总H计数:46。
示例性PROTAC
外消旋-N-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基)-6-(4-(5-(4-((2,6-二氧代哌啶-3-基)(乙基)氨基甲酰基)苯氧基)戊基)哌嗪-1-基)烟酰胺合成方案
步骤1:4-(5-羟基戊氧基)苯甲酸甲酯的合成
将4-羟基苯甲酸甲酯(3.0g,20mmol)、5-溴戊-1-醇(3.3g,20mmol)、碳酸钾(5.5g,40mmol)和碘化钾(0.3g,2mmol)在N,N-二甲基甲酰胺(20mL)中的混合物在110℃下加热过夜。将反应混合物冷却至室温。添加水(50mL)。用乙酸乙酯(50mL x 3)萃取,并且将合并的有机层用水(30mL x 2)和盐水(30mL x 2)洗涤,经无水硫酸钠干燥。浓缩溶剂,得到残余物,将其通过硅胶柱色谱(石油醚/乙酸乙酯=10/1)进行纯化,得到4-(5-羟基戊氧基)苯甲酸甲酯(2.2g,46%收率),呈白色固体。
LC-MS(Agilent LCMS 1200-6120,色谱柱:Waters X-Bridge C18(50mm x 4.6mmx 3.5μm);柱温:40℃;流速:2.0mL/min;流动相:在1.6分钟内从95%[水+10mM NH
步骤2:4-(5-羟基戊氧基)苯甲酸的合成
将4-(5-羟基戊氧基)苯甲酸甲酯(2.2g,9.2mmol)、氢氧化锂(1.6g,36.9mmol)在甲醇(10mL)和水(1mL)中的混合物在室温下搅拌过夜。真空去除溶剂,并添加水(5mL)。将其用乙酸乙酯萃取,用1N盐酸水溶液调节水相至pH=5-6。过滤并收集固体,将其真空干燥,得到4-(5-羟基戊氧基)苯甲酸(1.9g,90%收率),呈白色固体。
LC-MS(Agilent LCMS 1200-6120,色谱柱:Waters X-Bridge C18(50mm x 4.6mmx 3.5μm);柱温:40℃;流速:2.0mL/min;流动相:在1.6分钟内从95%[水+10mM NH
步骤3:3-(乙基氨基)哌啶-2,6-二酮的合成
将3-氨基哌啶-2,6-二酮盐酸盐(3.8g,23mmol)、乙醛(1.0g,23mmol)、氰基硼氢化钠(4.3g,69mmol)在甲醇(30mL)和冰醋酸(0.5mL)中的混合物在室温下搅拌过夜。添加水(10mL),并用二氯甲烷(50mL x 3)萃取。将合并的有机层用盐水(30mL x 2)洗涤,经无水硫酸钠干燥。浓缩溶剂,得到残余物,将其通过硅胶柱色谱(二氯甲烷/甲醇=10/1)进行纯化,得到3-(乙基氨基)哌啶-2,6-二酮(3.0g,33%收率),呈黄色油状物。
LC-MS(Agilent LCMS 1200-6120,色谱柱:Waters X-Bridge C18(50mm x 4.6mmx 3.5μm);柱温:40℃;流速:2.0mL/min;流动相:在1.6分钟内从95%[水+10mM NH
步骤4:N-(2,6-二氧代哌啶-3-基)-N-乙基-4-(5-羟基戊氧基)苯甲酰胺的合成
将3-(乙基氨基)哌啶-2,6-二酮(500mg,3.2mmol)、4-(5-羟基戊氧基)苯甲酸(3.3g,20mmol)、乙基二异丙胺(826mg,6.4mmol)和2-(7-氮杂-1H-苯并三唑-1-基)-1,1,3,3-四甲基脲六氟磷酸酯(1.8g,4.8mmol)在N,N-二甲基甲酰胺(5mL)中的混合物在室温下搅拌过夜。添加水(10mL)。用乙酸乙酯(20mL x 3)萃取,并且将合并的有机层用水(20mL x 2)和盐水(20mL x 2)洗涤,经无水硫酸钠干燥。浓缩溶剂,得到残余物,将其通过硅胶柱色谱(二氯甲烷/甲醇=10/1)进行纯化,得到N-(2,6-二氧代哌啶-3-基)-N-乙基-4-(5-羟基戊氧基)苯甲酰胺(108mg,9%收率),呈白色固体。
LC-MS(Agilent LCMS 1200-6120,色谱柱:Waters X-Bridge C18(50mm x 4.6mmx 3.5μm);柱温:40℃;流速:2.0mL/min;流动相:在1.6分钟内从95%[水+10mM NH
步骤5:N-(2,6-二氧代哌啶-3-基)-N-乙基-4-(5-氧代戊氧基)苯甲酰胺的合成
将N-(2,6-二氧代哌啶-3-基)-N-乙基-4-(5-羟基戊氧基)苯甲酰胺(108mg,0.3mmol)和戴斯-马丁过碘烷(254mg,0.6mmol)在二氯甲烷(10mL)中的混合物在室温下搅拌2小时。过滤反应混合物,并用二氯甲烷(10mL x 2)洗涤滤饼。浓缩滤液,并通过制备型TLC(二氯甲烷/甲醇=5/1)纯化残余物,得到N-(2,6-二氧代哌啶-3-基)-N-乙基-4-(5-氧代戊氧基)苯甲酰胺(97mg,90%收率),呈黄色固体。
LC-MS(Agilent LCMS 1200-6120,色谱柱:Waters X-Bridge C18(50mm x 4.6mmx 3.5μm);柱温:40℃;流速:2.0mL/min;流动相:在1.6分钟内从95%[水+10mM NH
步骤6:N-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基)-6-(4-(5-(4-((2,6-二氧代哌啶-3-基)(乙基)氨基甲酰基)苯氧基)戊基)哌嗪-1-基)烟酰胺的合成
将N-(2,6-二氧代哌啶-3-基)-N-乙基-4-(5-氧代戊氧基)苯甲酰胺(97mg,0.27mmol)、N-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基)-6-(哌嗪-1-基)烟酰胺盐酸盐(136mg,0.27mmol)、氰基硼氢化钠(34mg,0.54mmol)在甲醇(5mL)和冰醋酸(0.5mL)中的混合物在室温下搅拌过夜。添加水(10mL),并用二氯甲烷(20mL x 3)萃取。将合并的有机层用盐水(10mL x 2)洗涤,经无水硫酸钠干燥。浓缩溶剂,得到残余物,将其通过制备型HPLC纯化,得到N-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基)-6-(4-(5-(4-((2,6-二氧代哌啶-3-基)(乙基)氨基甲酰基)苯氧基)戊基)哌嗪-1-基)烟酰胺(55mg,25%收率),呈灰白色固体。
LC-MS(Agilent LCMS 1200-6120,色谱柱:Waters X-Bridge C18(50mm x4.6mm x3.5μm);柱温:40℃;流速:2.0mL/min;流动相:在3.0分钟内从95%[水+10mM NH
[M+H]
HPLC(Agilent HPLC 1200,色谱柱:Waters X-Bridge C18(150mm x
4.6mm x 3.5μm);柱温:40℃;流速:1.0mL/min;流动相:在10分钟内从95%[水+10mM NH
1
化学式:C
得自HNMR数据的总H计数:54。
示例性PROTAC
5-(3-(4-(5-(((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基)氨基甲酰基)吡啶-2-基)哌嗪-1-基)丙氧基)-N-(2,6-二氧代哌啶-3-基)吡啶酰胺
合成方案
步骤1:5-(3-羟基丙氧基)吡啶甲酸甲酯的合成
向5-羟基吡啶甲酸甲酯(5.0g,32.6mmol)的N,N-二甲基甲酰胺(60.0mL)溶液中添加3-溴丙-1-醇(5.45g,39.2mmol)、碳酸钾(9.03g,65.3mmol)。将该反应混合物在70℃下搅拌过夜。真空去除溶剂。通过硅胶色谱(二氯甲烷/甲醇=20:1)纯化残余物,得到5-(3-羟基丙氧基)吡啶甲酸甲酯(2.5g,36%),呈浅黄色固体。
1
化学式:C
得自HNMR数据的总H计数:13。
步骤2:5-(3-羟基丙氧基)吡啶甲酸的合成
向5-(3-羟基丙氧基)吡啶甲酸甲酯(2.5g,11.8mmol)的甲醇(50mL)溶液中添加氢氧化锂(1.49g,35.5mmol)。将该混合物在室温下搅拌3小时。去除溶剂并添加盐酸水溶液(0.5M)调节至PH=2~3。真空去除水,将残余物用二氯甲烷/甲醇(10:1)洗涤,过滤并真空浓缩,得到呈浅黄色固体的粗制物5-(3-羟基丙氧基)吡啶甲酸,将其用于下一步骤,无需进一步纯化。
步骤3:N-(2,6-二氧代哌啶-3-基)-5-(3-羟基丙氧基)吡啶酰胺的合成
将5-(3-羟基丙氧基)吡啶甲酸(粗制物,11.8mmol)、1-(3-二甲基氨基丙基)-3-乙基碳二亚胺盐酸盐(EDCI)(3.39g,17.7mmol)、1-羟基苯并三唑水合物(HOBt)(2.40g,17.7mmol)和乙基二异丙胺(4.58g,35.4mmol)的N,N-二甲基甲酰胺(DMF)(30mL)溶液搅拌30分钟,然后添加3-氨基哌啶-2,6-二酮(2.14g,13.0mmol)。将该混合物在室温下搅拌过夜,并添加水(100mL)。用乙酸乙酯(100mL x 3)萃取水层。将合并的有机层用盐水(20mL x4)洗涤,经无水硫酸钠干燥,过滤并真空浓缩。通过硅胶(二氯甲烷/甲醇=20:1)纯化残余物,得到N-(2,6-二氧代哌啶-3-基)-5-(3-羟基丙氧基)吡啶酰胺(2.1g,58%,两步),呈浅黄色固体。
步骤4:3-(6-(2,6-二氧代哌啶-3-基氨基甲酰基)吡啶-3-基氧基)丙基甲磺酸酯的合成
在氮气下,向N-(2,6-二氧代哌啶-3-基)-5-(3-羟基丙氧基)吡啶酰胺(500mg,1.63mmol)的二氯甲烷(50.0mL)溶液中添加三乙胺(329mg,3.25mmol)和甲磺酰氯(224mg,1.95mmol)。将所得反应混合物在0℃下搅拌1小时。然后添加水(20.0mL)并用二氯甲烷(20mL x 3)萃取,用盐水洗涤,干燥并真空浓缩,得到呈浅黄色油状物的粗制物3-(6-(2,6-二氧代哌啶-3-基氨基甲酰基)吡啶-3-基氧基)丙基甲磺酸酯,将其用于下一步骤,无需进一步纯化。
步骤5:6-氯烟酸叔丁酯的合成
将6-氯烟酸(31.6g,200mmol)和4-二甲基氨基吡啶(2.4g,20mmol)的THF(250mL)溶液回流3小时。然后逐滴添加二碳酸二叔丁酯(65.0g,300mmol)。添加后,将反应混合物回流3小时。反应完成后,将反应混合物冷却至室温。去除溶剂,并通过硅胶柱色谱(乙酸乙酯/石油醚=0~1/10)纯化残余物,得到6-氯烟酸叔丁酯(40g,94%收率),呈白色固体。
LC-MS(Agilent LCMS 1200-6120,色谱柱:Waters X-Bridge C18(50mm x4.6mm x3.5μm);柱温:40℃;流速:2.0mL/min;流动相:在1.6分钟内从95%[水+10mM NH
化学式:C
得自HNMR数据的总H计数:12。
步骤6:6-(哌嗪-1-基)烟酸叔丁酯的合成
将6-氯烟酸叔丁酯(20.0g,94mmol)和哌嗪(8.9g,103mmol)在N,N-二甲基乙酰胺(100mL)中的混合物在140℃下搅拌过夜。将反应混合物冷却至室温,并分批添加饱和碳酸钾水溶液(200mL)。过滤混合物,并用乙酸乙酯(600mL x 2)萃取滤液。将合并的有机层用水(600mL x 4)和盐水(600mL)洗涤,经无水硫酸钠干燥。减压浓缩溶剂,并通过硅胶柱色谱(二氯化物/甲醇=10/1)纯化残余物,得到6-(哌嗪-1-基)烟酸叔丁酯(6.5g,26%收率),呈黄色固体。
LC-MS(Agilent LCMS 1200-6120,色谱柱:Waters X-Bridge C18(50mm x4.6mm x3.5μm);柱温:40℃;流速:2.0mL/min;流动相:在3.0分钟内从95%[水+10mM NH
4.6mm x 3.5μm);柱温:40℃;流速:1.0mL/min;流动相:在10分钟内从95%[水+10mM NH
1
化学式:C
得自HNMR数据的总H计数:21。
步骤7:6-(4-(3-(6-(2,6-二氧代哌啶-3-基氨基甲酰基)吡啶-3-基氧基)丙基)哌嗪-1-基)烟酸叔丁酯的合成
向3-(6-(2,6-二氧代哌啶-3-基氨基甲酰基)吡啶-3-基氧基)丙基甲磺酸酯(粗制物,1.63mmol)的二甲基亚砜(5.0mL)溶液中添加6-(哌嗪-1-基)烟酸叔丁酯(472mg,1.79mmol)、乙基二异丙胺(632mg,4.89mmol)和碘化钾(27.1mg,0.163mmol)。将该反应混合物在45℃下搅拌过夜。然后添加水(20mL)并用乙酸乙酯(20mL x 3)萃取,用盐水(5mL x 4)洗涤。将合并的有机相经无水硫酸钠干燥,过滤并真空浓缩。通过制备型TLC(二氯甲烷/甲醇=10:1)纯化残余物,得到6-(4-(3-(6-(2,6-二氧代哌啶-3-基氨基甲酰基)吡啶-3-基氧基)丙基)哌嗪-1-基)烟酸叔丁酯(250mg,28%,两步),呈浅黄色固体。
1
m),2.54-2.64(6H,m),2.79-2.85(2H,m),3.69(4H,t,J=4.8Hz),4.17(2H,t,J=6.4Hz),4.76-4.82(1H,m),6.58(1H,d,J=9.2Hz),7.31(1H,dd,J=8.8Hz,3.2Hz),7.98(1H,dd,J=8.8Hz,2.4Hz),8.13(1H,d,J=8.8Hz),8.16(1H,brs),8.25(1H,d,J=2.8Hz),8.51(1H,d,J=6.8Hz),8.76(1H,d,J=2.0Hz)。
化学式:C
得自HNMR数据的总H计数:36。
步骤8:6-(4-(3-(6-(2,6-二氧代哌啶-3-基氨基甲酰基)吡啶-3-基氧基)丙基)哌嗪-1-基)烟酸的合成
向6-(4-(3-(6-(2,6-二氧代哌啶-3-基氨基甲酰基)吡啶-3-基氧基)丙基)哌嗪-1-基)烟酸叔丁酯(250mg,0.452mmol)的二氯甲烷(3.0mL)溶液中添加三氟乙酸(1mL)。将反应混合物在室温下搅拌2小时。然后真空去除溶剂,得到呈浅黄色油状物的6-(4-(3-(6-(2,6-二氧代哌啶-3-基氨基甲酰基)吡啶-3-基氧基)丙基)哌嗪-1-基)烟酸(粗制物),将其用于下一步骤,无需进一步纯化。
步骤9:5-(3-(4-(5-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基氨基甲酰基)吡啶-2-基)哌嗪-1-基)丙氧基)-N-(2,6-二氧代哌啶-3-基)吡啶酰胺的合成
向6-(4-(3-(6-(2,6-二氧代哌啶-3-基氨基甲酰基)吡啶-3-基氧基)丙基)哌嗪-1-基)烟酸(粗制物,0.452mmol)、1-(3-二甲基氨基丙基)-3-乙基碳二亚胺盐酸盐(EDCI)(130mg,0.678mmol)、1-羟基苯并三唑水合物(HOBt)(91.9mg,0.678mmol)和乙基二异丙胺(175mg,1.36mmol)的N,N-二甲基甲酰胺(DMF)(15mL)溶液搅拌30分钟,然后添加4-((1r,3r)-3-氨基-2,2,4,4-四甲基环丁氧基)-2-氯苯甲腈(139mg,0.497mmol)。将该混合物在室温下搅拌过夜,并添加水(20mL)。用乙酸乙酯(20mL x 3)萃取水层。将合并的有机层用盐水(5mL x 4)洗涤,经无水硫酸钠干燥,过滤并真空浓缩。通过制备型TLC(二氯甲烷/甲醇=10:1)和制备型HPLC纯化残余物,得到5-(3-(4-(5-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基氨基甲酰基)吡啶-2-基)哌嗪-1-基)丙氧基)-N-(2,6-二氧代哌啶-3-基)吡啶酰胺(57.7mg,17%,两步),呈白色固体。
LC-MS(Agilent LCMS 1200-6120,色谱柱:Waters X-Bridge C18
(50mm*4.6mm*3.5μm);柱温:40℃;流速:2.0mL/min;流动相:在3.0分钟内从95%[水+10mM NH
HPLC(Agilent HPLC 1200,色谱柱:Waters X-Bridge C18
(150mm*4.6mm*3.5μm);柱温:40℃;流速:1.0mL/min;流动相:在10分钟内从95%[水+10mM NH
1
化学式:C
得自HNMR数据的总H计数:45。
示例性PROTAC 53的合成
5-(4-((1-(5-(((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基)氨基甲酰基)吡啶-2-基)哌啶-4-基)甲基)哌嗪-1-基)-N-(2,6-二氧代哌啶-3-基)吡啶酰胺合成方案:
/>
步骤1:4-(6-(甲氧基羰基)吡啶-3-基)哌嗪-1-羧酸叔丁酯的合成
向5-溴吡啶甲酸甲酯(14.8g,68.5mmol)和哌嗪-1-羧酸叔丁酯(15.3g,82.2mmol)的甲苯(150mL)溶液中添加碳酸铯(55.8g,171.3mmol)、三(二亚苄基丙酮)二钯(0)(3.15g,3.44mmol)和(+/-)-2,2’-双(二苯基膦基)-1,1’-联萘(4.62g,7.42mmol),然后将其在100℃和氮气下搅拌过夜。冷却后,将其用水(100mL)淬灭并用乙酸乙酯(100mL x 3)萃取。将合并的有机层用盐水(200mL)洗涤,经无水硫酸钠干燥,过滤并真空浓缩。通过硅胶柱色谱(石油醚/乙酸乙酯=5/1)纯化残余物,得到4-(6-(甲氧基羰基)吡啶-3-基)哌嗪-1-羧酸叔丁酯(12.0g,55%收率),呈棕色固体。
LC-MS:(Agilent LCMS 1200-6120,色谱柱:Waters X-Bridge C18(30mm x 4.6mmx 3.5μm);柱温:40℃;流速:2.0mL/min;流动相:在1.0分钟内从90%[水+10mM NH
化学式:C
步骤2:5-(哌嗪-1-基)吡啶甲酸甲酯的合成
将4-(6-(甲氧基羰基)吡啶-3-基)哌嗪-1-羧酸叔丁酯(12.0g,37.4mmol)在HCl气体的1,4-二氧杂环己烷(100mL,4.0M)溶液中的混合物在30℃下搅拌1小时。将反应混合物真空浓缩,得到5-(哌嗪-1-基)吡啶甲酸甲酯(7.6g,93%收率),呈棕色固体。
化学式:C
步骤3:6-(4-甲酰基哌啶-1-基)烟酸叔丁酯的合成
将6-(4-(羟甲基)哌啶-1-基)烟酸叔丁酯(5.0g,17.1mmol)和戴斯-马丁过碘烷(21.8g,51.4mmol)在DCM(200mL)中的混合物在室温下搅拌4小时。过滤混合物,并真空浓缩滤液,得到6-(4-甲酰基哌啶-1-基)烟酸叔丁酯(3.5g,70%收率),呈黄色凝胶。
化学式:C
步骤4:5-(4-((1-(5-(叔丁氧基羰基)吡啶-2-基)哌啶-4-基)甲基)哌嗪-1-基)吡啶甲酸甲酯的合成
向6-(4-甲酰基哌啶-1-基)烟酸叔丁酯(3.5g,12.1mmol)和5-(哌嗪-1-基)吡啶甲酸甲酯(2.67g,12.1mmol)的MeOH(50mL)溶液中添加NaBH
化学式:C
步骤5:5-(4-((1-(5-(叔丁氧基羰基)吡啶-2-基)哌啶-4-基)甲基)哌嗪-1-基)吡啶甲酸的合成
向5-(4-((1-(5-(叔丁氧基羰基)吡啶-2-基)哌啶-4-基)甲基)哌嗪-1-基)吡啶甲酸甲酯(1.6g,2.35mmol)的THF(60mL)溶液中添加1mol/L NaOH水溶液(30mL),然后在30℃下搅拌2小时。将其用水(100mL)淬灭,并用DCM(50mL x 3)萃取。将合并的有机层用盐水(100mL)洗涤,经无水硫酸钠干燥,过滤并真空浓缩,得到5-(4-((1-(5-(叔丁氧基羰基)吡啶-2-基)哌啶-4-基)甲基)哌嗪-1-基)吡啶甲酸(1.5g,96%收率),呈棕色固体。
LC-MS:(Agilent LCMS 1200-6120,色谱柱:Waters X-Bridge C18(50mm x 4.6mmx 3.5μm);柱温:40℃;流速:2.0mL/min;流动相:在1.6分钟内从95%[水+10mM NH
化学式:C
步骤6:6-(4-((4-(6-(2,6-二氧代哌啶-3-基氨基甲酰基)吡啶-3-基)哌嗪-1-基)甲基)哌啶-1-基)烟酸叔丁酯的合成
将5-(4-((1-(5-(叔丁氧基羰基)吡啶-2-基)哌啶-4-基)甲基)哌嗪-1-基)吡啶甲酸(1.5g,3.1mmol)、3-氨基哌啶-2,6-二酮(0.56g,3.4mmol)、HATU(1.77g,4.65mmol)和DIEA(0.8g,6.2mmol)在DMF(50mL)中的混合物在室温下搅拌1小时。将该混合物倒入水(30mL)中,并用DCM(30mL x 3)萃取。浓缩合并的有机相,并通过制备型HPLC纯化残余物,得到6-(4-((4-(6-(2,6-二氧代哌啶-3-基氨基甲酰基)吡啶-3-基)哌嗪-1-基)甲基)哌啶-1-基)烟酸叔丁酯(1.0g,54%收率),呈白色固体。
LC-MS:(Agilent LCMS 1200-6120,色谱柱:Waters X-Bridge C18(50mm x 4.6mmx 3.5μm);柱温:40℃;流速:2.0mL/min;流动相:在1.6分钟内从95%[水+10mM NH
化学式:C
步骤7:6-(4-((4-(6-(2,6-二氧代哌啶-3-基氨基甲酰基)吡啶-3-基)哌嗪-1-基)甲基)哌啶-1-基)烟酸的合成
向6-(4-((4-(6-(2,6-二氧代哌啶-3-基氨基甲酰基)吡啶-3-基)哌嗪-1-基)甲基)哌啶-1-基)烟酸叔丁酯(500mg,0.85mmol)的DCM(10mL)溶液中添加TFA(5mL),然后在室温下搅拌2小时。将反应混合物真空浓缩,得到6-(4-((4-(6-(2,6-二氧代哌啶-3-基氨基甲酰基)吡啶-3-基)哌嗪-1-基)甲基)哌啶-1-基)烟酸(400mg,88%收率),呈白色固体。
LC-MS:(Agilent LCMS 1200-6120,色谱柱:Waters X-Bridge C18(50mm x 4.6mmx 3.5μm);柱温:40℃;流速:2.0mL/min;流动相:在1.6分钟内从95%[水+10mM NH
化学式:C
步骤8:5-(4-((1-(5-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基氨基甲酰基)吡啶-2-基)哌啶-4-基)甲基)哌嗪-1-基)-N-(2,6-二氧代哌啶-3-基)吡啶酰胺的合成
将6-(4-((4-(6-(2,6-二氧代哌啶-3-基氨基甲酰基)吡啶-3-基)哌嗪-1-基)甲基)哌啶-1-基)烟酸(400mg,0.75mmol)、4-((1r,3r)-3-氨基-2,2,4,4-四甲基环丁氧基)-2-氯苯甲腈(207.7mg,0.75mmol)、EDCI(158.4mg,0.825mmol)、HOBt(153mg,1.125mmol)和DIEA(290.25mg,2.25mmol)在DMF(10mL)中的混合物在室温下搅拌过夜。然后将反应混合物用水(20mL)淬灭,并用DCM(20mL x 3)萃取。将合并的有机层用盐水(30mL)洗涤,经无水硫酸钠干燥,过滤并真空浓缩。通过制备型HPLC纯化残余物,得到5-(4-((1-(5-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基氨基甲酰基)吡啶-2-基)哌啶-4-基)甲基)哌嗪-1-基)-N-(2,6-二氧代哌啶-3-基)吡啶酰胺(215mg,36%收率),呈白色固体。
LC-MS(Agilent LCMS 1200-6120,色谱柱:Waters X-Bridge C18(50mm x4.6mm x3.5μm);柱温:40℃;流速:2.0mL/min;流动相:在3.0分钟内从95%[水+10mM NH
[M+H]
HPLC(Agilent HPLC 1200,色谱柱:Waters X-Bridge C18(150mm x
4.6mm x 3.5μm);柱温:40℃;流速:1.0mL/min;流动相:在10分钟内从95%[水+10mM NH
1
化学式:C
得自HNMR数据的总H计数:50。
示例性PROTAC
N-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基)-6-(4-(4-(1-(2,6-二氧代哌啶-3-基)-6-氧代-1,6-二氢哒嗪-4-基)哌嗪-1-基)丁基)烟酰胺合成方案第1部分:
合成方案第2部分:
步骤1:6-(4-羟基丁-1-炔基)烟酸叔丁酯的合成
将6-氯烟酸叔丁酯(2.0g,9.39mmol)溶解于二甲氧基乙烷(50ml)中,依次添加水(30mL)、碳酸钾(5.18g,37.6mmol)、碘化亚铜(I)(0.1g,
0.5mmol)、三苯基膦(0.26g,1mmol)和10重量%钯碳(0.3g)。将反应混合物在室温下搅拌30分钟,然后添加2-甲基-3-丁炔-2-醇(5ml,50mmol),在80℃下加热5小时,然后冷却,通过硅藻土过滤,用水(150mL)稀释,并用乙酸乙酯(100mL x 2)萃取。将有机相用水洗涤,经硫酸钠干燥,过滤并真空浓缩。通过硅胶柱快速色谱纯化所得反应粗制物,得到6-(4-羟基丁-1-炔基)烟酸叔丁酯(1.7g,73%),呈无色油状物。
步骤2:6-(4-羟基丁基)烟酸叔丁酯的合成
在氢气(g)气氛下,将6-(4-羟基丁-1-炔基)烟酸叔丁酯(500mg,2.0mmol)、Pd/C(50mg)的叔丁醇(10mL)溶液在室温下搅拌过夜。通过硅藻土垫过滤该混合物以去除钯。真空蒸发溶剂,得到6-(4-羟基丁基)烟酸叔丁酯(450mg,88%收率),呈黄色油状物。将残余物用于下一步骤,无需进一步纯化。
步骤3:6-(4-羟基丁基)烟酸的合成
向6-(4-羟基丁基)烟酸酯(200mg,0.79mmol)的二氯甲烷(5mL)溶液中添加TFA(5mL),然后在室温下搅拌2小时。将其真空浓缩,得到呈黄色油状物的粗制物6-(4-羟基丁基)烟酸(130mg,84%收率),将其直接用于下一步骤,无需进一步纯化。
步骤4:N-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基)-6-(4-羟基丁基)烟酰胺的合成
向6-(4-羟基丁基)烟酸(570mg,粗制物,2.9mmol)、4-((1r,3r)-3-氨基-2,2,4,4-四甲基环丁氧基)-2-氯苯甲腈(400mg,1.4mmol)、EDCI(472mg,2.4mmol)和HOBt(332mg,2.4mmol)的DMF(10mL)溶液中添加DIEA(800mg,6.2mmol),然后在室温下搅拌两天。将其用水(20mL)稀释,并用乙酸乙酯(20mL x 2)萃取。将有机萃取物用水(40mL x 3)和盐水(40mL)洗涤,经无水Na
LCMS(Agilent LCMS 1200-6120,色谱柱:Waters X-Bridge C18(50mm x6mm x 5μm);柱温:40℃;流速:2.0mL/min;流动相:在1.6分钟内从90%[(总共10mM AcONH
/CH
化学式:C
步骤5:N-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基)-6-(4-氧代丁基)烟酰胺的合成
将N-(2,6-二氧代哌啶-3-基)-5-(5-羟基戊氧基)吡啶酰胺(240mg,
0.53mmol)和戴斯-马丁过碘烷(269mg,0.64mmol)在二氯甲烷(10mL)中的混合物在室温下搅拌1.5小时。过滤反应混合物,并用二氯甲烷(10mL x3)洗涤滤饼。将滤液浓缩并通过制备型TLC(DCM/MeOH=100/5)纯化,得到N-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基)-6-(4-氧代丁基)烟酰胺(100mg,42%收率),呈黄色固体。
LCMS(Agilent LCMS 1200-6120,色谱柱:Waters X-Bridge C18(50mm*4.6mm*3.5μm);柱温:40℃;流速:2.0mL/min;流动相:在1.6分钟内从90%[(总共10mM AcONH
[(总共10mM AcONH
/CH
步骤6:4-(5-氯-6-氧代-1,6-二氢哒嗪-4-基)哌嗪-1-羧酸叔丁酯的合成
向4,5-二氯哒嗪-3(2H)-酮(10g,60.6mmol)的N,N-二甲基甲酰胺(40mL)溶液中添加哌嗪-1-羧酸叔丁酯(22.5g,121.2mmol)和DIEA(25g,182mmol)。将该混合物在80℃下搅拌过夜。冷却至室温后,过滤混合物,并用乙酸乙酯(100mL x 3)和DCM(100mL x 3)洗涤残余物,得到化合物4-(5-氯-6-氧代-1,6-二氢哒嗪-4-基)哌嗪-1-羧酸叔丁酯(8g,42%收率),呈浅黄色固体。
LCMS(Agilent LCMS 1200-6120,色谱柱:Waters X-Bridge C18
(30mm*4.6mm*3.5μm);柱温:40℃;流速:2.0mL/min;流动相:在0.5分钟内从90%[水+10mM NH
化学式:C
步骤7:4-(5-氯-1-(2,6-二氧代哌啶-3-基)-6-氧代-1,6-二氢哒嗪-4-基)哌嗪-1-羧酸叔丁酯的合成
向1-溴-4-(5-溴戊氧基)苯(4g,12.7mmol)的DMSO(20mL)溶液中添加3-溴哌啶-2,6-二酮(4.8mg,25.4mmol)和碳酸钾(5.3g,38.1mmol)。将该混合物在40℃下搅拌两天。冷却至室温后,过滤该混合物,并用乙酸乙酯(20mL x 3)和DCM(20mL x 3)洗涤残余物。将合并的有机相经无水硫酸钠干燥,真空浓缩,并通过硅胶柱色谱(石油醚/乙酸乙酯=1:4)纯化,得到4-(5-氯-1-(2,6-二氧代哌啶-3-基)-6-氧代-1,6-二氢哒嗪-4-基)哌嗪-1-羧酸叔丁酯(3.7g,67%收率),呈浅黄色固体。
步骤8:4-(1-(2,6-二氧代哌啶-3-基)-6-氧代-1,6-二氢哒嗪-4-基)哌嗪-1-羧酸叔丁酯的合成
/>
在1atm氢气气氛下,将4-(5-氯-1-(2,6-二氧代哌啶-3-基)-6-氧代-1,6-二氢哒嗪-4-基)哌嗪-1-羧酸叔丁酯(300mg,0.7mmol)和10%活性炭载钯(90mg)在MeOH(30mL)中的混合物在37℃下搅拌过夜。过滤去除固体,真空浓缩滤液,得到3-(4-(3-羟基丙氧基)-6-氧代哒嗪-1(6H)-基)哌啶-2,6-二酮(190mg,93%收率),呈黄色固体。
LC-MS(Agilent LCMS 1200-6120,色谱柱:Waters X-Bridge C18(30mm x6mm x 5μm);柱温:40℃;流速:2.0mL/min;流动相:在0.5分钟内从90%[水+10mM NH
[M+H]
化学式:C
步骤9:3-(6-氧代-4-(哌嗪-1-基)哒嗪-1(6H)-基)哌啶-2,6-二酮的合成
将3-(4-(3-羟基丙氧基)-6-氧代哒嗪-1(6H)-基)哌啶-2,6-二酮(50mg,0.10mmol)在DCM(3mL)和三氟乙酸(3mL)中的溶液在室温下搅拌3小时。然后直接去除溶剂,得到3-(6-氧代-4-(哌嗪-1-基)哒嗪-1(6H)-基)哌啶-2,6-二酮(124mg,粗制物,88%收率),将其直接用于下一步骤,无需进一步纯化。
步骤10:N-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基)-6-(4-(4-(1-(2,6-二氧代哌啶-3-基)-6-氧代-1,6-二氢哒嗪-4-基)哌嗪-1-基)丁基)烟酰胺的合成
向3-(6-氧代-4-(哌嗪-1-基)哒嗪-1(6H)-基)哌啶-2,6-二酮(100mg,
0.34mmol)、N-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基)-6-(4-氧代丁基)烟酰胺(130mg,0.29mmol)的MeOH(6mL)溶液中添加乙酸(3滴),然后在室温下,在6小时内分7份添加NaBH
0.35mmol)。将所得混合物在室温下再搅拌1小时。将反应混合物浓缩,用盐水(15mL)稀释,并用CH
LC-MS(Agilent LCMS 1200-6120,色谱柱:Waters X-Bridge C18(50mm x4.6mm x3.5μm);柱温:40℃;流速:2.0mL/min;流动相:在3.0分钟内从95%[水+10mM NH
[M+H]
HPLC(Agilent HPLC 1200,色谱柱:Waters X-Bridge C18(150mm x
4.6mm x 3.5μm);柱温:40℃;流速:1.0mL/min;流动相:在10分钟内从95%[水+10mM NH
1
化学式:C
得自HNMR数据的总H计数:45。
示例性PROTAC
N-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基)-4-(4-((4-(4-(1-(2,6-二氧代哌啶-3-基)-4-甲基-5-氧代-4,5-二氢-1H-1,2,4-三唑-3-基)苯基)哌嗪-1-基)甲基)哌啶-1-基)苯甲酰胺
合成方案
步骤1:4-(4-(甲氧基羰基)苯基)哌嗪-1-羧酸叔丁酯的合成
将4-氟苯甲酸甲酯(3.1g,20.0mmol)、哌嗪-1-羧酸叔丁酯(3.7g,20.0mmol)和碳酸钾(2.7g,40.0mmol)在二甲基亚砜(30mL)中的混合物在120℃下加热24小时。将该混合物倒入水(100mL)中,并用乙酸乙酯(50mL x 3)萃取。将合并的有机相真空浓缩,得到4-(4-(甲氧基羰基)苯基)哌嗪-1-羧酸叔丁酯(5.1g,80%收率),呈白色固体。
化学式:C
步骤2:4-(4-(肼羰基)苯基)哌嗪-1-羧酸叔丁酯的合成
将4-(4-(甲氧基羰基)苯基)哌嗪-1-羧酸叔丁酯(3.2g,10.0mmol)和水合肼(1.0g,20.0mmol)在乙醇(30mL)中的混合物回流过夜。浓缩该混合物,得到呈白色固体的4-(4-(肼羰基)苯基)哌嗪-1-羧酸叔丁酯(2.6g,80%收率),将其直接使用。
LC-MS(Agilent LCMS 1200-6120,色谱柱:Waters X-Bridge C18(50mm x4.6mm x3.5μm);柱温:40℃;流速:2.0mL/min;流动相:在1.6分钟内从95%[水+10mM NH
[M+H]
化学式:C
步骤3:4-(4-(2-(甲基氨基甲酰基)肼羰基)苯基)哌嗪-1-羧酸叔丁酯的合成
将4-(4-(肼羰基)苯基)哌嗪-1-羧酸叔丁酯(2.0g,6.3mmol)和2,5-二氧代吡咯烷-1-基甲基氨基甲酸酯(1.1g,6.3mmol)在乙腈(30mL)中的混合物在室温下搅拌过夜。将该混合物倒入水(30mL)中并过滤,得到4-(4-(2-(甲基氨基甲酰基)肼羰基)苯基)哌嗪-1-羧酸叔丁酯(1.7g,70%),呈白色固体。化学式:C
步骤4:4-(4-(4-甲基-5-氧代-4,5-二氢-1H-1,2,4-三唑-3-基)苯基)哌嗪-1-羧酸叔丁酯的合成
将4-(4-(2-(甲基氨基甲酰基)肼羰基)苯基)哌嗪-1-羧酸叔丁酯(1.7g,
4.5mmol)和氢氧化钠(360mg,9.0mmol)在水(15mL)中的混合物回流3小时。将该混合物冷却至室温,并用盐酸(1.0N)将混合物的pH值调节至5-6。用二氯甲烷(30mL x 3)萃取混合物,并将合并的有机相真空浓缩,得到4-(4-(4-甲基-5-氧代-4,5-二氢-1H-1,2,4-三唑-3-基)苯基)哌嗪-1-羧酸叔丁酯(1.2g,75%收率),呈白色固体。
化学式:C
步骤5:4-(4-(1-(2,6-二氧代哌啶-3-基)-4-甲基-5-氧代-4,5-二氢-1H-1,2,4-三唑-3-基)苯基)哌嗪-1-羧酸叔丁酯的合成
将4-(4-(4-甲基-5-氧代-4,5-二氢-1H-1,2,4-三唑-3-基)苯基)哌嗪-1-羧酸叔丁酯(1.2g,3.3mmol)、3-溴哌啶-2,6-二酮(1.3g,6.6mmol)和叔丁醇钾(1.1g,9.9mmol)在乙腈(20mL)中的混合物回流过夜。将该混合物倒入饱和氯化铵溶液(30mL)中,并用二氯甲烷(30mL x 3)进行萃取。将合并的有机相真空浓缩,并通过制备型HPLC纯化残余物,得到4-(4-(1-(2,6-二氧代哌啶-3-基)-4-甲基-5-氧代-4,5-二氢-1H-1,2,4-三唑-3-基)苯基)哌嗪-1-羧酸叔丁酯(465mg,30%收率),呈白色固体。
化学式:C
步骤6:3-(4-甲基-5-氧代-3-(4-(哌嗪-1-基)苯基)-4,5-二氢-1H-1,2,4-三唑-1-基)哌啶-2,6-二酮的合成
将4-(4-(1-(2,6-二氧代哌啶-3-基)-4-甲基-5-氧代-4,5-二氢-1H-1,2,4-三唑-3-基)苯基)哌嗪-1-羧酸叔丁酯(465mg,0.99mmol)的无水盐酸盐/1,4-二氧杂环己烷(20mL,4.0N)溶液在室温下搅拌4小时。将该混合物真空浓缩,得到3-(4-甲基-5-氧代-3-(4-(哌嗪-1-基)苯基)-4,5-二氢-1H-1,2,4-三唑-1-基)哌啶-2,6-二酮(293mg,80%收率),呈白色固体。
化学式:C
步骤7:4-(4-(羟甲基)哌啶-1-基)苯甲酸叔丁酯的合成
向4-氟苯甲酸叔丁酯(23g,0.12mmol)的DMSO(100mL)溶液中添加哌啶-4-基甲醇(40.5g,0.35mmol)。在氮气下,将该混合物加热至120℃并保持过夜。冷却至室温后,将水(50mL)添加到反应混合物中,并用乙酸乙酯(20mL x 3)萃取。用盐水(15mL x 3)洗涤有机层。将合并的有机相经无水硫酸钠干燥,真空浓缩,并通过CC(PE/EA=10:1)纯化,得到化合物4-(4-(羟甲基)哌啶-1-基)苯甲酸叔丁酯(31g,91.2%),呈白色固体。
LCMS(Agilent LCMS 1200-6120,色谱柱:Waters X-Bridge C18(50mm x4.6mm x3.5μm);柱温:40℃;流速:2.0mL/min;流动相:在1.6分钟内从90%[(总共10mM AcONH
/CH
292.2[M+H]+。
HPLC(Agilent HPLC 1200,色谱柱:Waters X-Bridge C18(150mm x
4.6mm x 3.5μm);柱温:40℃;流速:1.0mL/min;流动相:在10分钟内从95%[水+10mM NH
1
化学式:C
得自HNMR数据的总H计数:25。
步骤8:4-(4-甲酰基哌啶-1-基)苯甲酸叔丁酯的合成
在0℃下,向4-(4-(羟甲基)哌啶-1-基)苯甲酸叔丁酯(300mg,1.03mmol)的二氯甲烷(20mL)溶液中缓慢添加戴斯-马丁过碘烷(1.31g,3.09mmol)。将反应混合物在室温下搅拌1小时。然后过滤,并真空浓缩,得到化合物4-(4-甲酰基哌啶-1-基)苯甲酸叔丁酯(240mg,81%),呈浅黄色固体。
步骤9:4-(4-((4-(4-(1-(2,6-二氧代哌啶-3-基)-4-甲基-5-氧代-4,5-二氢-1H-1,2,4-三唑-3-基)苯基)哌嗪-1-基)甲基)哌啶-1-基)苯甲酸叔丁酯的合成
将3-(4-甲基-5-氧代-3-(4-(哌嗪-1-基)苯基)-4,5-二氢-1H-1,2,4-三唑-1-基)哌啶-2,6-二酮(200mg,0.54mmol)、4-(4-甲酰基哌啶-1-基)苯甲酸叔丁酯(156mg,0.54mmol)、氰基硼氢化钠(100mg,1.6mmol)和乙酸(0.5mL)在甲醇(10mL)中的混合物在室温下搅拌过夜。将该混合物倒入水(20mL)中,并用二氯甲烷(20mL x 3)进行萃取。通过硅胶柱色谱(二氯甲烷/甲醇
=20/1)纯化合并的有机相,得到4-(4-((4-(4-(1-(2,6-二氧代哌啶-3-基)-4-甲基-5-氧代-4,5-二氢-1H-1,2,4-三唑-3-基)苯基)哌嗪-1-基)甲基)哌啶-1-基)苯甲酸叔丁酯(173mg,50%收率),呈棕色固体。
化学式:C
步骤10:4-(4-((4-(4-(1-(2,6-二氧代哌啶-3-基)-4-甲基-5-氧代-4,5-二氢-1H-1,2,4-三唑-3-基)苯基)哌嗪-1-基)甲基)哌啶-1-基)苯甲酸的合成
将4-(4-((4-(4-(1-(2,6-二氧代哌啶-3-基)-4-甲基-5-氧代-4,5-二氢-1H-1,2,4-三唑-3-基)苯基)哌嗪-1-基)甲基)哌啶-1-基)苯甲酸叔丁酯(150mg,
0.23mmol)和三氟乙酸(265mg,2.3mmol)在1,2-二氯乙烷(10mL)中的混合物搅拌2小时。将该混合物真空浓缩,得到呈棕色固体的4-(4-((4-(4-(1-(2,6-二氧代哌啶-3-基)-4-甲基-5-氧代-4,5-二氢-1H-1,2,4-三唑-3-基)苯基)哌嗪-1-基)甲基)哌啶-1-基)苯甲酸(95mg,70%收率),将其直接用于下一步骤,无需进一步纯化。
化学式:C
步骤11:N-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基)-4-(4-((4-(4-(1-(2,6-二氧代哌啶-3-基)-4-甲基-5-氧代-4,5-二氢-1H-1,2,4-三唑-3-基)苯基)哌嗪-1-基)甲基)哌啶-1-基)苯甲酰胺的合成
将4-(4-((4-(4-(1-(2,6-二氧代哌啶-3-基)-4-甲基-5-氧代-4,5-二氢-1H-1,2,4-三唑-3-基)苯基)哌嗪-1-基)甲基)哌啶-1-基)苯甲酸(95mg,0.16mmol)、4-((1r,3r)-3-氨基-2,2,4,4-四甲基环丁氧基)-2-氯苯甲腈(45mg,0.16mmol)、2-(7-氮杂苯并三唑-1-基)-N,N,N’,N’-四甲基脲六氟磷酸酯(91mg,
0.24mmol)和乙基二异丙胺(62mg,0.48mmol)在N,N-二甲基甲酰胺(5mL)中的混合物在室温下搅拌过夜。将该混合物倒入水(10mL)中,并用二氯甲烷(10mL x 3)进行萃取。将合并的有机相真空浓缩,并通过制备型HPLC纯化残余物,得到N-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基)-4-(4-((4-(4-(1-(2,6-二氧代哌啶-3-基)-4-甲基-5-氧代-4,5-二氢-1H-1,2,4-三唑-3-基)苯基)哌嗪-1-基)甲基)哌啶-1-基)苯甲酰胺(54mg,40%收率),呈白色固体。
LC-MS(Agilent LCMS 1200-6120,色谱柱:Waters X-Bridge C18(50mm x4.6mm x3.5μm);柱温:40℃;流速:2.0mL/min;流动相:在3.0分钟内从95%[水+10mM NH
[M+H]
HPLC(Agilent HPLC 1200,色谱柱:Waters X-Bridge C18(150mm x
4.6mm x 3.5μm);柱温:40℃;流速:1.0mL/min;流动相:在10分钟内从95%[水+10mM NH
1
化学式:C
得自HNMR数据的总H计数:54
示例性PROTAC 79的合成
N-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基)-6-(4-(5-((4-(2,4-二氧代-3,4-二氢嘧啶-1(2H)-基)异喹啉-7-基)氧基)戊基)哌嗪-1-基)烟酰胺
合成方案
步骤1:7-溴异喹啉的合成
向3-溴苯甲醛(50.0g,0.27mol)的甲苯(250mL)溶液中添加氨基乙醛缩二甲醇(31.1g,0.30mol),在室温下搅拌几分钟,然后在100℃下加热过夜。蒸发反应溶剂,得到呈黄色油状物的3-溴苯亚甲基氨基乙缩醛(70g,95%),将其直接用于下一步骤,无需进一步纯化。
将五氧化二磷(140g,2v)的浓硫酸(70mL,1v)溶液在室温下搅拌几分钟,然后在0℃下搅拌,向上述制备的混合物中缓慢添加3-溴苯亚甲基氨基乙缩醛(70g,0.26mol)。然后,将该混合物加热至160℃并保持30分钟。冷却至室温后,将反应混合物小心倒入冰水(100mL)中,同时剧烈搅拌,然后过滤,使用饱和氢氧化钠将pH进一步升至9并用二氯甲烷(100mL x 3)萃取,将合并的有机相经无水硫酸钠干燥,过滤,真空浓缩,并通过硅胶(石油醚/乙酸乙酯=6:1)纯化,得到7-溴异喹啉和5-溴异喹啉(15.0g,28%)的混合物,呈黄色固体。
步骤2:4,7-二溴异喹啉的合成
向7-溴异喹啉和5-溴异喹啉的混合物(15.0g,0.072mol)的乙酸(30mL)溶液中添加N-溴代琥珀酰亚胺(19.3g,0.11mol)。在氮气下,将该混合物加热至100℃并保持过夜。冷却至室温后,将水(10mL)添加到反应混合物中,并用饱和氢氧化钠中和,然后用乙酸乙酯(10mL x 3)萃取。将合并的有机相经无水硫酸钠干燥并真空浓缩,通过硅胶(石油醚/乙酸乙酯=15:1)纯化,得到化合物4,7-二溴异喹啉(6.0g,29%),呈黄色固体。
1
化学式:C
得自HNMR数据的总H计数:5。
步骤3:4-溴-7-甲氧基异喹啉的合成
向4,7-二溴异喹啉(1.0g,3.5mmol)的二甲基亚砜/甲醇(4:3)(10mL)溶液中添加甲醇钠(0.3g,5.6mmol)。将该混合物在140℃的微波反应器中加热1小时。向该混合物中添加水(5mL),并用乙酸乙酯(5mL x 3)萃取。将合并的有机层用盐水(5mL x 2)洗涤,经无水硫酸钠干燥,过滤并真空浓缩,并通过硅胶(石油醚/乙酸乙酯=10:1)纯化,得到4-溴-7-甲氧基异喹啉(180mg,22%),呈黄色固体。
1
化学式:C
得自HNMR数据的总H计数:8。
步骤4:4-溴异喹啉-7-醇的合成
在-20℃下,向4-溴-7-甲氧基异喹啉(110mg,0.46mmol)的二氯甲烷(2mL)溶液中添加BBr
LC-MS(Agilent LCMS 1200-6120,色谱柱:Waters X-Bridge C18(30mm x4.6mm x3.5μm);柱温:40℃;流速:1.5mL/min;流动相:在0.5分钟内从90%[水+10mM NH
步骤5:5-(4-溴异喹啉-7-基氧基)戊-1-醇的合成
向化合物4-溴异喹啉-7-醇(0.90g,4.02mmol)的DMF(10mL)溶液中添加5-溴戊-1-醇(0.66g,4.02mmol)和碳酸钾(0.74g,8.04mmol),然后在70℃下搅拌8小时。将该反应混合物倒入冷水中,并用二氯甲烷/甲醇(10mL x 3)进行萃取。将合并的有机层经无水硫酸钠干燥,过滤并真空浓缩,并通过制备型TLC(二氯甲烷/甲醇=15:1)纯化,得到5-(4-溴异喹啉-7-基氧基)戊-1-醇(1.0g,81%),呈黄色固体。
1
化学式:C
得自HNMR数据的总H计数:16。
步骤6:1-(7-(5-羟基戊氧基)异喹啉-4-基)嘧啶-2,4(1H,3H)-二酮的合成
在氩气气氛下,将5-(4-溴异喹啉-7-基氧基)戊-1-醇(100mg,0.32mmol)、嘧啶-2,4(1H,3H)-二酮(48mg,0.38mmol)、K
0.96mmol)、CuI(30mg,0.16mmol)、N-(2-氰基苯基)吡啶酰胺(22mg,0.16mmol)的DMSO(6mL)溶液在120℃下加热2小时。将该反应混合物冷却至室温,倒入冷水中,并用二氯甲烷/甲醇(10mL x 3)进行萃取。将合并的有机层经无水硫酸钠干燥,过滤并真空浓缩,并通过制备型TLC(二氯甲烷/甲醇=12:1)纯化,得到1-(7-(5-羟基戊氧基)异喹啉-4-基)嘧啶-2,4(1H,3H)-二酮(21mg,19%),呈黄色固体。
1
化学式:C
得自HNMR数据的总H计数:19。
步骤7:5-(4-(2,4-二氧代-3,4-二氢嘧啶-1(2H)-基)异喹啉-7-基氧基)戊醛的合成
向1-(7-(5-羟基戊氧基)异喹啉-4-基)嘧啶-2,4(1H,3H)-二酮(30mg,
0.088mmol)的二氯甲烷(10mL)溶液中添加戴斯-马丁过碘烷(112mg,0.26mmol)。将该混合物在室温下搅拌2小时。将该混合物添加到水(10.0mL)中,并用二氯甲烷(10.0mLx 2)进行萃取。将合并的有机层用盐水(20mL)洗涤,经无水硫酸钠干燥,过滤并真空浓缩,并通过制备型TLC(二氯甲烷/甲醇=12:1)纯化,得到5-(4-(2,4-二氧代-3,4-二氢嘧啶-1(2H)-基)异喹啉-7-基氧基)戊醛(20mg,67%),呈黄色固体。
步骤8:N-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基)-6-(4-(5-(4-(2,4-二氧代-3,4-二氢嘧啶-1(2H)-基)异喹啉-7-基氧基)戊基)哌嗪-1-基)烟酰胺的合成
向5-(4-(2,4-二氧代-3,4-二氢嘧啶-1(2H)-基)异喹啉-7-基氧基)戊醛(20mg,0.058mmol)的无水甲醇/1,2-二氯乙烷/HOAc(5mL/3mL/0.1mL)溶液中添加N-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基)-6-(哌嗪-1-基)烟酰胺(27mg,0.058mmol)。将该混合物在N
LC-MS(Agilent LCMS 1200-6120,色谱柱:Waters X-Bridge C18
(50mm*4.6mm*3.5μm);柱温:40℃;流速:2.0mL/min;流动相:在3.0分钟内从95%[水+10mM NH
HPLC(Agilent HPLC 1200,色谱柱:Waters X-Bridge C18
(150mm*4.6mm*3.5μm);柱温:40℃;流速:1.0mL/min;流动相:在10分钟内从95%[水+10mM NH
1
化学式:C
得自HNMR数据的总H计数:47。
示例性PROTAC
N-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基)-6-(4-(5-((3-(5-氰基-2,4-二氧代-3,4-二氢嘧啶-1(2H)-基)喹啉-6-基)氧基)戊基)哌嗪-1-基)烟酰胺合成方案
步骤1:5-(3-氨基喹啉-6-基氧基)戊-1-醇的合成
在氮气气氛下,向5-(3-溴喹啉-6-基氧基)戊-1-醇(1.1g,3.6mmol)、二苯甲酮亚胺(684mg,3.8mmol)和叔丁醇钠(691mg,7.2mmol)的甲苯(20mL)溶液中添加(+/-)-2,2’-双(二苯基膦基)-1,1’-联萘(448mg,0.7mmol)和三(二亚苄基丙酮)二钯(207mg,0.36mmol),并将该混合物回流2小时。将其冷却至室温后,添加水(20mL)。将所得混合物用乙酸乙酯(10mL x3)萃取,用盐水(20mL x 3)洗涤,经无水硫酸钠干燥,并过滤。然后向滤液中添加4NHCl(5mL),将该混合物搅拌一小时。分离各层,并用水(10mL x3)萃取有机层。然后将合并的水相用饱和NaHCO
LCMS(Agilent LCMS 1200-6120,色谱柱:Waters X-Bridge C18(50mm x4.6mm x3.5μm);柱温:40℃;流速:2.0mL/min;流动相:在1.6分钟内从95%[水+10mM NH
1
化学式:C
得自HNMR数据的总H计数:18。
步骤2:1-(6-(5-羟基戊氧基)喹啉-3-基)-2,4-二氧代-1,2,3,4-四氢嘧啶-5-甲腈的合成
将5-(3-氨基喹啉-6-基氧基)戊-1-醇(600mg,2.44mmol)、N-氨基甲酰基-2-氰基乙酰胺(1.2g,9.76mmol)和三甲氧基甲烷(1.0g,9.76mmol)的二甲基亚砜(10mL)溶液在80℃下搅拌过夜,并将反应混合物在120℃下继续搅拌2小时。当将其冷却至室温时,向混合物中添加水(30mL),得到白色固体。过滤所得混合物,并通过制备型HPLC纯化固体,得到1-(6-(5-羟基戊氧基)喹啉-3-基)-2,4-二氧代-1,2,3,4-四氢嘧啶-5-甲腈(110mg,12%收率),呈白色固体。
1
化学式:C
得自HNMR数据的总H计数:18。
步骤3:5-(3-(5-氰基-2,4-二氧代-3,4-二氢嘧啶-1(2H)-基)喹啉-6-基氧基)戊基甲磺酸酯的合成
在0℃下,向1-(6-(5-羟基戊氧基)喹啉-3-基)-2,4-二氧代-1,2,3,4-四氢嘧啶-5-甲腈(110mg,0.30mmol)和三乙胺(98mg,0.90mmol)的二氯甲烷(4mL)溶液中添加甲磺酰氯(51mg,0.45mmol),并将该混合物在室温下搅拌30分钟。然后向该混合物中添加水(5mL),并将所得混合物用二氯甲烷(5mL x3)萃取,用盐水(5mL x 3)洗涤,经无水Na
步骤4:1-(6-(5-碘代戊氧基)喹啉-3-基)-2,4-二氧代-1,2,3,4-四氢嘧啶-5-甲腈的合成
向5-(3-(5-氰基-2,4-二氧代-3,4-二氢嘧啶-1(2H)-基)喹啉-6-基氧基)戊基甲磺酸酯(150mg)的乙腈(3mL)溶液中添加碘化钾(50mg,0.3mmol),并将该混合物在90℃下搅拌4小时。当将其冷却至室温后,向该混合物中添加水(5mL),并且将所得混合物用二氯甲烷(5mL x 3)萃取,用盐水(5mL x 3)洗涤,经无水Na
=10/1)纯化残余物,得到所需产物(40mg,两步收率28%)。
LCMS(Agilent LCMS 1200-6120,色谱柱:Waters X-Bridge C18(50mm x4.6mm x3.5μm);柱温:40℃;流速:2.0mL/min;流动相:在1.6分钟内从90%[(总共10mM AcONH
/CH
步骤5:N-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基)-6-(4-(5-(3-(5-氰基-2,4-二氧代-3,4-二氢嘧啶-1(2H)-基)喹啉-6-基氧基)戊基)哌嗪-1-基)烟酰胺的合成
将1-(6-(5-碘代戊氧基)喹啉-3-基)-2,4-二氧代-1,2,3,4-四氢嘧啶-5-甲腈(40mg,0.08mmol)、N-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基)-6-(哌嗪-1-基)烟酰胺(39mg,0.08mmol)和乙基二异丙胺(30mg,0.25mmol)的乙腈(2mL)溶液在90℃下搅拌过夜。当将其冷却至室温后,添加水(5mL),并且将该混合物用乙酸乙酯(2mL x 3)萃取,用盐水(5mL x 3)洗涤,经无水Na
LCMS(Agilent LCMS 1200-6120,色谱柱:Waters X-Bridge C18
(50mm*4.6mm*3.5μm);柱温:40℃;流速:2.0mL/min;流动相:在3.0分钟内从95%[水+10mM NH
[M+H]
HPLC(Agilent HPLC 1200,色谱柱:Waters X-Bridge C18
(150mm*4.6mm*3.5μm);柱温:40℃;流速:1.0mL/min;流动相:在10分钟内从95%[水+10mM NH
1
得自HNMR数据的总H计数:46。
示例性PROTAC
N-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基)-4-(4-((4-(2-(2,6-二氧代哌啶-3-基)-4-亚甲基-1-氧代-1,2,3,4-四氢异喹啉-6-基)哌嗪-1-基)甲基)哌啶-1-基)苯甲酰胺
反应方案:
步骤1:4-溴-2-碘苯甲酸甲酯的合成
在1000mL 3颈圆底烧瓶中置入2-氨基-4-溴苯甲酸甲酯(5.0g,
21.73mmol,1.00当量)、硫酸(20%)(20mL)的水(100mL)溶液。然后,在0℃下搅拌1小时后,在0℃下一边搅拌一边逐滴添加NaNO
步骤2:4-溴-2-氰基苯甲酸甲酯的合成
在250mL圆底烧瓶中置入4-溴-2-碘苯甲酸甲酯(5.8g,17.01mmol,1.00当量)、NMP(60mL)、CuCN(1.82g,20.45mmol,1.20当量)。将所得溶液在60℃下在油浴中搅拌2小时。用乙酸乙酯(50mL x 2)萃取所得溶液,再合并有机层。用FeSO4(水溶液)(50mL x 2)洗涤所得混合物。将该混合物经无水硫酸钠干燥。将残余物施加到使用乙酸乙酯/石油醚(1/3)的硅胶柱上。这得到3.68g(90%)的4-溴-2-氰基苯甲酸甲酯,呈白色固体。
步骤3:6’-溴螺[环丙烷-1,1’-异吲哚啉]-3'-酮的合成
在用氮气惰性气氛吹扫并保持的100mL 3颈圆底烧瓶中置入4-溴-2-氰基苯甲酸甲酯(2.0g,8.33mmol,1.00当量)、乙醚(40mL)、2-(丙-2-基氧基)丙烷丙-2-醇丙-2-基钛二水合物(2.75mL,1.10当量)。然后在0℃下,一边搅拌一边逐滴添加EtMgBr(3M)(5.5mL,2.00当量)。将所得溶液在室温下搅拌3小时。然后通过添加20mL氯化氢(1M)来淬灭反应。用乙酸乙酯(50mL x 2)萃取所得溶液,并且将有机层合并,再经无水硫酸钠干燥。将残余物施加到使用乙酸乙酯/石油醚(7/3)的硅胶柱上。这得到409mg(21%)的6’-溴螺[环丙烷-1,1’-异吲哚啉]-3’-酮,呈黄色固体。
LC-MS(ES
步骤4:2-(6’-溴-3’-氧代螺[环丙烷-1,1’-异吲哚啉]-2'-基)戊二酸二甲酯的合成
在100mL圆底烧瓶中置入6’-溴螺[环丙烷-1,1’-异吲哚啉]-3’-酮(895.0mg,3.76mmol,1.00当量)、N,N-二甲基甲酰胺(15.0mL)、Cs
LC-MS(ES
步骤5:2-(6-(4-(叔丁氧基羰基)哌嗪-1-基)-4-亚甲基-1-氧代-3,4-二氢异喹啉-2(1H)-基)戊二酸二甲酯的合成
在用氮气惰性气氛吹扫并保持的20mL圆底烧瓶中置入2-(6’-溴-3’-氧代螺[环丙烷-1,1’-异吲哚啉]-2’-基)戊二酸二甲酯(740.0mg,1.87mmol,1.00当量)、甲苯(10mL)、哌嗪-1-羧酸叔丁酯(418.0mg,2.24mmol,1.20当量)、Cs
LC-MS(ES
步骤6:4-[2-(1-氨基甲酰基-4-甲氧基-4-氧代丁基)-4-亚甲基-1-氧代-1,2,3,4-四氢异喹啉-6-基]哌嗪-1-羧酸叔丁酯的合成在100mL圆底烧瓶中置入1,5-二甲基2-(6-[4-[(叔丁氧基)羰基]哌嗪-1-基]-4-亚甲基-1-氧代-1,2,3,4-四氢异喹啉-2-基)戊二酸酯(400mg,0.80mmol,1当量)、MeOH(50mL)、NH
步骤7:4-[2-(2,6-二氧代哌啶-3-基)-4-亚甲基-1-氧代-1,2,3,4-四氢异喹啉-6-基]哌嗪-1-羧酸叔丁酯的合成
在50mL圆底烧瓶中置入4-[2-(1-氨基甲酰基-4-甲氧基-4-氧代丁基)-4-亚甲基-1-氧代-1,2,3,4-四氢异喹啉-6-基]哌嗪-1-羧酸叔丁酯(188mg,
0.39mmol,1当量)、乙腈(20mL)、Cs
步骤8:3-[4-亚甲基-1-氧代-6-(哌嗪-1-基)-1,2,3,4-四氢异喹啉-2-基]哌啶-2,6-二酮(三氟乙酸盐)的合成
在50mL圆底烧瓶中置入4-[2-(2,6-二氧代哌啶-3-基)-4-亚甲基-1-氧代-1,2,3,4-四氢异喹啉-6-基]哌嗪-1-羧酸叔丁酯(120mg,0.26mmol,1当量)、二氯甲烷(20mL)、TFA(1.5mL)。将所得溶液在室温下搅拌2小时。真空浓缩所得混合物。这得到93mg(77.86%)的3-[4-亚甲基-1-氧代-6-(哌嗪-1-基)-1,2,3,4-四氢异喹啉-2-基]哌啶-2,6-二酮(TFA盐),呈黄色固体。
步骤9:N-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基)-4-(4-((4-(2-(2,6-二氧代哌啶-3-基)-4-亚甲基-1-氧代-1,2,3,4-四氢异喹啉-6-基)哌嗪-1-基)甲基)哌啶-1-基)苯甲酰胺的合成
在50mL圆底烧瓶中置入4-(4-甲酰基哌啶-1-基)-N-[(1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基]苯甲酰胺(83mg,0.17mmol,1当量)、二氯甲烷(20mL)、3-[4-亚甲基-1-氧代-6-(哌嗪-1-基)-1,2,3,4-四氢异喹啉-2-基]哌啶-2,6-二酮(TFA盐)(91.2mg、0.20mmol,1.2当量)、NaBH(OAc)
1
LC-MS(ES
化学式:C
得自HNMR数据的总H计数:54
示例性PROTAC
N-((1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基)-4-(4-((4-(2-(2,6-二氧代哌啶-3-基)-1-氧代-1,2-二氢异喹啉-6-基)哌嗪-1-基)甲基)哌啶-1-基)苯甲酰胺
合成方案
步骤1:4-(1-氧代-2H-异喹啉-6-基)哌嗪-1-羧酸叔丁酯的合成
将6-溴-2H-异喹啉-1-酮(2g,8.93mmol,1当量)、哌嗪-1-羧酸叔丁酯(2.49g,13.39mmol,1.5当量)、叔丁醇钠(2M,13.4mL,3当量)和[2-(2-氨基苯基)苯基]-氯-钯;[2-(2,6-二异丙氧基苯基)苯基]膦酸二环己酯(693mg,0.89mmol,0.1当量)在叔戊醇(30mL)中的混合物脱气并用氮气吹扫3次,然后在氮气气氛下,将该混合物在100℃下搅拌12小时。LCMS显示反应完成,并且可以检测到所需MS。将反应混合物用水(100mL)稀释,并用乙酸乙酯(50mL x 3)萃取。将合并的有机相用饱和盐水(50mL x 2)洗涤,用无水硫酸钠干燥,过滤并真空浓缩。通过硅胶色谱(石油醚:乙酸乙酯=20:1至3:1)纯化残余物,得到4-(1-氧代-2H-异喹啉-6-基)哌嗪-1-羧酸叔丁酯(2.3g,6.98mmol,78%收率),呈白色固体
LCMS:MS(ESI)m/z:330.1[M+1]
1
化学式:C
得自HNMR数据的总H计数:23。
步骤2:2-[6-(4-叔丁氧基羰基哌嗪-1-基)-1-氧代-2-异喹啉基]戊二酸二甲酯的合成
向4-(1-氧代-2H-异喹啉-6-基)哌嗪-1-羧酸叔丁酯(800mg,2.43mmol,1当量)的二甲基甲酰胺(16mL)溶液中添加碳酸铯(2.37g,7.29mmol,3当量)和2-溴戊二酸二甲酯(696mg,2.91mmol,1.2当量)。将该混合物在100℃下搅拌12小时。LCMS显示反应完成,并且可以检测到所需MS。用盐酸(1M)将反应混合物调节至pH4~5。将反应用水(60mL)稀释,并用乙酸乙酯(30mL x 3)萃取。将合并的有机相用饱和盐水(30mL x 2)洗涤,用无水硫酸钠干燥,过滤并真空浓缩,得到呈浅黄色油状物的粗产物2-[6-(4-叔丁氧基羰基哌嗪-1-基)-1-氧代-2-异喹啉基]戊二酸二甲酯(700mg,粗制物),将其用于下一步骤,无需进一步纯化。
LCMS:MS(ESI)m/z:474.1[M+1]
化学式:C
步骤3:2-[6-(4-叔丁氧基羰基哌嗪-1-基)-1-氧代-2-异喹啉基]戊二酸的合成
向2-[6-(4-叔丁氧基羰基哌嗪-1-基)-1-氧代-2-异喹啉基]戊二酸二甲酯(800mg,1.64mmol,1当量)在四氢呋喃(5mL)、甲醇(5mL)和水(5mL)中的溶液中添加氢氧化锂一水合物(413mg,9.85mmol,6当量)。将该混合物在30℃下搅拌12小时。LCMS显示反应完成,并且可以检测到所需MS。将反应用盐酸(1M)调节至pH4~5,并用水(25mL)稀释。用乙酸乙酯(15mL x3)萃取反应。将合并的有机相用无水硫酸钠干燥,过滤并真空浓缩,得到呈黄色固体的粗产物2-[6-(4-叔丁氧基羰基哌嗪-1-基)-1-氧代-2-异喹啉基]戊二酸(800mg,粗制物),将其用于下一步骤,无需进一步纯化。
LCMS:MS(ESI)m/z:460.1[M+1]
化学式:C
步骤4:4-[2-(2,6-二氧代-3-哌啶基)-1-氧代-6-异喹啉基)哌嗪-1-羧酸叔丁酯的合成
向2-[6-(4-叔丁氧基羰基哌嗪-1-基)-1-氧代-2-异喹啉基]戊二酸(800mg,1.74mmol,1当量)的N-甲基-2-吡咯烷酮(10mL)溶液中添加尿素(522mg,8.71mmol,5当量)。将该混合物在160℃下搅拌2小时。LCMS显示反应完成,并且可以检测到所需MS。将反应混合物用水(50mL)稀释,并用乙酸乙酯(25mL x 3)萃取。将合并的有机相用饱和盐水(30mL x2)洗涤,用无水硫酸钠干燥,过滤并真空浓缩。通过半制备型反相HPLC(色谱柱:PhenomenexSynergi Max-RP 250*50mm*10um;流动相:[水(0.225%FA)-ACN];B%:30ACN%-60ACN%,30分钟,50%分钟)纯化残余物。获得4-[2-(2,6-二氧代-3-哌啶基)-1-氧代-6-异喹啉基]哌嗪-1-羧酸叔丁酯(100mg,0.22mmol,13%收率),呈白色固体。
LCMS:MS(ESI)m/z:441.1[M+1]
化学式:C
步骤5:3-(1-氧代-6-哌嗪-1-基-2-异喹啉基)哌啶-2,6-二酮的合成
向4-[2-(2,6-二氧代-3-哌啶基)-1-氧代-6-异喹啉基]哌嗪-1-羧酸叔丁酯(100mg,0.22mmol,1当量)的二氯甲烷(3mL)溶液中添加4M盐酸的二氧杂环己烷(3mL,52.86当量)溶液。将该混合物在25℃下搅拌4小时。LCMS显示剩余14%的起始材料,并将反应再搅拌1小时。薄层色谱(二氯甲烷:甲醇=10:1)显示反应完成。减压浓缩反应混合物,以去除二氯甲烷、二氧杂环己烷和盐酸,得到粗产物3-(1-氧代-6-哌嗪-1-基-2-异喹啉基)哌啶-2,6-二酮(85mg,粗制物,盐酸盐),呈浅黄色固体。
LCMS:MS(ESI)m/z:341.0[M+1]
化学式:C
步骤6:N-[3-(3-氯-4-氰基-苯氧基)-2,2,4,4-四甲基-环丁基]-4-[4-[[4-[2-(2,6-二氧代-3-哌啶基)-1-氧代-6-异喹啉基]哌嗪-1-基]甲基]-1-哌啶基]苯甲酰胺的合成
向3-(1-氧代-6-哌嗪-1-基-2-异喹啉基)哌啶-2,6-二酮(85mg,0.22mmol,1当量,盐酸盐)的1,2-二氯乙烷(4mL)溶液中添加三乙胺(0.9mmol,
0.12mL,4当量)和N-[3-(3-氯-4-氰基-苯氧基)-2,2,4,4-四甲基-环丁基]-4-(4-甲酰基-1-哌啶基)苯甲酰胺(111mg,0.22mmol,1当量)。将该混合物在20℃下搅拌0.5小时。添加三乙酰氧基硼氢化钠(95mg,0.45mmol,2当量),并将该混合物在20℃下搅拌12小时。LCMS显示反应完成,并且可以检测到所需MS。减压浓缩反应混合物,以去除1,2-二氯乙烷。将残余物溶解于二甲基甲酰胺(3mL)中并过滤。通过半制备型反相HPLC(色谱柱:Phenomenex Synergi C18 150*25*10um;流动相:[水(0.05%HCl)-ACN];
B%:23%-53%,10分钟)纯化滤液,得到N-[3-(3-氯-4-氰基-苯氧基)-2,2,4,4-四甲基-环丁基]-4-[4-[[4-[2-(2,6-二氧代-3-哌啶基)-1-氧代-6-异喹啉基]哌嗪-1-基]甲基]-1-哌啶基]苯甲酰胺(50.9mg,0.05mmol,25%收率,95.8%纯度,盐酸盐),呈白色固体。
LCMS:MS(ESI)m/z:818.4[M+1]
1
化学式:C
得自HNMR数据的总H计数:53。
示例性PROTAC
3-[3-[4-[4-[[1-[4-[(1R,2S)-6-羟基-2-苯基-四氢萘-1-基]苯基]-4-哌啶基]甲基]哌嗪-1-基]苯基]-5-氧代-2H-吡咯-1-基]哌啶-2,6-二酮
步骤1:6-叔丁氧基四氢萘-1-酮的制备
在0℃下,向6-羟基四氢萘-1-酮(50g,308.29mmol,1当量)在无水二氯甲烷(2000mL)中的搅拌溶液中添加2,2,2-三氯乙酰亚胺酸叔丁酯(67.36g,308.29mmol,55mL,1当量)和对甲苯磺酸吡啶盐(7.75g,30.83mmol,0.1当量)。将反应混合物在10℃下搅拌3小时。再添加一份2,2,2-三氯乙酰亚胺酸叔丁酯(67.36g,308.29mmol,55mL,1当量)和对甲苯磺酸吡啶盐(7.75g,30.83mmol,0.1当量),并将反应混合物在10℃下搅拌15小时。此过程重复三次。薄层色谱(石油醚:乙酸乙酯=3:1,R
步骤2:(6-叔丁氧基-3,4-二氢萘-1-基)三氟甲磺酸酯的制备
在-70℃下,向6-叔丁氧基四氢萘-1-酮(40g,183.24mmol,1当量)的四氢呋喃(500mL)溶液中添加二异丙基氨基锂(2M,137mL,1.5当量)。将该混合物在-70℃下搅拌1小时,然后将1,1,1-三氟-N-苯基-N-(三氟甲基磺酰基)甲烷磺酰胺(72.01g,201.56mmol,1.1当量)的四氢呋喃(200mL)溶液逐滴添加到混合物中。将反应混合物在20℃下搅拌2小时。薄层色谱(石油醚:乙酸乙酯=5:1)显示反应完成。向混合物中添加饱和氯化铵(300mL),分离有机相。向混合物中添加乙酸乙酯(500mL x 3),用盐水(1000mL x 2)洗涤所得混合物。将合并的有机相经硫酸钠干燥,过滤并真空浓缩。通过硅胶色谱(石油醚:乙酸乙酯=1:0至50:1)纯化残余物,得到(6-叔丁氧基-3,4-二氢萘-1-基)三氟甲磺酸酯(52g,144.64mmol,78%收率,97%纯度),呈黄色油状物。LC-MS(ESI)m/z:294.9[M+1-56]
步骤3:4-(6-叔丁氧基-3,4-二氢萘-1-基)苯酚的制备
在氮气下,向(6-叔丁氧基-3,4-二氢萘-1-基)三氟甲磺酸酯(52g,148.42mmol,1当量)、(4-羟基苯基)硼酸(24.57g,178.11mmol,1.2当量)在二氧杂环己烷(800mL)和水(150mL)的溶液中添加碳酸钾(41.03g,296.84mmol,2当量)和(1,1’-双(二苯基膦基)二茂铁)钯(II)二氯化物(10.86g,14.84mmol,0.1当量)。将反应混合物在100℃下搅拌10小时。薄层色谱(石油醚:乙酸乙酯=5:1)显示反应完成。将残余物用水(500mL)稀释,并用乙酸乙酯(500mL x 2)萃取。将合并的有机层用盐水(1000mL x 2)洗涤,用无水硫酸钠干燥,过滤并真空浓缩。通过硅胶色谱(石油醚:四氢呋喃=50:1至20:1)纯化残余物,得到4-(6-叔丁氧基-3,4-二氢萘-1-基)苯酚(43g,131.46mmol,88%收率,90%纯度),呈黄色油状物。LCMS(ESI)m/z:239.1[M+1-56]
步骤4:4-(2-溴-6-叔丁氧基-3,4-二氢萘-1-基)苯酚的制备
向4-(6-叔丁氧基-3,4-二氢萘-1-基)苯酚(1g,3.06mmol,1当量)的乙腈(20mL)溶液中分三份添加N-溴代琥珀酰亚胺(489mg,2.75mmol,0.9当量)。将反应混合物在20℃下搅拌1.5小时。LC-MS显示反应完成。将残余物用水(20mL)稀释,并用乙酸乙酯(20mL x 2)萃取。将合并的有机层用盐水(20mL x 2)洗涤,用无水硫酸钠干燥,过滤并真空浓缩。通过硅胶色谱(石油醚:乙酸乙酯=1:0至20:1)纯化残余物,得到4-(2-溴-6-叔丁氧基-3,4-二氢萘-1-基)苯酚(1g,2.46mmol,80%收率,91%纯度),呈黄色油状物。LC-MS(ESI)m/z:316.9[M+1-56]
步骤5:4-(6-叔丁氧基-2-苯基-3,4-二氢萘-1-基)苯酚的制备
在氮气下,向4-(2-溴-6-叔丁氧基-3,4-二氢萘-1-基)苯酚(1g,2.46mmol,1当量)、苯基硼酸(314mg,2.58mmol,1.05当量)在二氧杂环己烷(10mL)和水(2mL)的溶液中添加碳酸钾(678mg,4.91mmol,2当量)和(1,1’-双(二苯基膦基)二茂铁)钯(II)二氯化物(179mg,0.24mmol,0.1当量)。将反应混合物在100℃下搅拌12小时。LC-MS显示反应完成。将残余物用水(20mL)稀释,并用乙酸乙酯(20mL x 2)萃取。将合并的有机层用盐水(20mL x3)洗涤,用无水硫酸钠干燥,过滤并真空浓缩。通过硅胶色谱(石油醚:乙酸乙酯=1:0至10:1)纯化残余物,得到4-(6-叔丁氧基-2-苯基-3,4-二氢萘-1-基)苯酚(930mg,2.35mmol,95收率,93%纯度),呈橙色油状物。LCMS(ESI)m/z:314.1[M+1-56]
步骤6:4-(6-叔丁氧基-2-苯基-四氢萘-1-基)苯酚的制备
在氮气下,向4-(6-叔丁氧基-2-苯基-3,4-二氢萘-1-基)苯酚(930mg,
2.35mmol,1当量)在四氢呋喃(20mL)和甲醇(4mL)中的溶液中添加活性炭载钯催化剂(100mg,10%纯度)。将悬浮液真空脱气并用氢气吹扫三次。将混合物在氢气(50psi)下在30℃下搅拌36小时。LC-MS显示反应完成。过滤反应混合物并浓缩溶液。将所得物质直接用于下一步骤,无需进一步纯化,得到顺-4-(6-叔丁氧基-2-苯基-四氢萘-1-基)苯酚(870mg,2.14mmol,91%收率,91%纯度),呈白色固体。LC-MS(ESI)m/z:317.0[M+1-56]
步骤7:WX-ARV-HD-012-E1,4-[(1S,2R)-6-叔丁氧基-2-苯基-四氢萘-1-基]苯酚的制备
使4-(6-叔丁氧基-2-苯基-四氢萘-1-基)苯酚(870mg,2.13mmol,1当量)经受超临界流体色谱以进行手性分离(色谱柱:AD,250mm x 30mm,5um;流动相:0.1%氢氧化铵的甲醇溶液,20%-20%,每次运行4.2分钟),得到作为第一级分的4-[(1S,2R)-6-叔丁氧基-2-苯基-四氢萘-1-基]苯酚(420mg,1.04mmol,97%收率,92%纯度)以及作为第二级分的4-[(1R,2S)-6-叔丁氧基-2-苯基-四氢萘-1-基]苯酚(420mg,1.04mmol,97%收率,92%纯度)。级分1:[ ]
级分2:[α]
步骤8:4-(6-苄氧基-2-苯基-3,4-二氢萘-1-基)苯基]1,1,2,2,3,3,4,4,4-九氟丁烷-1-磺酸酯的制备
向4-[(1R,2S)-6-叔丁氧基-2-苯基-四氢萘-1-基]苯酚(1g,2.68mmol,1当量)和1,1,2,2,3,3,4,4,4-九氟丁烷-1-磺酰氟(811mg,2.68mmol,1当量)在四氢呋喃(5mL)和乙腈(5mL)中的溶液中添加碳酸钾(557mg,4.03mmol,1.5当量)。将反应混合物在25℃下搅拌16小时。TLC(石油醚:乙酸乙酯
=10:1)显示起始材料被完全消耗,并且形成一个新斑点。减压浓缩反应混合物。通过硅胶色谱(石油醚:乙酸乙酯=1:0至50:1)纯化残余物。获得所需化合物[4-[(1R,2S)-6-叔丁氧基-2-苯基-四氢萘-1-基]苯基]1,1,2,2,3,3,4,4,4-九氟丁烷-1-磺酸酯(1.6g,2.44mmol,91%收率),呈无色油状物。
步骤9:1-[4-(6-苄氧基-2-苯基-3,4-二氢萘-1-基)苯基]-4-(二甲氧基甲基)哌啶的制备
使[4-[(1R,2S)-6-叔丁氧基-2-苯基-四氢萘-1-基]苯基]1,1,2,2,3,3,4,4,4-九氟丁烷-1-磺酸酯(1.6g,2.44mmol,1当量)、4-(二甲氧基甲基)哌啶(584mg,3.67mmol,1.5当量)、叔丁醇钠(705mg,7.33mmol,3当量)、乙酸钯
(82mg,0.37mmol,0.15当量)和二环己基膦基-2’,4’,6’-三异丙基联苯(233mg,0.49mmol,0.2当量)在甲苯(30mL)中的混合物脱气并用氮气吹扫3次,然后在氮气气氛下,将该混合物在90℃下搅拌16小时。LC-MS显示检测到具有所需MS的一个主峰。TLC(石油醚:乙酸乙酯=10:1)显示起始材料被完全消耗,并且形成一个新斑点。将混合物冷却,用乙酸乙酯(50mL)稀释,在硅藻土塞上过滤,用乙酸乙酯(30mL)洗涤滤饼。浓缩滤液。通过硅胶色谱(石油醚:乙酸乙酯=100:1至10:1)纯化残余物。获得所需化合物1-[4-[(1R,2S)-6-叔丁氧基-2-苯基-四氢萘-1-基]苯基]-4-(二甲氧基甲基)哌啶(1.1g,2.14mmol,87%收率),呈白色固体。LCMS(ESI)m/z:514.3
[M+1]
步骤10:1-[4-[4-(二甲氧基甲基)-1-哌啶基]苯基]-2-苯基-四氢萘-6-醇的制备
向1-[4-[(1R,2S)-6-叔丁氧基-2-苯基-四氢萘-1-基]苯基]-4-(二甲氧基甲基)哌啶(1.1g,2.14mmol,1当量)的四氢呋喃(45mL)溶液中添加硫酸(2M,43mL,40当量)。将反应混合物在70℃下搅拌1小时。薄层色谱(石油醚:乙酸乙酯=3:1)显示起始材料被完全消耗,并且形成一个新斑点。通过添加饱和碳酸氢钠溶液至pH=7~8使反应混合物淬灭,并用乙酸乙酯(20mL x 2)萃取。将合并的有机层用盐水(20mL)洗涤,经硫酸钠干燥,过滤并减压浓缩。将残余物用于下一步骤,无需进一步纯化。获得所需化合物1-[4-[(1R,2S)-6-羟基-2-苯基-四氢萘-1-基]苯基]哌啶-4-甲醛(900mg,
2.14mmol,99%收率,97%纯度),呈浅黄色固体。LCMS MS(ESI)m/z:412.1[M+1]
步骤11:(Z)-3-(4-溴苯基)丁-2-烯酸乙酯的制备
向冷却至0℃的氢化钠(2.41g,60.29mmol,60%纯度,1.2当量)在四氢呋喃(100mL)中的悬浮液中缓慢添加2-二乙氧基磷酰乙酸乙酯(13.52g,60.29mmol,12mL,1.2当量),并将反应混合物在25℃下搅拌1小时。逐滴添加1-(4-溴苯基)乙酮(10g,50.24mmol,1当量)的四氢呋喃(100mL)溶液,并将该混合物在25℃下搅拌12小时。向该混合物中添加饱和氯化铵水溶液(50mL)。用乙酸乙酯(100mL x 3)萃取混合物。将有机层经无水硫酸钠干燥并浓缩。用制备型HPLC(乙腈:水=50:1至5:1)纯化残余物。获得呈黄色油状物的(Z)-3-(4-溴苯基)丁-2-烯酸乙酯(6.6g,24.52mmol,48.9%收率),还获得呈黄色油状物的(E)-3-(4-溴苯基)丁-2-烯酸乙酯(2.6g,9.66mmol,19.3%收率)。LC/MS(ESI)m/z:270.0[M+1]
步骤12:4-[4-[(Z)-3-乙氧基-1-甲基-3-氧代-丙-1-烯基]苯基]哌嗪-1-羧酸叔丁酯的制备
将(Z)-3-(4-溴苯基)丁-2-烯酸乙酯(2.0g,7.43mmol,1当量)、哌嗪-1-羧酸叔丁酯(2.08g,11.15mmol,1.5当量)、碳酸铯(4.84g,14.86mmol,2当量)、乙酸钯(334mg,1.49mmol,0.2当量)和XPhos(708mg,1.49mmol,0.2当量)在甲苯(30mL)中的混合物脱气并用氮气吹扫三次。在氮气气氛下,将该混合物在100℃下搅拌12小时。将所得混合物过滤并减压浓缩。将残余物用饱和盐水(30mL x 2)洗涤,并用乙酸乙酯(30mL x 2)萃取。将合并的有机层经无水硫酸钠干燥,过滤并减压浓缩。通过半制备型反相HPLC(色谱柱:PhenomenexSynergi Max-RP 250x 50mm,10um;流动相:[水(0.225%甲酸)-乙腈];B%:50%乙腈-80%乙腈,30分钟)纯化残余物。获得4-[4-[(Z)-3-乙氧基-1-甲基-3-氧代-丙-1-烯基]苯基]哌嗪-1-羧酸叔丁酯(2.24g,5.83mmol,78%收率,97%纯度),呈白色固体。LC/MS(ESI)m/z:375.1[M]
步骤13:4-[4-[(E)-1-(溴甲基)-3-乙氧基-3-氧代-丙-1-烯基]苯基]哌嗪-1-羧酸叔丁酯的制备
向4-[4-[(Z)-3-乙氧基-1-甲基-3-氧代-丙-1-烯基]苯基]哌嗪-1-羧酸叔丁酯(1.0g,2.60mmol,1当量)和1-溴吡咯烷-2,5-二酮(462.93mg,
2.60mmol,1当量)的二氯乙烷(10mL)溶液中添加过氧化苯甲酰(189mg,0.78mmol,0.3当量)。将该混合物脱气并用氮气吹扫3次。在氮气气氛下,将该混合物在70℃下搅拌12小时。LC-MS显示检测到约24%的所需化合物。将反应混合物用饱和盐水(25mL x 2)洗涤,并用二氯甲烷(40mL x 2)萃取。将合并的有机层经无水硫酸钠干燥,过滤并减压浓缩。通过硅胶色谱(石油醚/乙酸乙酯=50/1至25:1)纯化残余物。获得4-[4-[(E)-1-(溴甲基)-3-乙氧基-3-氧代-丙-1-烯基]苯基]哌嗪-1-羧酸叔丁酯(0.3g,
0.43mmol,16%收率,65%纯度),呈黄色油状物。LC/MS(ESI)m/z:453.0[M+1]
步骤14:4-[4-[1-(2,6-二氧代-3-哌啶基)-5-氧代-2H-吡咯-3-基]苯基]哌嗪-1-羧酸叔丁酯的制备
向3-氨基哌啶-2,6-二酮(84.95mg,0.52mmol,1.2当量,HCl盐)在二甲基甲酰胺(3mL)中的混合物中添加N,N-二异丙基乙胺(556mg,4.30mmol,0.7mL,10当量)。将该混合物在20℃下搅拌1小时。然后向反应中添加4-[4-[(E)-1-(溴甲基)-3-乙氧基-3-氧代-丙-1-烯基]苯基]哌嗪-1-羧酸叔丁酯(0.3g,0.43mmol,1当量),并将该混合物在50℃下搅拌0.5小时。将所得混合物进一步加热至120℃并搅拌12小时。LC-MS显示检测到所需化合物。将反应混合物冷却,用乙酸乙酯稀释,用饱和盐水(25mL x 2)洗涤,并用乙酸乙酯(30mL x 2)萃取。将合并的有机层经无水硫酸钠干燥,过滤并减压浓缩。通过用甲基叔丁基醚(15mL)研磨来纯化残余物。获得产物4-[4-[1-(2,6-二氧代-3-哌啶基)-5-氧代-2H-吡咯-3-基]苯基]哌嗪-1-羧酸叔丁酯(175mg,0.23mmol,52%收率,58%纯度),呈棕色固体。LC/MS(ESI)m/z:455.1[M+1]
步骤15:3-[5-氧代-3-(4-哌嗪-1-基苯基)-2H-吡咯-1-基]哌啶-2,6-二酮的制备
向4-[4-[1-(2,6-二氧代-3-哌啶基)-5-氧代-2H-吡咯-3-基]苯基]哌嗪-1-羧酸叔丁酯(175mg,0.22mmol,1当量)溶液中添加HCl的二氧杂环己烷(4M,5mL)溶液。将该混合物在20℃下搅拌1小时。真空浓缩反应混合物,得到残余物。获得3-[5-氧代-3-(4-哌嗪-1-基苯基)-2H-吡咯-1-基]哌啶-2,6-二酮(260mg,粗制物,HCl盐),呈棕色固体。LC/MS(ESI)m/z:355.1[M+1]
步骤16:3-[3-[4-[4-[[1-[4-[(1R,2S)-6-羟基-2-苯基-四氢萘-1-基]苯基]-4-哌啶基]甲基]哌嗪-1-基]苯基]-5-氧代-2H-吡咯-1-基]哌啶-2,6-二酮(示例性PROTAC89)的制备
向3-[5-氧代-3-(4-哌嗪-1-基苯基)-2H-吡咯-1-基]哌啶-2,6-二酮(260mg,0.66mmol,1当量,HCl盐)的二氯乙烷(3mL)溶液中添加三乙胺(202mg,2.00mmol,0.3mL,3当量)和1-[4-[(1R,2S)-6-羟基-2-苯基-四氢萘-1-基]苯基]哌啶-4-甲醛(109mg,0.26mmol,0.4当量)。将该混合物在25℃下搅拌15分钟,然后添加乙酸硼氢化钠(282mg,1.33mmol,2当量)。将该混合物在25℃下再搅拌11.5小时。LC-MS显示检测到约74%的所需化合物。将反应混合物用二氯甲烷稀释,用饱和盐水(20mL x 2)洗涤,并用二氯甲烷(30mL x 2)萃取。将合并的有机层经无水硫酸钠干燥,过滤并减压浓缩,得到残余物。通过制备型HPLC(色谱柱:Phenomenex Synergi C18 150x25mm,10um;流动相:[水(0.225%甲酸)-乙腈];B%:在10分钟内从22%到43%)纯化残余物。获得产物3-[3-[4-[4-[[1-[4-[(1R,2S)-6-羟基-2-苯基-四氢萘-1-基]苯基]-4-哌啶基]甲基]哌嗪-1-基]苯基]-5-氧代-2H-吡咯-1-基]哌啶-2,6-二酮(38.7mg,0.04mmol,7%收率,95%纯度,甲酸盐),呈棕色固体。
LC/MS(ESI)m/z:750.3[M+1]
1
示例性PROTAC
3-[4-[4-[[1-[4-[(1R,2S)-6-羟基-2-苯基-四氢萘-1-基]苯基]-4-哌啶基]甲基]哌嗪-1-基]-6-氧代-哒嗪-1-基]哌啶-2,6-二酮
步骤1:4-(5-氯-6-氧代-1H-哒嗪-4-基)哌嗪-1-羧酸叔丁酯的制备
向4,5-二氯-1H-哒嗪-6-酮(5g,30.31mmol,1当量)的二甲基亚砜(100mL)溶液中添加二异丙基乙胺(11.75g,90.92mmol,3当量)和哌嗪-1-羧酸叔丁酯盐酸盐(6.75g,30.31mmol,1当量)。将该混合物在120℃下搅拌3小时。将所得混合物冷却至室温,过滤并通过添加水(500mL)来淬灭,然后用乙酸乙酯(100mL x 3)萃取。将合并的有机相用盐水(100mL)洗涤,经无水硫酸钠干燥,过滤并减压浓缩。通过硅胶色谱(二氯甲烷:甲醇=200:1至100:1)纯化残余物。获得4-(5-氯-6-氧代-1H-哒嗪-4-基)哌嗪-1-羧酸叔丁酯(8.18g,24.95mmol,82%收率,96%纯度),呈黄色固体。LC/MS(ESI)m/z:315.1[M+1]
步骤2:4-(6-氧代-1H-哒嗪-4-基)哌嗪-1-羧酸叔丁酯的制备
在氮气下,向4-(5-氯-6-氧代-1H-哒嗪-4-基)哌嗪-1-羧酸叔丁酯(1g,3.18mmol,1当量)在四氢呋喃(1mL)和甲醇(9mL)中的溶液中添加钯/活性炭催化剂(200mg,10%纯度)。将悬浮液真空脱气并用氢气吹扫数次。将混合物在氢气(45psi)下在25℃下搅拌0.5小时。用三乙胺碱化反应,然后过滤,并浓缩滤液。将残余物用于下一步骤,无需进一步纯化。获得4-(6-氧代-1H-哒嗪-4-基)哌嗪-1-羧酸叔丁酯(1g,粗制物),呈白色固体。LC/MS(ESI)m/z:281.1[M+1]
步骤3:4-[1-(2,6-二氧代-3-哌啶基)-6-氧代-哒嗪-4-基]哌嗪-1-羧酸叔丁酯的制备
在25℃下,向4-(6-氧代-1H-哒嗪-4-基)哌嗪-1-羧酸叔丁酯(950mg,3.39mmol,1当量)的二甲基亚砜(15mL)溶液中添加氢化钠(271mg,6.78mmol,60%纯度,2当量),然后添加3-溴哌啶-2,6-二酮(650mg,3.39mmol,1当量)。将该混合物在25℃下搅拌12小时。将所得混合物过滤并通过添加水(200mL)来淬灭,然后用乙酸乙酯(50mL x 3)萃取。将合并的有机相用盐水(50mL x 3)洗涤,经无水硫酸钠干燥,过滤并减压浓缩。通过半制备型反相HPLC(色谱柱:Phenomenex luna C18 250x50mm,10um;流动相:[水(0.225%甲酸)-ACN];B%:在30分钟内从16%到46%)纯化残余物。获得4-[1-(2,6-二氧代-3-哌啶基)-6-氧代-哒嗪-4-基]哌嗪-1-羧酸叔丁酯(190mg,0.48mmol,14%收率),呈白色固体。LC/MS(ESI)m/z:392.1[M+1]
步骤4:3-(6-氧代-4-哌嗪-1-基-哒嗪-1-基)哌啶-2,6-二酮的制备
向4-[1-(2,6-二氧代-3-哌啶基)-6-氧代-哒嗪-4-基]哌嗪-1-羧酸叔丁酯(190mg,0.48mmol,1当量)的二氯甲烷(2mL)溶液中添加盐酸的二氧杂环己烷(4M,10mL,78当量)溶液。将该混合物在25℃下搅拌0.5小时。减压浓缩所得混合物,以去除二氧杂环己烷。将粗产物用于下一步骤,无需进一步纯化。获得化合物3-(6-氧代-4-哌嗪-1-基-哒嗪-1-基)哌啶-2,6-二酮(120mg,0.36mmol,75%收率,盐酸盐),呈浅黄色固体。LC/MS(ESI)m/z:292.0[M+1]
步骤5:3-[4-[4-[[1-[4-[(1R,2S)-6-羟基-2-苯基-四氢萘-1-基]苯基]-4-哌啶基]甲基]哌嗪-1-基]-6-氧代-哒嗪-1-基]哌啶-2,6-二酮(示例性PROTAC 102)的制备
向3-(6-氧代-4-哌嗪-1-基-哒嗪-1-基)哌啶-2,6-二酮(57mg,0.17mmol,1.2当量,盐酸盐)和1-[4-[(1R,2S)-6-羟基-2-苯基-四氢萘-1-基]苯基]哌啶-4-甲醛(60mg,0.14mmol,1当量,参见步骤10,示例性PROTAC 89的合成)的1,2-二氯乙烷(3mL)溶液中添加三乙胺(30mg,0.29mmol,2当量),并将该混合物在25℃下搅拌0.5小时。然后添加三乙酰氧基硼氢化钠(93mg,0.43mmol,3当量)。将该混合物在25℃下进一步搅拌0.5小时。减压浓缩反应混合物,以去除1,2-二氯乙烷。通过制备型HPLC(色谱柱:Luna C18150x 25mm,5um;流动相:[水(0.225%甲酸)-ACN];B%:在7.8分钟内从18%到38%)纯化残余物。获得化合物3-[4-[4-[[1-[4-[(1R,2S)-6-羟基-2-苯基-四氢萘-1-基]苯基]-4-哌啶基]甲基]哌嗪-1-基]-6-氧代-哒嗪-1-基]哌啶-2,6-二酮(33mg,0.04mmol,30%收率,99%纯度,甲酸盐),呈白色固体。LC/MS(ESI)m/z:687.3[M+1]
示例性PROTAC
3-(4-(3-(1-(3-(4-((1R,2S)-6-羟基-2-苯基-1,2,3,4-四氢萘-1-基)苯氧基)丙基)哌啶-4-基)苯氧基)-2-氧代-2,5-二氢-1H-吡咯-1-基)哌啶-2,6-二酮
合成方案第1部分:
步骤1:4-(3-羟基苯基)-3,6-二氢-2H-吡啶-1-羧酸叔丁酯的制备
在氮气下,向4-(4,4,5,5-四甲基-1,3,2-二氧杂戊硼烷-2-基)-3,6-二氢-2H-吡啶-1-羧酸叔丁酯(7.00g,22.64mmol,1.00当量)和3-碘苯酚(4.98g,22.64mmol,1.00当量)在二氧杂环己烷(100mL)和水(10mL)中的混合物中添加碳酸钾(6.26g,45.28mmol,2.00当量)和(二苯基)膦酸环戊酯;二氯化钯;铁(1.66g,2.26mmol,0.10当量)。在氮气下,将该混合物在90℃下搅拌4小时。LC-MS显示起始材料被完全消耗,并且检测到一个具有所需MS的主峰。将反应混合物倒入水(500mL)中并过滤,将滤液用乙酸乙酯
(200mL)稀释并用乙酸乙酯(300mL×3)萃取,将合并的有机相用饱和盐水(150mL)洗涤,经无水硫酸钠干燥,过滤并减压浓缩。通过硅胶柱色谱(石油醚/乙酸乙酯=10/1至8/1)纯化残余物。获得4-(3-羟基苯基)-3,6-二氢-2H-吡啶-1-羧酸叔丁酯(4.00g,14.53mmol,64%收率),呈白色固体。
LCMS:MS(ESI)m/z:298.1[M+23]
化学式:C
步骤2:4-(3-羟基苯基)哌啶-1-羧酸叔丁酯的制备
在氮气下,向4-(3-羟基苯基)-3,6-二氢-2H-吡啶-1-羧酸叔丁酯(4.00g,14.53mmol,1.00当量)的甲醇(4mL)溶液中添加活性炭载钯催化剂(1.00g,10%纯度)。将悬浮液真空脱气并用氢气吹扫数次。将混合物在氢气(40psi)下在30℃下搅拌4小时。LC-MS显示起始材料被完全消耗,并且检测到一个具有所需MS的主峰。将反应混合物过滤并减压浓缩。将残余物用于下一步骤,无需进一步纯化。获得粗制的4-(3-羟基苯基)哌啶-1-羧酸叔丁酯(4.00g,粗制物),呈灰白色固体。
LCMS:MS(ESI)m/z:300[M+23]
化学式:C
步骤3:4-[3-[(E)-3-甲氧基-1-甲基-3-氧代-丙-1-烯氧基]苯基]哌啶-1-羧酸叔丁酯的制备
向4-(3-羟基苯基)哌啶-1-羧酸叔丁酯(2.00g,7.21mmol,1.00当量)和丁-2-炔酸甲酯(1.06g,10.82mmol,1.50当量)的异丙醇(20mL)溶液中添加1,4-二氮杂双环[2.2.2]辛烷(808mg,7.21mmol,1.00当量)。将该混合物在15℃下搅拌12小时。LC-MS显示起始材料被完全消耗,并且检测到一个具有所需MS的主峰。在15℃下用水20mL淬灭反应混合物,并用乙酸乙酯(20mL x 3)萃取。将合并的有机层用饱和盐水(20mL×2)洗涤,经无水硫酸钠干燥,过滤并减压浓缩。通过硅胶柱色谱(石油醚/乙酸乙酯=20:1至10:1)纯化残余物。获得4-[3-[(E)-3-甲氧基-1-甲基-3-氧代-丙-1-烯氧基]苯基]哌啶-1-羧酸叔丁酯(1.72g,4.58mmol,63%收率),呈白色固体。LCMS:MS(ESI)m/z:398.1[M+23]
化学式:C
步骤4:(E)-4-(3-((1-溴-4-甲氧基-4-氧代丁-2-烯-2-基)氧基)苯基)哌啶-1-羧酸叔丁酯的制备
向4-[3-[(E)-3-甲氧基-1-甲基-3-氧代-丙-1-烯氧基]苯基]哌啶-1-羧酸叔丁酯(1.2g,3.20mmol,1.00当量)在二氯乙烷(50mL)中的混合物中添加N-溴代琥珀酰亚胺(853mg,4.79mmol,1.5当量)和过氧化苯甲酰(232mg,0.96mmol,0.3当量)。将该混合物在70℃下搅拌12小时。LC-MS显示起始材料被完全消耗,并且检测到一个具有所需MS的主峰。将混合物通过添加水(200mL)淬灭,用乙酸乙酯(20mL)稀释并用乙酸乙酯(30mL×3)萃取,将合并的有机相用饱和盐水(30mL)洗涤,经无水硫酸钠干燥,过滤并减压浓缩。通过硅胶柱色谱(石油醚/乙酸乙酯=100:1至40:1)纯化残余物。获得4-[3-[(E)-1-(溴甲基)-3-甲氧基-3-氧代-丙-1-烯氧基]苯基]哌啶-1-羧酸叔丁酯(960mg,粗制物),呈黄色油状物。
LCMS:MS(ESI)m/z:477.9[M+23]
化学式:C
步骤5:4-[3-[[1-(2,6-二氧代-3-哌啶基)-5-氧代-2H-吡咯-3-基]氧基]苯基]哌啶-1-羧酸叔丁酯的制备
向3-氨基哌啶-2,6-二酮(1.56g,9.46mmol,5当量,盐酸盐)在二甲基甲酰胺(20mL)中的混合物中添加二异丙基乙胺(2.45g,18.93mmol,10当量)。将该混合物在14℃下搅拌1小时。向反应中添加4-[3-[(E)-1-(溴甲基)-3-甲氧基-3-氧代-丙-1-烯氧基]苯基]哌啶-1-羧酸叔丁酯(860mg,
1.89mmol,1当量)。然后将该混合物在50℃下搅拌0.5小时。然后将该混合物加热至100℃并保持12小时。LC-MS显示起始材料溴化物被完全消耗,并且检测到一个具有所需MS的主峰。将混合物通过添加水(200mL)淬灭,用乙酸乙酯(50mL)稀释并用乙酸乙酯(50mL×3)萃取,将合并的有机相用饱和盐水(50mL×3)洗涤,经无水硫酸钠干燥,过滤并减压浓缩。通过用甲基叔丁基醚(30mL)研磨来纯化残余物。获得4-[3-[[1-(2,6-二氧代-3-哌啶基)-5-氧代-2H-吡咯-3-基]氧基]苯基]哌啶-1-羧酸叔丁酯(376mg,0.80mmol,42%收率),呈棕色固体
LCMS:MS(ESI)m/z:492.2[M+23]
化学式:C
步骤6:3-[5-氧代-3-[3-(4-哌啶基)苯氧基]-2H-吡咯-1-基]哌啶-2,6-二酮的制备
向4-[3-[[1-(2,6-二氧代-3-哌啶基)-5-氧代-2H-吡咯-3-基]氧基]苯基]哌啶-1-羧酸叔丁酯(420mg,0.89mmol,1当量)在二氯甲烷(10mL)中的混合物中添加氯化氢/二氧杂环己烷(4M,4mL,20当量)。将该混合物在14℃下搅拌0.5小时。LC-MS显示起始材料被完全消耗,并且检测到一个具有所需MS的主峰。减压浓缩反应物。将残余物用于下一步骤,无需进一步纯化。获得粗制物3-[5-氧代-3-[3-(4-哌啶基)苯氧基]-2H-吡咯-1-基]哌啶-2,6-二酮(400mg,粗制物,盐酸盐),呈棕色固体
LCMS:MS(ESI)m/z:370[M+1]
化学式:C
合成方案第2部分:
步骤7:(顺)-6-苄氧基-1-[4-(3-溴丙氧基)苯基]-2-苯基-四氢化萘的制备
向4-[(1R,2S)-6-苄氧基-2-苯基-四氢萘-1-基]苯酚(1.00g,2.46mmol,1.00当量)的丙酮(20mL)溶液中添加碳酸钾(1.02g,7.38mmol,3.00当量)和1,3-二溴丙烷(2.48g,12.30mmol,1.3mL,5.00当量)。将该混合物在70℃下搅拌12小时。LC-MS显示起始材料被完全消耗,并且检测到一个具有所需MS的主峰。在15℃下通过添加水(40mL)淬灭反应混合物,并用乙酸乙酯(20mL X 3)萃取。将合并的有机层用乙酸乙酯(20mL X 2)洗涤,经无水硫酸钠干燥,过滤并减压浓缩。通过制备型HPLC(色谱柱:Phenomenex Synergi Max-RP 250*50mm*10um;流动相:[水(0.225%FA)-ACN];B%:70%-100%,30;52%分钟)纯化残余物。获得(顺)-6-苄氧基-1-[4-(3-溴丙氧基)苯基]-2-苯基-四氢化萘(850mg,1.61mmol,65%收率,99%纯度),呈白色固体。
LCMS:MS(ESI)m/z:527.2[M+1]
化学式:C
步骤8:(1S,2R)-6-苄氧基-1-[4-(3-溴丙氧基)苯基]-2-苯基-四氢化萘和(1R,2S)-6-苄氧基-1-[4-(3-溴丙氧基)苯基]-2-苯基-四氢化萘的制备。
使用超临界流体色谱分离(顺)6-苄氧基-1-[4-(3-溴丙氧基)苯基]-2-苯基-四氢化萘(850mg,1.61mmol,1.00当量)的对映体。通过超临界流体色谱(色谱柱:OJ(250mm*30mm,10um);流动相:[0.1%NH3H2O MEOH];
B%:60%-60%,20.9min;300minmin)分离残余物,流速:2mL/min,波长:220nm。
获得(1S,2R)-6-苄氧基-1-[4-(3-溴丙氧基)苯基]-2-苯基-四氢化萘(350mg,0.65mmol,81%收率,97%纯度),呈白色固体。
获得(1R,2S)-6-苄氧基-1-[4-(3-溴丙氧基)苯基]-2-苯基-四氢化萘(350mg,0.66mmol,82%收率,99%纯度),呈白色固体。
化学式:C
步骤9:3-[3-[3-[1-[3-[4-[(1R,2S)-6-苄氧基-2-苯基-四氢萘-1-基]苯氧基]丙基]-4-哌啶基]苯氧基]-5-氧代-2H-吡咯-1-基]哌啶-2,6-二酮的制备
向(1R,2S)-6-苄氧基-1-[4-(3-溴丙氧基)苯基]-2-苯基-四氢化萘(164mg,0.31mmol,1.1当量)和3-[5-氧代-3-[3-(4-哌啶基)苯氧基]-2H-吡咯-1-基]哌啶-2,6-二酮(115mg,0.28mmol,1当量,盐酸盐)在乙腈(5mL)中的混合物中添加二异丙基乙胺(110mg,0.85mmol,3当量)和碘化钾(47mg,0.28mmol,1当量)。将该混合物在100℃下搅拌1.5小时。LC-MS显示胺起始材料被完全消耗,并且检测到一个具有所需MS的主峰。将混合物通过添加水(100mL)淬灭,用乙酸乙酯(15mL)稀释并用乙酸乙酯(20mL X 4)萃取,将合并的有机相用饱和盐水(20mL)洗涤,经无水硫酸钠干燥,过滤并减压浓缩。通过制备型TLC(二氯甲烷:甲醇=10:1)纯化残余物。获得3-[3-[3-[1-[3-[4-[(1R,2S)-6-苄氧基-2-苯基-四氢萘-1-基]苯氧基]丙基]-4-哌啶基]苯氧基]-5-氧代-2H-吡咯-1-基]哌啶-2,6-二酮(100mg,0.12mmol,43%收率),呈棕色固体。
LCMS:MS(ESI)m/z:816.4[M+1]
化学式:C
步骤10:3-[3-[3-[1-[3-[4-[(1R,2S)-6-羟基-2-苯基-四氢萘-1-基]苯氧基]丙基]-4-哌啶基]苯氧基]-5-氧代-2H-吡咯-1-基]哌啶-2,6-二酮的制备
在-68℃下,向3-[3-[3-[1-[3-[4-[(1R,2S)-6-苄氧基-2-苯基-四氢萘-1-基]苯氧基]丙基]-4-哌啶基]苯氧基]-5-氧代-2H-吡咯-1-基]哌啶-2,6-二酮(100mg,0.12mmol,1当量)在二氯甲烷(5mL)中的混合物中添加三溴化硼(92mg,0.37mmol,3当量)。将该混合物在-68℃下搅拌30分钟。LC-MS显示起始材料被完全消耗,并且检测到一个具有所需MS的主峰。将残余物用水(20mL)稀释,并用乙酸乙酯(20mL X 2)萃取。将合并的有机层用饱和盐水(20mL X 3)洗涤,用无水硫酸钠干燥,过滤并真空浓缩。通过制备型HPLC(色谱柱:BostonGreen ODS 150*30 5u;流动相:[水(0.225%FA)-ACN];B%:34%-55%,10分钟)纯化残余物。获得3-[3-[3-[1-[3-[4-[(1R,2S)-6-羟基-2-苯基-四氢萘-1-基]苯氧基]丙基]-4-哌啶基]苯氧基]-5-氧代-2H-吡咯-1-基]哌啶-2,6-二酮(16mg,0.02mmol,16%收率,97%纯度,甲酸盐),呈白色固体
LCMS:MS(ESI)m/z:726.3[M+1]
1
δ=10.92(s,1H),9.48-8.87(m,1H),8.21(s,1H),7.41-7.34(m,1H),7.23-7.06(m,6H),6.82(d,J=6.8Hz,2H),6.66-6.57(m,2H),6.55-6.44(m,3H),6.25(d,J=8.8Hz,2H),4.91-4.82(m,2H),4.18-3.97(m,3H),3.84(t,J=6.4Hz,2H),3.30-3.27(m,2H),3.02-2.82(m,5H),2.55-2.52(m,3H),2.39(t,J=6.9Hz,2H),2.26(dd,J=4.8,13.6Hz,1H),2.11-1.58(m,11H)
化学式:C
示例性PROTAC
3-(8-((2-(4-(2-(4-((2-(4-溴苯基)-6-羟基苯并[b]噻吩-3-基)氧基)苯氧基)乙基)哌嗪-1-基)乙基)氨基)-2-甲基-4-氧代喹唑啉-3(4H)-基)哌啶-2,6-二酮合成方案第1部分:
合成方案第2部分:
步骤1:4-[2-(4-苄氧基苯氧基)乙基]哌嗪-1-羧酸叔丁酯的合成
在氮气下,向4-(2-氯乙基)哌嗪-1-羧酸叔丁酯(1.00g,4.02mmol,1.00当量)、4-苄氧基苯酚(965mg,4.82mmol,1.20当量)的N,N-二甲基甲酰胺(20mL)溶液中添加碳酸铯(1.57g,4.82mmol,1.20当量)和碘化钾(66mg,0.4mmol,0.10当量)。将反应在80℃下搅拌10小时。TLC(石油醚/乙酸乙酯=3/1)和LCMS显示大部分起始材料被消耗。向混合物中添加水(100mL),用乙酸乙酯(50mL x 3)萃取所得混合物。将合并的有机相用盐水(80mL)洗涤,经硫酸钠干燥,过滤并真空浓缩。通过硅胶色谱(石油醚/乙酸乙酯=50/1至3/1)纯化残余物,获得4-[2-(4-苄氧基苯氧基)乙基]哌嗪-1-羧酸叔丁酯(1.4g,3.39mmol,84%收率),呈无色油状物。
化学式:C24H32N2O4,分子量:412.5
得自HNMR数据的总H计数:32
1
δ:7.46-7.29(m,5H),6.95-6.88(m,2H),6.88-6.81(m,2H),5.02(s,2H),4.07(t,J=5.8Hz,2H),3.51-3.42(m,4H),2.80(t,J=5.8Hz,2H),2.56-2.48(m,4H),1.47(s,9H)
步骤2:4-[2-(4-羟基苯氧基)乙基]哌嗪-1-羧酸叔丁酯的合成
在氮气下,向4-[2-(4-苄氧基苯氧基)乙基]哌嗪-1-羧酸叔丁酯(1.40g,3.39mmol,1.00当量)的甲醇(20mL)溶液中添加炭载钯(200mg,10%纯度)。将悬浮液真空脱气并用氢气吹扫数次。将混合物在氢气(50psi)下在20℃下搅拌4小时。TLC(石油醚/乙酸乙酯=1/1)显示大部分起始材料被消耗。过滤反应混合物并真空浓缩滤液。获得4-[2-(4-羟基苯氧基)乙基]哌嗪-1-羧酸叔丁酯(1g,3.07mmol,90%收率,99%纯度),呈浅黄色固体。
化学式:C17H26N2O4,分子量:322.4
得自HNMR数据的总H计数:26
1
δ:6.74(s,4H),4.04(t,J=5.6Hz,2H),3.54-3.38(m,5H),2.79(t,J=5.6Hz,2H),2.53(s,4H),1.46(s,9H)
步骤3:4-(2-(4-((2-(4-溴苯基)-6-甲氧基-1-氧代苯并[b]噻吩-3-基)氧基)苯氧基)乙基)哌嗪-1-羧酸叔丁酯的合成
在0℃下,向4-[2-(4-羟基苯氧基)乙基]哌嗪-1-羧酸叔丁酯(234mg,0.72mmol,1.00当量)的N,N-二甲基甲酰胺(5mL)溶液中添加NaH(29mg,0.72mmol,60%矿物油,1.00当量)。将该混合物在20℃下搅拌0.5小时。添加3-溴-2-(4-溴苯基)-6-甲氧基-1-氧代-苯并噻吩-1-鎓(300mg,0.72mmol,1.00当量),然后将该混合物在20℃下搅拌1小时。LCMS显示反应完成,并且可以检测到所需MS。将反应混合物用水
(10mL)淬灭,并用乙酸乙酯(10mL x 3)萃取。将合并的有机相用饱和盐水(10mL x2)洗涤,用无水硫酸钠干燥,过滤并真空浓缩,得到呈黄色固体的4-[2-[4-[2-(4-溴苯基)-6-甲氧基-1-氧代-苯并噻吩-1-鎓-3-基]氧基苯氧基]乙基]哌嗪-1-羧酸叔丁酯(430mg,0.66mmol,90%收率),将其直接用于下一步骤,无需进一步纯化。
LCMS:MS(ESI)m/z:657.0[M+1]
1
δ:7.65(d,J=8.4Hz,2H),7.52-7.46(m,3H),7.05-6.89(m,4H),6.81(d,J=8.4Hz,2H),4.05(t,J=5.6Hz,2H),3.89(s,3H),3.50-3.42(m,4H),2.81(t,J=5.6Hz,2H),2.52(s,4H),1.47(s,9H)
化学式:C
得自HNMR数据的总H计数:35。
步骤4:4-(2-(4-((2-(4-溴苯基)-6-甲氧基苯并[b]噻吩-3-基)氧基)苯氧基)乙基)哌嗪-1-羧酸叔丁酯的合成
向4-[2-[4-[2-(4-溴苯基)-6-甲氧基-1-氧代-苯并噻吩-1-鎓-3-基]氧基苯氧基]乙基]哌嗪-1-羧酸叔丁酯(370mg,0.56mmol,1.00当量)的乙腈(6mL)溶液中添加碘化钠(254mg,1.69mmol,3.00当量)和三甲基氯硅烷(123mg,1.13mmol,2.00当量)。将该混合物在20℃下搅拌1小时。LCMS显示反应完成,并且可以检测到所需MS。将反应混合物用饱和亚硫酸钠(2mL)淬灭,用水(15mL)稀释,并用乙酸乙酯(10mL x 2)萃取。将合并的有机相用饱和盐水(10mL x 2)洗涤,用无水硫酸钠干燥,过滤并真空浓缩,得到呈黄色油状物的4-[2-[4-[2-(4-溴苯基)-6-甲氧基-苯并噻吩-3-基]氧基苯氧基]乙基]哌嗪-1-羧酸叔丁酯(350mg,粗制物),将其直接用于下一步骤,无需进一步纯化。
LCMS:MS(ESI)m/z:639.0[M+1]
化学式:C
步骤5:2-(4-溴苯基)-3-(4-(2-(哌嗪-1-基)乙氧基)苯氧基)苯并[b]噻吩-6-醇的合成
在0℃下,向4-[2-[4-[2-(4-溴苯基)-6-甲氧基-苯并噻吩-3-基]氧基苯氧基]乙基]哌嗪-1-羧酸叔丁酯(350mg,0.55mmol,1.00当量)的二氯甲烷(6mL)溶液中添加三溴化硼(410mg,1.64mmol,0.16mL,3.00当量)。将该混合物在20℃下搅拌1小时。LCMS显示反应完成,并且可以检测到所需MS。在0℃下,将反应混合物用饱和碳酸氢钠(5mL)淬灭,用水(10mL)稀释,并用二氯甲烷(10mL x 3)萃取。将合并的有机相用饱和盐水(5mL x 2)洗涤,用无水硫酸钠干燥,过滤并真空浓缩,得到呈黄色固体的2-(4-溴苯基)-3-[4-(2-哌嗪-1-基乙氧基)苯氧基]苯并噻吩-6-醇(250mg,粗制物),将其直接用于下一步骤,无需进一步纯化。
LCMS:MS(ESI)m/z:527.0[M+1]
1
δ:7.65-7.56(m,4H),7.31(d,J=2.0Hz,1H),7.14(d,J=8.4Hz,1H),6.86(s,4H),6.83(dd,J=2.0,8.4Hz,1H),5.75(s,1H),3.97(t,J=5.6Hz,2H),2.78-2.66(m,4H),2.61(t,J=5.6Hz,2H),2.40(s,4H),2.45-2.34(m,1H)
化学式:C
得自HNMR数据的总H计数:25。
步骤6:2-甲基-8-硝基-4H-苯并[d][1,3]噁嗪-4-酮的合成
将2-氨基-3-硝基-苯甲酸(2g,10.98mmol,1.00当量)在乙酸酐(10mL)中的混合物在120℃下再搅拌16小时。TLC(石油醚:乙酸乙酯)表明形成了新斑点。浓缩反应混合物以去除溶剂。将残余物用石油醚:乙酸乙酯=2:1(30mL)研磨,然后过滤。获得滤饼,为所需产物2-甲基-8-硝基-3,1-苯并噁嗪-4-酮(600mg,2.91mmol,26%收率)。
1
δ:8.42-8.31(m,2H),7.72(t,J=8.0Hz,1H),3.42(s,3H)。
化学式:C
得自HNMR数据的总H计数:6。
步骤7:3-(2-甲基-8-硝基-4-氧代喹唑啉-3(4H)-基)哌啶-2,6-二酮的合成
向2-甲基-8-硝基-3,1-苯并噁嗪-4-酮(1g,4.85mmol,1.00当量)和3-氨基哌啶-2,6-二酮(956mg,5.82mmol,1.20当量,盐酸盐)的N,N-二甲基甲酰胺(15mL)溶液中添加亚磷酸三苯酯(2.26g,7.27mmol,1.9mL,1.50当量)。将该混合物在100℃下搅拌14小时。LCMS显示反应完成,并且可以检测到所需MS。将反应混合物用水(40mL)稀释,用乙酸乙酯(30mLx 2)萃取。将合并的有机相用盐水(30mL x 3)洗涤,用无水硫酸钠干燥,过滤并真空浓缩,得到粗产物3-(2-甲基-8-硝基-4-氧代-喹唑啉-3-基)哌啶-2,6-二酮(450mg,粗制物),将其用于下一步骤,无需进一步纯化。
LCMS:MS(ESI)m/z:316.9[M+1]
化学式:C
步骤8:3-(8-氨基-2-甲基-4-氧代喹唑啉-3(4H)-基)哌啶-2,6-二酮的合成
在氮气气氛下,向3-(2-甲基-8-硝基-4-氧代-喹唑啉-3-基)哌啶-2,6-二酮(450mg,1.42mmol,1.00当量)的四氢呋喃(50mL)溶液中添加钯/C催化剂(100mg,0.14mmol,10%纯度)。将悬浮液脱气并用氢气吹扫3次。将混合物在氢气(15Psi)下在20℃下搅拌16小时。LCMS显示反应完成,并且可以检测到所需MS。过滤反应混合物并浓缩滤液,得到粗产物3-(8-氨基-2-甲基-4-氧代-喹唑啉-3-基)哌啶-2,6-二酮(380mg,1.33mmol,94%收率),将其用于下一步骤,无需进一步纯化。
LCMS:MS(ESI)m/z:287.1[M+1]
1
δ:11.01(s,1H),7.20-7.10(m,2H),6.97(dd,J=2.0,7.2Hz,1H),5.67(s,2H),5.27-5.18(m,1H),2.91-2.79(m,1H),2.70-2.58(m,5H),2.21-2.10(m,1H)化学式:C
得自HNMR数据的总H计数:14。
步骤9:2-(4-溴苯基)-3-(4-(2-(4-(2,2-二甲氧基乙基)哌嗪-1-基)乙氧基)苯氧基)苯并[b]噻吩-6-醇的合成
将2-(4-溴苯基)-3-[4-(2-哌嗪-1-基乙氧基)苯氧基]苯并噻吩-6-醇(250mg,0.33mmol,1.00当量,氢溴酸盐)、二异丙基乙胺(213mg,1.65mmol,0.3mL,5.00当量)和2-溴-1,1-二甲氧基-乙烷(112mg,0.66mmol,0.1mL,2.00当量)溶解于微波管中的N-甲基-2-吡咯烷酮(3.00mL)中。将密封管在微波下在150℃下加热1小时。TLC(二氯甲烷:甲醇
=10:1,R
LCMS:MS(ESI)m/z:615.0[M+1]
化学式:C
步骤10:2-(4-(2-(4-((2-(4-溴苯基)-6-羟基苯并[b]噻吩-3-基)氧基)苯氧基)乙基)哌嗪-1-基)乙醛的合成
向2-(4-溴苯基)-3-[4-[2-[4-(2,2-二甲氧基乙基)哌嗪-1-基]乙氧基]苯氧基]苯并噻吩-6-醇(120mg,0.20mmol,1.00当量)的二氧杂环己烷(2mL)溶液中添加盐酸(2M,2mL,20.45当量)。将该混合物在50℃下搅拌2小时。LCMS显示反应完成,并且可以检测到所需MS。减压浓缩反应混合物以去除二氧杂环己烷和水,得到粗产物2-[4-[2-[4-[2-(4-溴苯基)-6-羟基-苯并噻吩-3-基]氧基苯氧基]乙基]哌嗪-1-基]乙醛(100mg,粗制物),将其用于下一步骤,无需进一步纯化。
LCMS:MS(ESI)m/z:585.0[M+18]
化学式:C
步骤11:3-(8-((2-(4-(2-(4-((2-(4-溴苯基)-6-羟基苯并[b]噻吩-3-基)氧基)苯氧基)乙基)哌嗪-1-基)乙基)氨基)-2-甲基-4-氧代喹唑啉-3(4H)-基)哌啶-2,6-二酮的合成
向2-[4-[2-[4-[2-(4-溴苯基)-6-羟基-苯并噻吩-3-基]氧基苯氧基]乙基]哌嗪-1-基]乙醛(1000mg,0.18mmol,1.00当量)的甲醇(2mL)溶液中添加乙酸(0.2mL)和3-(8-氨基-2-甲基-4-氧代-喹唑啉-3-基)哌啶-2,6-二酮(50mg,0.18mmol,1.00当量)。将该混合物在20℃下搅拌0.5小时。添加硼烷;2-甲基吡啶(38mg,0.35mmol,2.00当量),然后将该混合物在20℃下搅拌2小时。LCMS显示反应完成,并且可以检测到所需MS。通过制备型HPLC(色谱柱:Boston Green ODS 150*30 5u;流动相:[水(0.225%FA)-ACN];B%:25%-55%,10分钟)纯化反应混合物。将收集的级分浓缩以去除大部分乙腈,并且添加盐酸(1M,2mL)。将溶液冻干成3-[8-[2-[4-[2-[4-[2-(4-溴苯基)-6-羟基-苯并噻吩-3-基]氧基苯氧基]乙基]哌嗪-1-基]乙基氨基]-2-甲基-4-氧代-喹唑啉-3-基]哌啶-2,6-二酮(10mg,0.01mmol,7%收率,盐酸盐),呈黄色固体。
LCMS:MS(ESI)m/z:839.0[M+1]
1
δ:11.01(s,1H),10.04(s,1H),7.62(s,4H),7.33(d,J=2.0Hz,1H),7.31-7.23(m,1H),7.21-7.11(m,2H),7.01(br d,J=8.0Hz,1H),6.92(q,J=8.8Hz,4H),6.85(dd,J=2.0,8.8Hz,1H),5.25(dd,J=5.2,13.2Hz,1H),4.29(s,2H),3.68-3.45(m,14H),2.87-2.79(m,1H),2.69-2.61(m,5H),2.19-2.10(m,1H)化学式:C
得自HNMR数据的总H计数:40。
示例性PROTAC
3-(8-(2-(4-(2-(4-((2-(4-溴苯基)-6-羟基苯并[b]噻吩-3-基)氧基)苯氧基)乙基)哌嗪-1-基)乙氧基)-2-甲基-4-氧代喹唑啉-3(4H)-基)哌啶-2,6-二酮
合成方案:
步骤1:3-(烯丙氧基)-2-硝基苯甲酸烯丙酯的合成
向3-羟基-2-硝基-苯甲酸(1g,5.46mmol,1.00当量)的N,N-二甲基甲酰胺(15mL)溶液中添加碳酸钾(3g,21.84mmol,4.00当量)和3-溴丙-1-烯(2.64g,21.84mmol,4.00当量)。将该混合物在20℃下搅拌15小时。LCMS显示反应完成,并且可以检测到所需MS。将残余物用水(100mL)稀释,并用乙酸乙酯(30mL x 3)萃取。将合并的有机相用盐水(30mL x 2)洗涤,用无水硫酸钠干燥,过滤并真空浓缩,得到3-烯丙氧基-2-硝基-苯甲酸烯丙酯(1.30g,粗制物),呈黄色油状物。
LCMS:MS(ESI)m/z:286.0[M+23]
化学式:C
步骤2:3-(烯丙氧基)-2-硝基苯甲酸的合成
向3-烯丙氧基-2-硝基-苯甲酸烯丙酯(1.44g,5.47mmol,1.00当量)的四氢呋喃(40mL)溶液中添加氢氧化锂一水合物(2M,11mL,4.00当量)。将该混合物在20℃下搅拌12小时。LCMS显示反应完成,并且可以检测到所需MS。将反应混合物用盐酸(2M,10mL)调节至pH=(4~5),用水(50mL)稀释,并用乙酸乙酯(30mL x 3)萃取。将合并的有机相用饱和盐水(40mL x 2)洗涤,用无水硫酸钠干燥,过滤并真空浓缩,得到3-烯丙氧基-2-硝基-苯甲酸(1.20g,粗制物),将其用于下一步骤,无需进一步纯化。LCMS:MS(ESI)m/z:246.0[M+23]
1
δ:7.70(d,J=8.0Hz,1H),7.52(t,J=8.0Hz,1H),7.32(d,J=8.0Hz,1H),6.07-5.93(m,1H),5.47-5.29(m,2H),4.70(d,J=5.2Hz,2H)
化学式:C
得自HNMR数据的总H计数:8。
步骤3:3-(烯丙氧基)-2-氨基苯甲酸的合成
在20℃下,向3-烯丙氧基-2-硝基-苯甲酸(1.2g,5.38mmol,1.00当量)在甲醇(20mL)和水(5mL)中的溶液中缓慢添加铁(1.2g,21.52mmol,4.00当量)、氯化铵(1.44g,26.90mmol,5.00当量)。将该混合物在80℃下搅拌2小时。LCMS显示反应完成,并且可以检测到所需MS。过滤反应混合物并浓缩滤液,得到3-烯丙氧基-2-氨基-苯甲酸(850mg,粗制物),将其用于下一步骤,无需进一步纯化。
LCMS:MS(ESI)m/z:194.1[M+1]
1
δ:7.54(s,1H),6.99-6.44(m,2H),6.07(s,2H),5.39(s,2H),4.59(s,3H),4.76-4.40(m,1H)
化学式:C
得自HNMR数据的总H计数:11。
步骤4:2-乙酰胺基-3-(烯丙氧基)苯甲酸的合成
向3-烯丙氧基-2-氨基-苯甲酸(800mg,4.14mmol,1.00当量)的乙腈(10mL)溶液中添加咪唑(282mg,4.14mmol,1.00当量)和乙酰氯(650mg,8.28mmol,2.00当量)。将该混合物在20℃下搅拌12小时。LCMS显示反应完成,并且可以检测到所需MS。将反应用水(30mL)稀释,并用乙酸乙酯(15mL x 3)萃取。将合并的有机相用饱和盐水(20mL x 2)洗涤,用无水硫酸钠干燥,过滤并真空浓缩,得到呈黄色固体的2-乙酰胺基-3-烯丙氧基-苯甲酸(900mg,粗制物),将其直接用于下一步骤,无需进一步纯化。
LCMS:MS(ESI)m/z:236.1[M+1]
化学式:C
步骤5:3-(8-(烯丙氧基)-2-甲基-4-氧代喹唑啉-3(4H)-基)哌啶-2,6-二酮的合成
向2-乙酰胺基-3-烯丙氧基-苯甲酸(800mg,3.40mmol,1.00当量)和3-氨基哌啶-2,6-二酮(672mg,4.08mmol,1.20当量,盐酸盐)的N,N-二甲基甲酰胺(15mL)溶液中添加亚磷酸三苯酯(1.58g,5.10mmol,1.50当量)和咪唑(232mg,92.60mmol,27.23当量)。将该混合物在100℃下搅拌16小时。LCMS显示反应完成,并且可以检测到所需MS。将反应混合物用水(40mL)稀释,并用乙酸乙酯(20mL x 2)萃取。将合并的有机相用饱和盐水(20mL x 2)洗涤,用无水硫酸钠干燥,过滤并真空浓缩。通过硅胶柱色谱(二氯甲烷:甲醇=100:1至20:1)纯化残余物,得到3-(8-烯丙氧基-2-甲基-4-氧代-喹唑啉-3-基)哌啶-2,6-二酮(420mg,1.28mmol,38%收率),呈浅黄色固体。
LCMS:MS(ESI)m/z:328.2[M+1]
1
δ:11.03(s,1H),7.58(dd,J=1.6,7.6Hz,1H),7.43-7.32(m,2H),6.17-6.01(m,1H),5.45(dd,J=1.6,17.2Hz,1H),5.34-5.25(m,2H),4.74(d,J=4.8Hz,2H),2.88-2.79(m,1H),2.70-2.55(m,5H),2.20-2.12(m,1H)
化学式:C
得自HNMR数据的总H计数:17。
步骤6:2-((3-(2,6-二氧代哌啶-3-基)-2-甲基-4-氧代-3,4-二氢喹唑啉-8-基)氧基)乙醛的合成
在-70℃下,将臭氧鼓泡到3-(8-烯丙氧基-2-甲基-4-氧代-喹唑啉-3-基)哌啶-2,6-二酮(200mg,0.61mmol,1.00当量)在二氯甲烷(8mL)和甲醇(2mL)中的溶液中并保持30分钟。用氮气吹扫过量的臭氧后,在-70℃下添加二甲基硫醚(380mg,6.11mmol,10.00当量)。将该混合物在20℃下搅拌16小时。LCMS显示反应完成,并且可以检测到所需MS。减压浓缩反应混合物以去除甲醇、二氯甲烷和二甲基硫醚,得到2-[3-(2,6-二氧代-3-哌啶基)-2-甲基-4-氧代-喹唑啉-8-基]氧基乙醛(220mg,粗制物),呈棕色固体。LCMS:MS(ESI)m/z:362.0[M+23]
化学式:C
步骤7:3-(8-(2-(4-(2-(4-((2-(4-溴苯基)-6-羟基苯并[b]噻吩-3-基)氧基)苯氧基)乙基)哌嗪-1-基)乙氧基)-2-甲基-4-氧代喹唑啉-3(4H)-基)哌啶-2,6-二酮的合成
向2-[3-(2,6-二氧代-3-哌啶基)-2-甲基-4-氧代-喹唑啉-8-基]氧基乙醛(120mg,0.36mmol,1.00当量)的甲醇(4mL)溶液中添加2-(4-溴苯基)-3-[4-(2-哌嗪-1-基乙氧基)苯氧基]苯并噻吩-6-醇(110mg,0.18mmol,0.50当量,氢溴酸盐,示例性PROTAC 107的合成的中间产物,参见上文)和乙酸(44mg,0.72mmol,2.00当量)。将该混合物在20℃下搅拌0.5小时。在20℃下添加氰基硼氢化钠(44mg,0.73mmol,2.00当量),然后将该混合物在20℃下搅拌2小时。LCMS显示反应完成,并且可以检测到所需MS。减压浓缩反应混合物,以去除甲醇。通过制备型HPLC(色谱柱:Phenomenex Synergi C18 150*30mm*4um;流动相:[水(0.225%FA)-ACN];B%:25%-55%,12分钟)纯化残余物。将收集的级分浓缩以去除大部分乙腈,并且添加盐酸(1M,2mL)。将溶液冻干,得到3-[8-[2-[4-[2-[4-[2-(4-溴苯基)-6-羟基-苯并噻吩-3-基]氧基苯氧基]乙基]哌嗪-1-基]乙氧基]-2-甲基-4-氧代-喹唑啉-3-基]哌啶-2,6-二酮(18mg,0.02mmol,5%收率,91%纯度,盐酸盐),呈白色固体。
LCMS:MS(ESI)m/z:840.2[M+1]
1
δ:11.06(s,1H),9.99(s,1H),7.66(d,J=7.2Hz,1H),7.63(s,4H),7.54-7.42(m,1H),7.52-7.42(m,1H),7.33(d,J=2.0Hz,1H),7.15(d,J=8.8Hz,1H),6.97-6.91(m,4H),6.84(dd,J=2.0,8.8Hz,1H),5.28(dd,J=5.2,13.2Hz,1H),4.54(s,2H),4.27(s,4H),3.56-3.49(m,10H),2.82-2.80(m,1H),2.65-2.59(m,5H),2.21-2.14(m,1H)
化学式:C
得自HNMR数据的总H计数:40。
示例性PROTAC
2-(2,6-二氧代哌啶-3-基)-8-(14-((5-(5-甲基-5H-吡啶并[4,3-b]吲哚-7-基)吡啶-2-基)氧基)-3,6,9,12-四氧杂十四烷基)-2,8-二氮杂螺[4.5]癸烷-1,3-二酮
反应方案:
步骤1:4-甲基4-(2-乙氧基-2-氧代乙基)-哌啶-1,4-二羧酸1-叔丁酯的制备
在-78℃下,向2-溴乙酸乙酯(8.65g,51.80mmol,5.7mL,1当量)的四氢呋喃(1000mL)溶液中添加二异丙基氨基锂(2M,39mL,1.5当量)。将该混合物在-78℃下搅拌1小时。然后添加O4-甲基哌啶-1,4-二羧酸O1-叔丁酯(20g,82.2mmol,1.59当量),并将该混合物在该温度下搅拌1小时。然后,将该混合物在15℃下再搅拌24小时。薄层色谱(石油醚:乙酸乙酯=5:1)表明剩余50%的反应物1,并且检测到一个具有较低极性的主要新斑点(R
1
化学式:C
2.步骤:1-(叔丁氧基羰基)-4-(羧甲基)哌啶-4-羧酸的制备
向O4-甲基4-(2-乙氧基-2-氧代-乙基)哌啶-1,4-二羧酸O1-叔丁酯(3.8g,11.50mmol,1当量)在四氢呋喃(20mL)、水(15mL)中的溶液中添加氢氧化钠(2.3g,57.7mmol,5当量)和甲醇(10mL)。将该混合物在25℃下搅拌36小时。高效液相色谱-质谱显示反应物1被完全消耗。将反应混合物用水20mL稀释并减压浓缩,以去除四氢呋喃和甲醇。将水层用石油醚(30mL×2)洗涤,然后用盐酸溶液酸化至pH~5,用乙酸乙酯(30mL×3)萃取。将合并的有机层用盐水60mL洗涤,经硫酸钠干燥,过滤并减压浓缩。获得1-叔丁氧基羰基-4-(羧甲基)哌啶-4-羧酸(2.9g,10mmol,87%收率),呈棕色固体。
LCMS:MS(ESI)m/z:286。
1
化学式:C
3.步骤:2-(2,6-二氧代哌啶-3-基)-1,3-二氧代-2,8-二氮杂螺[4.5]癸烷-8-羧酸叔丁酯的制备
将1-叔丁氧基羰基-4-(羧甲基)哌啶-4-羧酸(1.9g,6.61mmol,1当量)和乙酸酐(21.80g,213.54mmol,20mL,32.29当量)的混合物脱气并用氮气吹扫3次,然后在氮气气氛下,将该混合物在120℃下搅拌0.5小时。减压浓缩反应混合物,以去除乙酸酐。用吡啶(20mL)稀释残余物,并添加3-氨基哌啶-2,6-二酮(1.31g,7.94mmol,1.2当量,盐酸盐)。在氮气气氛下,将该混合物在140℃下搅拌12小时。高效液相色谱-质谱显示反应物1被完全消耗,并且检测到一个具有所需质量的主峰。减压浓缩反应混合物。用水(10mL×3)洗涤残余物,得到产物。获得2-(2,6-二氧代-3-哌啶基)-1,3-二氧代-2,8-二氮杂螺[4.5]癸烷-8-羧酸叔丁酯(1.2g,3.2mmol,47%收率),呈灰色固体。
LCMS:MS(ESI)m/z:402[M+23]
1
化学式:C
4.步骤:2-(2,6-二氧代哌啶-3-基)-2,8-二氮杂螺[4.5]癸烷-1,3-二酮的制备
向2-(2,6-二氧代-3-哌啶基)-1,3-二氧代-2,8-二氮杂螺[4.5]癸烷-8-羧酸叔丁酯(1.2g,3.16mmol,1当量)的二氧杂环己烷(15mL)溶液中添加盐酸溶液(4M的二氧杂环己烷溶液,20mL,25.3当量)。将该混合物在15℃下搅拌3小时。减压浓缩反应混合物。获得2-(2,6-二氧代-3-哌啶基)-2,8-二氮杂螺[4.5]癸烷-1,3-二酮(1.2g,盐酸盐),呈灰色固体。
1
化学式:C
5.步骤:2-[2-[2-[2-[2-[叔丁基(二苯基)甲硅烷基]氧基乙氧基]乙氧基]乙氧基]乙氧基]乙醇的制备
向2-[2-[2-[2-(2-羟基乙氧基)乙氧基]乙氧基]乙氧基]乙醇(2g,8.40mmol,1当量)的二氯甲烷(20mL)溶液中添加咪唑(1.92g,12.6mmol,1.9mL,1.5当量)和叔丁基-氯-二苯基-硅烷(2.42g,8.8mmol,2.3mL,1.05当量)。将该混合物在15℃下搅拌3小时。薄层色谱(乙酸乙酯)表明剩余10%的反应物1,并且检测到具有一个较低极性的主要新斑点(R
LCMS:MS(ESI)m/z:494[M+18]
HNMR:(400MHz,CDCl
化学式:C
6.步骤:2-[2-[2-[2-[2-[[5-(5-甲基吡啶并[4,3-b]吲哚-7-基)-2-吡啶基]氧基]乙氧基]乙氧基]乙氧基]乙氧基]乙醇的制备
在0℃下,向2-[2-[2-[2-[2-[叔丁基(二苯基)甲硅烷基]氧基乙氧基]乙氧基]乙氧基]乙氧基]乙醇(258mg,0.54mmol,1.5当量)的N,N-二甲基甲酰胺(5mL)溶液中添加氢化钠(29mg,0.72mmol,60%纯度的矿物油,2当量)。将该混合物在15℃下搅拌1小时。然后添加7-(6-氟-3-吡啶基)-5-甲基-吡啶并[4,3-b]吲哚(0.1g,361umol,1当量)。将该混合物在15℃下搅拌12小时。高效液相色谱-质谱显示反应物1被完全消耗,并且检测到一个具有所需MS的主峰。通过在0℃下添加水(15mL)来淬灭反应混合物,然后用乙酸乙酯45mL(15mL*3)萃取。将合并的有机层经硫酸钠干燥,过滤并减压浓缩,得到残余物。通过柱色谱(二氯甲烷:甲醇=20:1,R
LCMS:MS(ESI)m/z:496.0[M+1]
HNMR:(400MHz,CDCl
化学式:C
7.步骤:2-[2-[2-[2-[2-[[5-(5-甲基吡啶并[4,3-b]吲哚-7-基)-2-吡啶基]氧基]乙氧基]乙氧基]乙氧基]乙氧基]乙基4-甲基苯磺酸酯的制备
向2-[2-[2-[2-[2-[[5-(5-甲基吡啶并[4,3-b]吲哚-7-基)-2-吡啶基]氧基]乙氧基]乙氧基]乙氧基]乙氧基]乙醇(90mg,0.18mmol,1当量)的二氯甲烷(5mL)溶液中添加三乙胺(37mg,0.36mmol,2当量),然后添加对甲苯磺酰氯(139mg,0.73mmol,4当量)。将该混合物在15℃下搅拌12小时。LCMS显示反应物1被完全消耗,并且检测到一个具有所需质量数的主峰。减压浓缩反应混合物。通过制备型薄层色谱(二氯甲烷:甲醇=10:1,产物R
LCMS:MS(ESI)m/z:650[M+1]
化学式:C
8.步骤:2-(2,6-二氧代哌啶-3-基)-8-(14-((5-(5-甲基-5H-吡啶并[4,3-b]吲哚-7-基)吡啶-2-基)氧基)-3,6,9,12-四氧杂十四烷基)-2,8-二氮杂螺[4.5]癸烷-1,3-二酮的制备
将2-[2-[2-[2-[2-[[5-(5-甲基吡啶并[4,3-b]吲哚-7-基)-2-吡啶基]氧基]乙氧基]乙氧基]乙氧基]乙氧基]乙基4-甲基苯磺酸酯(50mg,0.07mmol,1当量)、2-(2,6-二氧代-3-哌啶基)-2,8-二氮杂螺[4.5]癸烷-1,3-二酮(32mg,0.10mmol,1.33当量,盐酸盐)、碘化钾(19mg,0.12mmol,1.5当量)、N,N-二异丙基乙胺(30mg,0.23mmol,3当量)在乙腈(5mL)中的混合物脱气并用氮气吹扫3次,然后在氮气气氛下,将该混合物在100℃下搅拌12小时。LCMS显示反应物1被完全消耗,并且检测到一个具有所需MS的主峰。减压浓缩反应混合物。通过半制备型反相HPLC(色谱柱:Phenomenex Synergi C18 150*25*10um;流动相:[水(0.05%HCl)-ACN];
B%:0%-30%,10分钟)纯化残余物。残余物的纯度为90%。通过半制备型反相HPLC(色谱柱:Phenomenex Synergi C18 150*30mm*4um;流动相:[水(0.225%FA)-ACN];B%:0%-26%,10.5分钟;流速(ml/min):25)纯化残余物。获得2-(2,6-二氧代-3-哌啶基)-8-[2-[2-[2-[2-[2-[[5-(5-甲基吡啶并[4,3-b]吲哚-7-基)-2-吡啶基]氧基]乙氧基]乙氧基]乙氧基]乙氧基]乙基]-2,8-二氮杂螺[4.5]癸烷-1,3-二酮(12.9mg,0.01mmol,20%收率,99%纯度,双甲酸盐),呈黄色固体。
LCMS:MS(ESI)m/z:757.3[M+1]
HNMR:(400MHz,DMSO-d
化学式:C
蛋白质水平控制
本说明书还提供了通过细胞控制蛋白质水平的方法。这基于使用如本文所述的化合物,已知所述化合物与特定靶蛋白相互作用,使得体内靶蛋白的降解将导致控制生物系统中蛋白质的量,优选地实现特定目标蛋白质治疗有益效果。
以下实施例用于帮助描述本发明,但不应视为以任何方式限制本发明。
本公开的示例性实施方案
本公开涵盖以下具体实施方案。如所指出的那些,以下这些实施方案可包括前述实施方案中所述的所有特征。适用时,以下实施方案还可包括任何前述实施方案中包含的或替代方案所述的特征
一个方面公开了一种具有以下化学结构的双官能化合物:
CLM―L―PTM,或其药学上可接受的盐、对映体、立体异构体、溶剂化物、多晶型物或前药,其中:PTM是包含蛋白质靶向部分的小分子;L是共价偶联CLM和PTM的键或化学连接部分;并且CLM是小分子人小脑蛋白E3泛素连接酶结合部分,其结合或靶向人小脑蛋白E3泛素连接酶并且具有选自以下的化学结构:
/>
/>
/>
其中:
W独立地选自CH
Q
R
R
R
R
R
X是C、CH、C=O或N;
X
R’选自H、卤素、胺、烷基(例如,C1-C3烷基)、取代的烷基(例如,取代的C1-C3烷基)、烷氧基(例如,C1-C3烷氧基)、取代的烷氧基(例如,取代的C1-C3烷氧基)、NR
n是0-4;
并且
在本文所述的任何方面或实施方案中,CLM经由W、X、R
在本文所述的任何方面或实施方案中,PTM是结合Brd4、Tau蛋白、雌激素受体(ER)或雄激素受体(AR)的部分。
在本文所述的任何方面或实施方案中,所述化合物还包含通过接头基团偶联的第二E3泛素连接酶结合部分。
在本文所述的任何方面或实施方案中,第二E3泛素连接酶结合部分结合或靶向选自希佩尔-林道蛋白(VLM)、人小脑蛋白(CLM)、小鼠双微体同源物2(MLM)和凋亡抑制蛋白(ILM)的E3泛素连接酶。
在本文所述的任何方面或实施方案中,CLM由选自以下的化学结构表示:
/>
/>
其中:
W独立地选自CH
R
R
R
R
R是H或卤素;
R’是H或者PTM、PTM’、化学接头基团(L)、ULM、CLM、CLM’的附接点,
Q1和Q2各自独立地是被独立地选自H或C1-C3烷基的基团取代的C或N;
是单键或双键;并且
Rn包含官能团或原子。
在本文所述的任何方面或实施方案中,CLM由选自以下的化学结构表示:
/>
其中R’是卤素。
在本文所述的任何方面或实施方案中,CLM由选自以下的化学结构表示:
在本文所述的任何方面或实施方案中,接头(L)包含由下式表示的化学结构单元:
-(A
其中:
(A
q是大于或等于1的整数;
每个A
R
在本文所述的任何方面或实施方案中,L选自:
-N(R)-(CH2)
-O-(CH2)
-O-(CH2)
-N(R)-(CH2)
-(CH2)
-(CH2)
/>
/>
所述接头的m、n、o、p、q和r独立地是0、1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20;
当数目为零时,不存在N-O或O-O键
所述接头的R是H、甲基和乙基;
所述接头的X是H和F
其中所述接头的m可以是2、3、4、5
/>
/>
/>
/>
/>
/>
/>
/>
其中所述接头的n和m可以是0、1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20。
在本文所述的任何方面或实施方案中,L选自:
/>
/>
/>
其中每个m和n独立地选自0、1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20。
在本文所述的任何方面或实施方案中,接头(L)选自:
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
其中每个m、n、o、p、q和r独立地是0、1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19或20。
在本文所述的任何方面或实施方案中,接头(L)选自:
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
在本文所述的任何方面或实施方案中,接头(L)选自:
/>
/>
/>
其中:
上述结构中的“X”可以是具有2至14个原子的直链,并且所述链可含有杂原子,例如氧;并且
上述结构中的“Y”可以是O、N、S(O)
在本文所述的任何方面或实施方案中,接头(L)包含选自以下的结构:
其中:
W
Y
n是0-10;并且
虚线指示与所述PTM或CLM部分的附接点。
在本文所述的任何方面或实施方案中,接头(L)包含选自以下的结构:
其中:
W
Y
Q
R
n是0-10;并且
虚线指示与所述PTM或CLM部分的附接点。
在本文所述的任何方面或实施方案中,接头(L)是包含1至10个乙二醇单元的任选被芳基或苯基取代的聚乙烯氧基基团。
在本文所述的任何方面或实施方案中,化合物包含多个ULM、多个CLM、多个PTM、多个接头或它们的任何组合。
在本文所述的任何方面或实施方案中,PTM具有包含(A)、(B)、(C)、(D)、(E)或它们的组合中的至少一者的化学结构:
(A)包含PTM-I或PTM-II的雌激素受体结合部分(EBM):
其中:
X
X
R
R
R
每个相应环上的P
(B)由以下化学结构表示的雌激素受体蛋白质靶向部分:
式(II
其中:
每个X
指示所述接头、所述CLM、CLM’或它们的组合中的至少一者的附接位点;
每个R
每个R
每个R
R
(C)雄激素受体(AR)结合部分(ABM)包含选自以下的结构:
其中:
W
Y
Y
Q是具有0-4个杂原子的3-6元环,其任选地被0-6个R
R
W
每个R
R
虚线指示所述接头、所述CLM、CLM’或它们的组合中的至少一者的附接位点;
(D)由式I-XI中的至少一个表示的Tau蛋白靶向部分:
其中:
A、B、C、D、E和F独立地选自任选被取代的5或6元芳基或杂芳基环、任选被取代的4至7元环烷基或杂环烷基,其中圆圈之间的接触指示环稠合;
L
R
(E)三环二氮杂环庚三烯或氮杂环庚三烯BET/BRD4结合配体,其包含根据化学结构PTM-a的基团:
其中:
Y
A和B各自独立地选自5元芳环、6元芳环、杂芳环、碳环、噻吩、吡咯环、吡啶、嘧啶、吡嗪、吡唑环,各自任选地被烷基、烷氧基、卤素、芳族和杂芳族环取代;其中环A稠合至中心氮杂环庚三烯(Y1=C)或二氮杂环庚三烯(Y1=N)部分;并且
Z1选自甲基或烷基,并且
其中虚线指示所述接头、所述CLM、CLM’或它们的组合中的至少一者的附接位点;
在本文所述的任何方面或实施方案中,在Tau蛋白靶向部分中,以下中的至少一者:
A、B、C、F或它们的组合中的至少一者选自任选被取代的5或6元芳基或杂芳基环;
PTM的A、B、C、D和E的芳基和杂芳基环任选被1至8个取代基取代,所述取代基各自独立地选自烷基、烯基、卤代烷基、卤素、羟基、烷氧基、氟代烷氧基、氨基、烷基氨基、二烷基氨基、酰基氨基、三氟甲基和氰基,其中所述烷基和烯基进一步任选被取代;或者
它们的组合。
在本文所述的任何方面或实施方案中,PTM是式I,并且:
A、B和C环独立地是5或6元稠合芳基或杂芳基环;
L
D选自6元芳基、杂芳基或杂环烷基,
其中A、B、C和D任选被烷基、卤代烷基、卤素、羟基、烷氧基、氨基、烷基氨基、二烷基氨基、三氟甲基或氰基取代。
在本文所述的任何方面或实施方案中,PTM是式I,并且:
A和C是苯基或6元杂芳基环;
B是5元杂芳基环;
L
D是6元杂芳基或6元杂环烷基环,
其中每个A、B、C和D任选独立地被烷基、卤代烷基、卤素、羟基、烷氧基、氨基、二烷基氨基、三氟甲基或氰基取代,并且其中A、B、C和D环中任何一个的氮原子并非与杂原子或碳原子直接连接,另一个杂原子与所述杂原子或碳原子直接附接。
在本文所述的任何方面或实施方案中,PTM是式III或IV,并且:
A、B和C是5或6元稠合芳基或杂芳基环;
L
D和E是5或6元稠合芳基或杂芳基环;
其中A、B、C、D和E任选被烷基、卤代烷基、卤素、羟基、烷氧基、氨基、烷基氨基、二烷基氨基、三氟甲基或氰基取代。
在本文所述的任何方面或实施方案中,PTM具有选自以下的结构:
/>
/>
其中R或接头是将所述CLM偶联至所述PTM的键或化学接头部分,包括其药学上可接受的盐形式。
在本文所述的任何方面或实施方案中,化合物选自PROTAC-1至PROTAC-112。
在本文所述的任何方面或实施方案中,化合物选自:
4-{3-[4-({1-[5-氯-1-(2,6-二氧代哌啶-3-基)-6-氧代-1,6-二氢哒嗪-4-基]-1,4,7,10-四氧杂十二烷-12-基}氧基)苯基]-4,4-二甲基-5-氧代-2-亚磺酰基咪唑啉-1-基}-2-(三氟甲基)苯甲腈;
4-{3-[4-(2-{2-[4-(2-{[1-(2,6-二氧代哌啶-3-基)-6-氧代-1,6-二氢哒嗪-4-基]氧基}乙基)哌嗪-1-基]乙氧基}乙氧基)苯基]-4,4-二甲基-5-氧代-2-亚磺酰基咪唑啉-1-基}-2-(三氟甲基)苯甲腈;
4-[3-(4-{2-[4-(2-{[5-氯-1-(2,6-二氧代哌啶-3-基)-6-氧代-1,6-二氢哒嗪-4-基]氧基}乙基)哌嗪-1-基]乙氧基}苯基)-4,4-二甲基-5-氧代-2-亚磺酰基咪唑啉-1-基]-2-(三氟甲基)苯甲腈;
6-{4-[5-({6-[(2,6-二氧代哌啶-3-基)氨基甲酰基]吡啶-3-基}氧基)戊基]哌嗪-1-基}-N-[(1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基]吡啶-3-甲酰胺;
6-[4-(5-{[3-(2,6-二氧代哌啶-3-基)-2-甲基-4-氧代-1,2,3,4-四氢喹唑啉-8-基]氧基}戊基)哌嗪-1-基]-N-[(1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基]吡啶-3-甲酰胺;
6-[4-(6-{[1-(2,6-二氧代哌啶-3-基)-6-氧代-1,6-二氢哒嗪-4-基]氧基}己基)哌嗪-1-基]-N-[(1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基]吡啶-3-甲酰胺;
6-[4-(5-{[3-(2,6-二氧代哌啶-3-基)-2-甲基-4-氧代-3,4-二氢喹唑啉-8-基]氧基}戊基)哌嗪-1-基]-N-[(1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基]吡啶-3-甲酰胺;
5-(5-{4-[2-(4-{3-[4-氰基-3-(三氟甲基)苯基]-5,5-二甲基-4-氧代-2-亚磺酰基咪唑啉-1-基}苯氧基)乙基]哌嗪-1-基}-1,3-二氧代-2,3-二氢-1H-异吲哚-2-基)-6-氧代-1,6-二氢吡啶-2-甲腈;
4-[3-(4-{2-[4-({1-[5-(2,4-二氧代-1,2,3,4-四氢嘧啶-1-基)吡啶-3-基]哌啶-4-基}甲基)哌嗪-1-基]乙氧基}苯基)-4,4-二甲基-5-氧代-2-亚磺酰基咪唑啉-1-基]-2-(三氟甲基)苯甲腈;
4-[3-(4-{[3-(3-{[3-(2,4-二氧代-1,2,3,4-四氢嘧啶-1-基)喹啉-5-基]氧基}丙氧基)丙基]氨基}苯基)-4,4-二甲基-5-氧代-2-亚磺酰基咪唑啉-1-基]-2-(三氟甲基)苯甲腈;
4-[3-(4-{[3-(3-{[3-(2,4-二氧代-1,2,3,4-四氢嘧啶-1-基)喹啉-5-基]氧基}丙氧基)丙基]氨基}苯基)-4,4-二甲基-5-氧代-2-亚磺酰基咪唑啉-1-基]-2-(三氟甲基)苯甲腈;
4-[4-(2-{2-[(2-{[2-(2,4-二氧代-1,3-二氮杂环己烷-1-基)乙基]氨基甲酰基}苯基)氨基]乙氧基}乙基)哌嗪-1-基]-N-[(1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基]苯甲酰胺;
5-(4-{2-[(1,3-二氧代-2-{6-氧代-2-氧杂-5-氮杂螺[3.5]壬酰-9-基}-2,3-二氢-1H-异吲哚-4-基)氨基]乙基}哌嗪-1-基)-N-[(1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基]吡啶-2-甲酰胺;
4-(4,4-二甲基-3-{4-[4-(3-{[2-(1-甲基-2,4-二氧代-1,2,3,4-四氢嘧啶-5-基)-1,3-二氧代-2,3-二氢-1H-异吲哚-5-基]氧基}丙基)哌嗪-1-基]苯基}-5-氧代-2-亚磺酰基咪唑啉-1-基)-2-(三氟甲基)苯甲腈;
5-[4-(2-{[2-(5,5-二甲基-2,4-二氧代咪唑啉-1-基)-3-氧代-八氢吲哚嗪-6-基]氨基}乙基)哌嗪-1-基]-N-[(1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基]吡啶-2-甲酰胺;
4-[3-(4-{[3-(3-{[4-(2,4-二氧代-1,2,3,4-四氢嘧啶-1-基)异喹啉-7-基]氧基}丙氧基)丙基]氨基}苯基)-4,4-二甲基-5-氧代-2-亚磺酰基咪唑啉-1-基]-2-(三氟甲基)苯甲腈;
4-[3-(4-{1-[3-(2,4-二氧代-1,2,3,4-四氢嘧啶-1-基)-4-甲基喹啉-7-基]-1,4,7-三氧杂-10-氮杂癸烷-10-基}苯基)-4,4-二甲基-5-氧代-2-亚磺酰基咪唑啉-1-基]-2-(三氟甲基)苯甲腈;
4-[2-(2-{[3-(2,4-二氧代-1,3-二氮杂环己烷-1-基)-4-甲基喹啉-7-基]氧基}乙氧基)乙氧基]-N-[(1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基]苯甲酰胺;
5-{3-[4-(1,3-二氧代-2-{6-氧代-2-氧杂-5-氮杂螺[3.5]壬酰-9-基}-2,3-二氢-1H-异吲哚-5-基)哌嗪-1-基]丙基}-N-[(1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基]吡啶-2-甲酰胺;
4-{4-[2-(2-{[1-(2,6-二氧代哌啶-3-基)-6-氧代-1,6-二氢哒嗪-4-基]氨基}乙氧基)乙基]哌嗪-1-基}-N-[(1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基]苯甲酰胺;
4-[4-({1-[5-(2,4-二氧代-1,2,3,4-四氢嘧啶-1-基)吡啶-3-基]哌啶-4-基}甲基)哌嗪-1-基]-N-[(1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基]苯甲酰胺;
4-(4-{2-[4-(2-{[1-(2,6-二氧代哌啶-3-基)-6-氧代-1,6-二氢哒嗪-4-基]氧基}乙基)哌嗪-1-基]乙氧基}丁氧基)-N-[(1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基]苯甲酰胺;
2-[(2-{2-[4-(4-{3-[4-氰基-3-(三氟甲基)苯基]-5,5-二甲基-4-氧代-2-亚磺酰基咪唑啉-1-基}苯基)哌嗪-1-基]乙氧基}乙基)氨基]-N-[2-(2,4-二氧代-1,3-二氮杂环己烷-1-基)乙基]苯甲酰胺;
2-{[2-(2-{[4-(4-{3-[4-氰基-3-(三氟甲基)苯基]-5,5-二甲基-4-氧代-2-亚磺酰基咪唑啉-1-基}苯基)苯基]氨基}乙氧基)乙基]氨基}-N-[2-(2,4-二氧代-1,3-二氮杂环己烷-1-基)乙基]苯甲酰胺;
4-{4-[2-({1,3-二氧代-2-[2-氧代-6-(三氟甲基)哌啶-3-基]-2,3-二氢-1H-异吲哚-4-基}氨基)乙基]哌嗪-1-基}-N-[(1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基]苯甲酰胺;
4-{4-[2-({1,3-二氧代-2-[2-氧代-6-(三氟甲基)哌啶-3-基]-2,3-二氢-1H-异吲哚-5-基}氧基)乙基]哌嗪-1-基}-N-[(1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基]苯甲酰胺;
4-{4-[2-({1,3-二氧代-2-[2-氧代-6-(三氟甲基)-1,2-二氢吡啶-3-基]-2,3-二氢-1H-异吲哚-4-基}氨基)乙基]哌嗪-1-基}-N-[(1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基]苯甲酰胺;
4-{4-[2-({1,3-二氧代-2-[2-氧代-6-(三氟甲基)-1,2-二氢吡啶-3-基]-2,3-二氢-1H-异吲哚-5-基}氧基)乙基]哌嗪-1-基}-N-[(1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基]苯甲酰胺;
4-[3-(4-{2-[4-(2-{[2-(2,6-二氧代哌啶-3-基)-1,1,3-三氧代-2,3-二氢-1λ6,2-苯并噻唑-6-基]氨基}乙基)哌嗪-1-基]乙氧基}苯基)-4,4-二甲基-5-氧代-2-亚磺酰基咪唑啉-1-基]-2-(三氟甲基)苯甲腈;
4-[3-(4-{2-[4-(2-{[2-(2,6-二氧代哌啶-3-基)-1,1,3-三氧代-2,3-二氢-1λ6,2-苯并噻唑-6-基]氧基}乙基)哌嗪-1-基]乙氧基}苯基)-4,4-二甲基-5-氧代-2-亚磺酰基咪唑啉-1-基]-2-(三氟甲基)苯甲腈;
6-[4-(5-{[2-(2,6-二氧代哌啶-3-基)-1,1,3-三氧代-2,3-二氢-1λ6,2-苯并噻唑-6-基]氧基}戊基)哌嗪-1-基]-N-[(1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基]吡啶-3-甲酰胺;
6-[4-(5-{[2-(2,6-二氧代哌啶-3-基)-1,1,3-三氧代-2,3-二氢-1λ6,2-苯并噻唑-6-基]氨基}戊基)哌嗪-1-基]-N-[(1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基]吡啶-3-甲酰胺;
6-[4-(5-{[2-(2,6-二氧代哌啶-3-基)-1,1,3-三氧代-2,3-二氢-1λ6,2-苯并噻唑-7-基]氨基}戊基)哌嗪-1-基]-N-[(1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基]吡啶-3-甲酰胺;
6-[4-(5-{[2-(2,6-二氧代哌啶-3-基)-1,1,3-三氧代-2,3-二氢-1λ6,2-苯并噻唑-7-基]氧基}戊基)哌嗪-1-基]-N-[(1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基]吡啶-3-甲酰胺;
4-[3-(4-{2-[2-(2-{[2-(2,6-二氧代哌啶-3-基)-1,1,3-三氧代-2,3-二氢-1λ6,2-苯并噻唑-6-基]氧基}乙氧基)乙氧基]乙氧基}苯基)-4,4-二甲基-5-氧代-2-亚磺酰基咪唑啉-1-基]-2-(三氟甲基)苯甲腈;以及
6-[3-(3-{[2-(2,6-二氧代哌啶-3-基)-1,1,3-三氧代-2,3-二氢-1λ6,2-苯并噻唑-6-基]氧基}丙氧基)丙氧基]-N-[(1r,3r)-3-(3-氯-4-氰基苯氧基)-2,2,4,4-四甲基环丁基]吡啶-3-甲酰胺,包括其药学上可接受的盐形式。
另一方面公开了一种组合物,其包含有效量的本公开的双官能化合物以及药学上可接受的载体。
在本文所述的任何方面或实施方案中,该组合物还包含另外的生物活性剂或本公开的另一种双官能化合物中的至少一种。
在本文所述的任何方面或实施方案中,所述另外的生物活性剂是抗癌剂、抗神经变性剂、抗微生物剂、抗病毒剂、抗HIV剂或抗真菌剂。
另一方面公开了一种组合物,其包含有效量的本公开的至少一种化合物以及药学上可接受的载体、添加剂和/或赋形剂,该组合物用于治疗受试者的疾病或病症,方法包括将该组合物施用于有此需要的受试者,其中所述化合物能够有效治疗或改善疾病或病症的至少一种症状。
在本文所述的任何方面或实施方案中,所述疾病或病症与靶蛋白的累积和/或聚集相关。
在本文所述的任何方面或实施方案中,所述疾病或病症选自哮喘、自体免疫疾病如多发性硬化症、各种癌症、纤毛病、腭裂、糖尿病、心脏病、高血压、发炎性肠病、智力迟钝、情绪障碍、肥胖症、屈光不正、不育症、Angelman综合征、Canavan病、乳糜泻、夏-马-图三氏病(Charcot-Marie-Tooth disease)、囊肿性纤维化、杜氏肌营养不良、血色病、血友病、克氏综合征、神经纤维瘤病、苯丙酮尿症、多囊性肾病(PKD1)或4(PKD2)、Prader-Willi综合征、镰状细胞病、Tay-Sachs病、Turner综合征。
在本文所述的任何方面或实施方案中,所述疾病或病症选自阿尔茨海默病、肌萎缩性脊髓侧索硬化症(Lou Gehrig病)、神经性厌食症、焦虑症、动脉粥样硬化、注意力不足过动症、自闭症,双相情感障碍、慢性疲劳综合症、慢性阻塞性肺病、克罗恩病、冠心病、痴呆、抑郁症、1型糖尿病、2型糖尿病、癫痫、格林-巴利综合征、肠易激综合征、狼疮、代谢综合征、多发性硬化、心肌梗死、肥胖症、强迫症、恐慌症、帕金森病、牛皮癣、类风湿性关节炎、结节病、精神分裂症、中风、血栓闭塞性脉管炎、妥瑞症、血管炎。
在本文所述的任何方面或实施方案中,所述疾病或病症选自铜蓝蛋白缺乏症、软骨成长不全II型、软骨发育不全、尖头、2型戈谢病、急性间歇性卟啉症、卡纳万病、腺瘤性结肠息肉病、ALA脱水酶缺乏症、腺苷酸琥珀酸裂解酶缺乏症、肾上腺生殖综合征、肾上腺脑白质营养不良、ALA-D卟啉症、ALA脱水酶缺乏症、黑尿症、亚历山大病、黑尿性褐黃病、α1-抗胰蛋白酶缺乏症、α-1蛋白酶抑制剂、肺气肿、肌萎缩性脊髓侧索硬化症、
在本文所述的任何方面或实施方案中,其中组合物还包含另外的生物活性剂。
在本文所述的任何方面或实施方案中,所述另外的生物活性剂是抗癌剂、抗神经变性剂、抗微生物剂、抗病毒剂、抗HIV剂、抗真菌剂或它们的组合中的至少一种。
在本文所述的任何方面或实施方案中,抗癌剂选自依维莫司、曲贝替定、阿布拉生、TLK 286、AV-299、DN-101、帕唑帕尼、GSK690693、RTA 744、ON 0910.Na、AZD 6244(ARRY-142886)、AMN-107、TKI-258、GSK461364、AZD 1152、恩扎妥林、凡德他尼、ARQ-197、MK-0457、MLN8054、PHA-739358、R-763、AT-9263、FLT-3抑制剂、VEGFR抑制剂、EGFR TK抑制剂、极光激酶抑制剂、PIK-1调节剂、Bcl-2抑制剂、HDAC抑制剂、c-MET抑制剂、PARP抑制剂、Cdk抑制剂、EGFR TK抑制剂、IGFR-TK抑制剂、抗HGF抗体、PI3激酶抑制剂、AKT抑制剂、mTORC1/2抑制剂、JAK/STAT抑制剂、检查点-1或2抑制剂、病灶粘附激酶抑制剂、Map激酶(mek)抑制剂、VEGF截获抗体、培美曲塞、埃罗替尼、达沙替尼、尼罗替尼、德卡坦尼、帕尼单抗、氨柔比星、奥戈伏单抗、Lep-etu、诺拉曲特、azd2171、巴他布林、奥法木单抗、扎木单抗、艾特咔林(edotecarin)、汉防己碱、鲁比替康、替米利芬、奥利默森、替西木单抗、伊匹单抗、棉子酚、Bio 111、131-I-TM-601、ALT-110、BIO140、CC 8490、西仑吉肽、吉马替康、IL13-PE38QQR、INO 1001、IPdR
另一方面公开了一种用于诱导细胞中靶蛋白降解的方法,该方法包括将有效量的本公开化合物施用于细胞,其中所述化合物实现靶蛋白的降解。
又一方面公开了一种包含有效量的本公开化合物的组合物,其用于治疗癌症的方法中,所述方法包括将所述组合物施用于有此需要的患者,其中所述组合物实现患者的癌症的至少一种症状的治疗或缓解。
在本文所述的任何方面或实施方案中,所述癌症是鳞状细胞癌、基底细胞癌、腺癌、肝细胞癌和肾细胞癌;膀胱、肠道、乳房、子宫颈、结肠、食道、头、肾、肝、肺、颈、卵巢、胰腺、前列腺和胃的癌症;白血病;良性和恶性淋巴瘤,尤其是伯基特氏淋巴瘤(Burkitt’slymphoma)和非霍奇金淋巴瘤(Non-Hodgkin’s lymphoma);良性和恶性黑素瘤;骨髓增生性疾病;多发性骨髓瘤,肉瘤,包括尤文氏肉瘤(Ewing’s sarcoma)、血管内皮瘤、卡波西肉瘤(Kaposi’s sarcoma)、脂肪肉瘤、肌肉瘤、外周神经上皮瘤、滑膜肉瘤、神经胶质瘤、星形细胞瘤、少突神经胶质瘤、室管膜瘤、胶质母细胞瘤、神经母细胞瘤、节细胞神经瘤、神经节细胞胶质瘤、髓母细胞瘤、松果体细胞肿瘤、脑膜瘤、脑膜肉瘤、神经纤维瘤和许旺细胞瘤(Schwannomas);肠道癌、乳癌、前列腺癌、子宫颈癌、子宫癌、肺癌、卵巢癌、睾丸癌、甲状腺癌、星形细胞瘤、食道癌、胰脏癌、胃癌、肝癌、结肠癌、黑素瘤;癌肉瘤、霍奇金病、维尔姆斯瘤或畸胎癌、T谱系急性淋巴母细胞白血病(T-ALL)、T谱系淋巴母细胞淋巴瘤(T-LL)、外周T细胞淋巴瘤、成人T细胞白血病、Pre-B ALL、Pre-B淋巴瘤、大B细胞淋巴瘤、Burkitts淋巴瘤、B细胞ALL、费城染色体阳性ALL和费城染色体阳性CML。
在本文所述的任何方面或实施方案中,L包含非直链、脂族或芳族或杂芳族环状部分。
在本文所述的任何方面或实施方案中,L选自:
/>
/>
其中:
“X”是具有2至14个原子的直链,其任选被取代以含有杂原子;并且“Y”独立地选自O、N、S(O)
实施例
缩写:
ACN:乙腈
ADDP:1,1'-(偶氮二羰基)二哌啶
BAST:N,N-双(2-甲氧基乙基)氨基三氟化硫
BPO:过氧化苯甲酰
Cbz:羰基苄氧基
DAST:二乙氨基三氟化硫
DBE:1,2-二溴乙烷
DCM:二氯甲烷
DEAD:偶氮二甲酸二乙酯
DIAD:偶氮二甲酸二异丙酯
DIBAL:二异丁基氢化铝
DIEA或DIPEA:二异丙基乙胺
DMA:N,N-二甲基乙酰胺
DMF:N,N-二甲基甲酰胺
DMP:戴斯-马丁过碘烷
EA:乙酸乙酯
EDCI:1-乙基-3-(3-二甲基氨基丙基)碳二亚胺
HBTU:N,N,N’N’-四甲基-O-(1H-苯并三唑-1-基)脲六氟磷酸酯
HMDS:双(三甲基甲硅烷基)胺
HMPA:六甲基磷酰胺
LDA:二异丙基氨基锂
MCPBA:间-氯过氧苯甲酸
MsCl:甲磺酰氯
M.W:微波
NBS:N-溴代琥珀酰亚胺
NMP:N-甲基吡咯烷酮
PCC:氯铬酸吡啶鎓
Pd-118或Pd(dtpf)Cl
Pd(dppf)Cl
Pd(dba)
Pd
PPTS:对甲苯磺酸吡啶鎓
PTSA:对甲苯磺酸
RuPhos-Pd-G3:XPhos-Pd-G3:[(2-二环己基膦基-2’,6’-二异丙氧基-1,1’-联苯基)-2-(2’-氨基-1,1’-联苯基)]钯(II)甲磺酸盐
RuPhos-Pd-G2:氯[(2-二环己基膦基-2',6'-二异丙氧基-1,1'-联苯基)-2-(2'-氨基-1,1'-联苯基)]钯(II)
SFC:超临界流体色谱
t-BuXPhos-Pd-G3:[(2-二叔丁基膦基-2’,4’,6’-三异丙基-1,1’-联苯基)-2-(2’-氨基-1,1’-联苯基)]钯(II)甲磺酸盐
TEA:三甲胺
TFA:三氟乙酸
TLC:薄层色谱
TMP:2,2,6,6-四甲基哌啶
TEMPO:2,2,6,6-四甲基哌啶-N-氧化物
TosCl或TsCl:对甲苯磺酰氯
TsOH:对甲苯磺酸
XantPhos:4,5-双(二苯基膦基)-9,9-二甲基呫吨
XPhos:2-二环己基膦基-2'4'6'-三异丙基联苯
XPhos-Pd-G3:[(2-二环己基膦基-2',4',6'-三异丙基-1,1'-联苯基)-2-(2'-氨基-1,1'-联苯基)]钯(II)甲磺酸盐
12354-85-7:双(五甲基环戊二烯基二氯化铑)
A.人CRBN和DDB1的克隆、表达和纯化。该过程对于本领域技术人员来说是标准的,如Lopez-Girona等人的描述所示。(人小脑蛋白是来那度胺和泊马度胺的免疫调节和抗增殖活性的直接蛋白质靶标,A Lopez-Girona、D Mendy、T Ito、K Miller、A K Gandhi、JKang、S Karasawa、GCarmel、P Jackson、M Abbasian、A Mahmoudi、B Cathers、E Rychak、SGaidarova、R Chen、P H Schafer、H Handa、T O Daniel、J F Evans和RChopra,Leukemia26:2326-2335,2012)。
可使用作为聚合酶的Pfusion(NEB)和以下引物序列通过PCR扩增CRBN和DDB1基因的cDNA:
使用不依赖于连接的克隆26,可将CRBN克隆到pBV-ZZ-HT-LIC、pBV-GST-LIC、pMA-HT-LIC中,并将DDB1克隆到pBV-notag-LIC中。为了克隆到哺乳动物载体pMA-HT-LIC中,CRBN-Flag-Reverse寡核苷酸增添用于免疫检测的C末端FLAG标记。DDB1-Rev增添了StrepTag 27。ZZ标记28是实现可溶性CRBN高表达所必需的;没有它,His-CRBN以低水平表达,而GST-CRBN导致蛋白质聚集。使用得自Invitrogen的Sf9昆虫细胞中的Bac-to-Bac杆状病毒表达系统生成并扩增ZZ-His-CRBN和DDB1-StrepTag(ST)的重组杆状病毒。在27℃下,使用得自Expression Systems的未补充的ESF921培养基,在10L波浪袋(wave bag)内的High Five(Tni)昆虫中共表达ZZ-His-CRBN和DDB1-ST。感染后48小时通过离心收获细胞,并将糊状物重悬于PBS plus5X蛋白酶抑制剂混合物(Roche,Indianapolis,IN)中。
所有后续蛋白质纯化步骤均在4℃下进行。将冷冻的细胞解冻,重悬于5体积的裂解缓冲液(50mM Tris HCl pH 8.0,0.5M NaCl,10%甘油,2mM DTT)和20mM咪唑和蛋白酶抑制剂中,裂解并离心,得到澄清的上清液。使用Nickel-Sepharose和S200 Sephacryl色谱在
2.测量化合物与重组CRBN的结合的荧光热熔测定
该测定对于本领域技术人员来说是标准的,如Lopez-Girona等人的描述所示。(人小脑蛋白是来那度胺和泊马度胺的免疫调节和抗增殖活性的直接蛋白质靶标,A Lopez-Girona、D Mendy、T Ito、K Miller、A KGandhi、J Kang、S Karasawa、G Carmel、P Jackson、M Abbasian、AMahmoudi、B Cathers、E Rychak、S Gaidarova、R Chen、P H Schafer、HHanda、T O Daniel、J F Evans和R Chopra,Leukemia 26:2326-2335,2012)。
根据Pantoliano等,在存在或不存在测试化合物的情况下CRBN-DDB1的热稳定性以微量板形式在Sypro Orange存在下根据Pantoliano等人(Pantoliano MW、Petrella EC、Kwasnoski JD、Lobanov VS、Myslik J、Graf E等人,High-density miniaturized thermalshift assays as a general strategy for drug discovery.J Biomol Screen 2001;6:429–440)进行测定。将20ml测定缓冲液(25mM Tris HCl,pH 8.0,150mM NaCl,2uMSyproOrange)中的两毫克蛋白质从20℃逐步升温至70℃,并且每升1℃,在ABIPrism7900HT(Applied Biosystems,Carlsbad,CA,USA)上读取一次荧光。将化合物溶解于DMSO(在测定中最终浓度为1%),并在30nM至1000μM的浓度范围内一式四份进行测试;对照仅含1%DMSO。
3.LCMS方法:
分析在Poroshell 120EC C18柱(50mm x 3.0mm内径,2.7μm填充直径)上、在45℃下进行。
所采用的溶剂是:
A=0.1%v/v甲酸的水溶液。
B=0.1%v/v甲酸的乙腈溶液。
所采用的梯度如下:
UV检测是波长210nm至350nm的平均信号,并且使用正模式电喷雾电离在质谱仪上记录质谱。
下文说明了化合物通过制备型HPLC进行纯化时使用的流动相和梯度。
4.制备型HPLC(甲酸改性剂)
HPLC分析在X Bridge RP18 OBD柱(150mm x 19mm内径,5μm填充直径)上、在环境温度下进行。
所采用的溶剂是:
A=0.1%v/v甲酸的水溶液。
B=乙腈。
5.制备型HPLC(碳酸氢铵改性剂)
HPLC分析在X Bridge RP18 OBD柱(150mm x 19mm内径,5μm填充直径)上、在环境温度下进行。
所采用的溶剂是:
A=10mM碳酸氢铵水溶液。
B=乙腈。
对于每种制备型纯化,无论所用改性剂如何,所用梯度取决于在分析LCMS中记录的经历纯化的特定化合物的保留时间。流速为20mL/min。
UV检测是来自254nm或220nm波长的信号。
虽然本文已经示出和描述了本发明的优选实施方案,但应当理解,这些实施方案仅作为示例提供。在不脱离本发明的实质的情况下,本领域技术人员将想到许多变型、改变和替代。因此,所附权利要求旨在覆盖落入本发明的实质和范围内的所有这些变化。
B.合成
下面包含的实施例的合成细节代表了更广泛的实施例集的合成的一般工序。
1.N-(3-(5-溴-2-氯嘧啶-4-基氨基)丙基)-N-甲基环丁烷甲酰胺
步骤1:N-{3-[(5-溴-2-氯嘧啶-4-基)氨基]丙基}-N-甲基氨基甲酸叔丁酯
将N-(3-氨基丙基)-N-甲基氨基甲酸叔丁酯(826mg,4.40mmol)和5-溴-2,4-二氯嘧啶(400mg,1.76mmol)在MeOH(10mL)中的混合物在室温下搅拌1小时。然后将反应混合物真空浓缩,并使用Teledyne ISCO色谱[0→35%EtOAc/庚烷]纯化残余物,得到N-{3-[(5-溴-2-氯嘧啶-4-基)氨基]丙基}-N-甲基氨基甲酸叔丁酯(615mg,92%收率)。LC-MS(ES
步骤2:{3-[(5-溴-2-氯嘧啶-4-基)氨基]丙基}(甲基)胺
在室温下,向N-{3-[(5-溴-2-氯嘧啶-4-基)氨基]丙基}-N-甲基氨基甲酸叔丁酯(615mg,1.62mmol)的DCM(5mL)溶液中添加三氟乙酸(0.54mL,6.5mmol)。将该混合物搅拌1小时后,将其真空浓缩。使用Teledyne ISCO色谱[0→15%甲醇的DCM溶液]纯化残余物,得到{3-[(5-溴-2-氯嘧啶-4-基)氨基]丙基}(甲基)胺(371mg,82%收率)。LC-MS(ES
步骤3:N-{3-[(5-溴-2-氯嘧啶-4-基)氨基]丙基}-N-甲基环丁烷甲酰胺
在室温下,向{3-[(5-溴-2-氯嘧啶-4-基)氨基]丙基}(甲基)胺(371mg,1.33mmol)和环丁烷碳酰氯(188mg,1.60mmol)的DCM(10mL)溶液中添加三乙胺(0.41mL,2.92mmol)。将反应混合物在室温下搅拌16小时,然后真空浓缩。使用Teledyne ISCO色谱[0→100%EtOAc/庚烷]纯化残余物,得到N-{3-[(5-溴-2-氯嘧啶-4-基)氨基]丙基}-N-甲基环丁烷甲酰胺(268mg,56%)。LC-MS(ES
2.(S)-2-(4-(4-氯苯基)-2,3,9-三甲基-6H-噻吩并[3,2-f][1,2,4]三唑并[4,3-a][1,4]二氮杂环庚三烯-6-基)乙酸
根据WO2011/143660中描述的工序制备标题化合物
3.(Z)-4-(4-((2,4-二氧代噻唑烷-5-亚基)甲基)-2-甲氧基苯氧基)-3-(三氟甲基)苯甲腈
根据Patch,R.J.等人,J.Med.Chem.2011,54,788–808中描述的工序制备标题化合物。
4.4-[3-(4-羟基苯基)-4,4-二甲基-5-氧代-2-亚磺酰基咪唑啉-1-基]-2-(三氟甲基)苯甲腈
根据Jung,M.E.等人,J.Med.Chem.2010,53,2779-2796中描述的工序制备标题化合物。
5.2-氯-4-(反-3-氨基-2,2,4,4-四甲基环丁氧基)苯甲腈氯化氢盐
根据Guo,C.等人,J.Med.Chem.2011,54,7693-7704中描述的工序制备标题化合物。
C.蛋白质降解生物测定:
以下生物测定使用本文所公开的代表性化合物评估在各种细胞类型中观察到的蛋白质降解水平。
在每种生物测定中,用不同量的本公开所涵盖的化合物处理细胞。可评估以下蛋白质的降解:TANK结合激酶1(TBK1)、雌激素受体α(ERα)、含溴结构域蛋白4(BRD4)、雄激素受体(AR)、c-Myc和tau蛋白。
1.表2中的化合物的ERE荧光素酶测定。
将T47D-KBluc细胞(
2.在MCF-7细胞中使用蛋白质印迹法的表5中雌激素受体-α(ERα)降解测定。
经由蛋白质印迹评价示例性新型ERα降解剂在MCF-7细胞中降解ERα的活性。该测定在10%FBS或者高百分比的人或小鼠血清的存在下进行。蛋白质印迹分析的方案描述如下。
使MCF7细胞在含有10%FBS的DMEM/F12中生长,并且以每孔24,000个细胞以100μl接种到96孔透明组织培养板内。第二天,用PROTAC以7点浓度曲线处理细胞,其中100nM是最高浓度,并连续稀释以制备其他浓度(30nM、10nM、3nM、1nM和0.3nM)。在所有浓度下,0.01%DMSO是孔中的最终浓度。第二天,抽吸板,用50μl冷PBS洗涤。用50μl/孔4℃细胞裂解缓冲液(产品目录号9803;Cell Signaling Technology,Danvers,MA)(20mM Tris-HCL(pH 7.5),150mM NaCl,1mM Na
可替代地,使MCF7细胞在含有10%FBS的DMEM/F12中生长,并且以每孔24,000个细胞以500μl接种到24孔透明组织培养板内。第二天,在0.01%DMSO存在下,用PROTAC以5点浓度曲线(100nM、33nM、11nM、3.7nM和1.2nM)处理细胞。72小时后,抽吸孔并用500μl PBS洗涤。用100μl/孔4℃细胞裂解缓冲液(产品目录号9803;Cell Signaling Technology,Danvers,MA)(20mM Tris-HCL(pH 7.5),150mM NaCl,1mM Na
3.使用In-Cell Western
使用In-Cell Western
4.BRD4 Western方案
VCaP细胞购自ATCC,将其在补充有10%FBS(ATCC)和青霉素/链霉素(LifeTechnologies)的Dulbecco’s改良Eagle’s培养基(ATCC)中培养。在12孔板中进行DMSO对照和化合物处理(0.003μM、0.01μM、0.03μM和0.1μM)16小时。收获细胞,并在补充有蛋白酶和磷酸酶抑制剂的RIPA缓冲液(50mM Tris pH8,150mM NaCl,1%Tx-100,0.1%SDS,0.5%脱氧胆酸钠)中裂解。将裂解物以16,000g澄清10分钟,并测定蛋白质浓度。将等量的蛋白质(20μg)进行SDS-PAGE分析,然后根据标准方案进行免疫印迹。所用抗体是BRD4(CellSignaling产品目录号13440)和肌动蛋白(Sigma产品目录号5441)。检测试剂是ClarityWestern ECL底物(Bio-rad产品目录号170-5060)。
5.AR ELISA测定方案
使用类似方案,在LNCaP和/或VCaP细胞中通过此测定评估化合物。结合VCaP细胞使用的方案描述如下。使用PathScan AR夹心ELISA(Cell Signaling产品目录号12850),根据以下分析步骤进行雄激素受体ELISA分析:
将VCaP细胞在VCaP分析培养基[不含酚红的RPMI(Gibco产品目录号11835-030);5%炭处理(聚葡萄糖处理)的FBS(Omega Scientific,产品目录号FB-04);1%青霉素/链霉素(Life Technologies,Gibco产品目录号:10378-016)]中以每孔40,000个细胞、每孔100μL的体积接种于Corning3904板中。将细胞孵育至少3天。向细胞中添加稀释于0.01%DMSO中的PROTAC,并药物处理5小时。
AR ELISA(Cell Signaling)如下进行。制备1x Cell Signaling细胞溶解缓冲液(产品目录号9803;随试剂盒提供)。吸出处理过的孔中的培养基,并且每孔添加100μL lx细胞裂解缓冲液。将细胞置于振荡器上,在4℃下保持10分钟。将二十微升裂解物转移至ELISA板中的100μl稀释液(0.15μg/ml–0.075μg/ml)中。将裂解物-稀释剂混合物在37℃下振荡30分钟。使小鼠AR抗体、抗小鼠抗体、TMB和终止溶液达到室温。制备试剂盒中包含的1xELISA缓冲液并将其装入储库中。弃去板中的培养基,将ELISA板在纸巾上轻敲,并使用洗板器洗涤4x 200μl ELISA洗涤缓冲液。
添加一百(100)μL/孔的小鼠AR检测抗体;盖上板并在37℃下振荡1小时;弃去板中的培养基,将板在纸巾上轻敲,用洗板器用200μL ELISA洗涤缓冲液洗涤4次。
添加一百(100)μL/孔的抗小鼠-HRP缀合抗体(随试剂盒提供);盖上板并在37℃下振荡30分钟;使TMB试剂达到室温;弃去板中的培养基,将板在纸巾上轻敲,用200μL ELISA洗涤缓冲液洗涤4次;将板在纸巾上轻敲。添加一百(100)μL的TMB,将板振荡2分钟,同时观察显色。当出现浅蓝色时,添加一百(100)μL终止溶液。振荡培养板且在450nM处读取。
经抗雄激素疗法治疗的患者的前列腺癌进展通常涉及雄激素受体(AR)信号传导增强的若干机理之一,包括增加的瘤内雄激素合成、增加的AR表达和AR突变。使用同时结合所选标靶和E3连接酶的双官能分子的PROTAC(靶向蛋白分解的嵌合体)通过诱导靶向病理蛋白的接近和降解来引起泛素化。与作为竞争过程的传统靶标抑制不同,降解是渐进过程。因此,内源性配体、靶标表达或靶标中的突变不大容易增加。因此,此技术对于解决前列腺癌患者的AR抗性机理似乎是理想的。使用GraphPad Prism软件分析数据并作图。
6.BRD4人c-Myc ELISA测定方案
将22RV-1细胞以30,000个细胞/孔、以75μL/孔的体积接种在96孔板内含有10%FBS的RPMI培养基中,并在37℃下生长过夜。向细胞中添加稀释于0.4%DMSO中的4倍浓度的化合物;将化合物以1:3连续稀释,得到8点剂量曲线。将二十五(25)ul化合物添加到细胞中,最终浓度为0.1%DMSO中的300nM-0.3nM或1μM-1nM,并孵育18小时。吸出培养基,用PBS洗涤细胞1次并吸出。在补充有蛋白酶和磷酸酶抑制剂的50ul RIPA缓冲液(50mM TrispH8,150mM NaCl,1%Tx-100,0.1%SDS,0.5%脱氧胆酸钠)中裂解细胞。将板在冰上孵育15分钟,然后在4℃下以4000rpm离心10分钟。将来自96孔测定板的五十(50)μl澄清裂解物添加到96孔c-myc ELISA板(Novex,Life Technologies产品目录号KH02041)中。用标准稀释缓冲液重构c-myc标准品;制备333pg/ml-0pg/ml的标准曲线范围,以1:2稀释为8点剂量曲线。其余的测定按照c-myc ELISA试剂盒的方案进行。使用GraphPad Prism软件分析数据并作图。分析本公开中所述的化合物,且c-myc抑制效力列于表4中。
7.BRD4免疫印迹
22Rv1和VCaP细胞系购自ATCC。BRD2(#5848)、BRD4(#13440)、PARP(#9532)、c-Myc(#5605)抗体购自cell signaling。BRD3(sc-81202)抗体购自Santa Cruz Biotech。用于免疫组织化学的抗体是c-MYC(abcam#ab32072)和BRD4(Bethyl Laboratories#a301-985a50)。肌动蛋白和微管蛋白抗体购自Sigma。
在补充有蛋白酶抑制剂(Pierce
8.BRD4细胞增殖测定
将22RV-1细胞以5,000个细胞/孔、以75μL/孔的体积接种在96孔板内的RPMI+10%FBS培养基中,并在37℃下生长过夜。向细胞中添加稀释于0.4%DMSO中的四倍浓度的化合物;将化合物以1:3连续稀释,得到10点剂量曲线。将二十五(25)ul化合物添加到细胞中,最终浓度为0.1%DMSO中的300nM-0.3nM,并孵育72小时。在另一个板中,将100μl 5,000个细胞/孔接种在8个孔中,添加100μl的CellTiter-Glo(
9.BRD4细胞凋亡测定
将22RV-1细胞以5,000个细胞/孔、以75μL/孔的体积接种在96孔板内的RPMI+10%FBS培养基中,并在37℃下生长过夜。向细胞中添加稀释于0.4%DMSO中的4倍浓度的化合物;将化合物以1:3连续稀释,得到8点剂量曲线。将二十五(25)ul化合物添加到细胞中,最终浓度为0.1%DMSO中的300nM-0.3nM,并孵育48小时。48小时后,添加100μl
10.Tau蛋白体外降解测定
为了测定PROTAC对tau蛋白质降解的影响,在化合物添加之前,将SK-N-SH细胞接种在24孔组织培养处理板中并保持至少18小时。通过在含有蛋白酶抑制剂的RIPA缓冲液中裂解细胞,然后与tau PROTAC孵育72小时,从而评估Tau PROTAC的tau降解。将细胞裂解物在标准SDS-PAGE凝胶上进行电泳,并且使用得自Abcam(Cambridge,UK)的结合所有形式的人tau的Tau-13抗体,通过蛋白质印迹检测tau水平。数据示于表6中。
小分子抑制剂已成为肿瘤药物开发的基石,并且通常通过抑制酶活性(例如激酶抑制剂)或通过干扰蛋白质-蛋白质相互作用(例如BRD4抑制剂)起作用。鉴于大多数小分子抑制剂的可逆结合,通常需要较大的全身药物浓度以确保足够的功能抑制。另外,实现和维持体内功效所需的高全身药物水平已被证明对许多靶标具有挑战性。
BRD4是溴结构域和额外末端结构域(BET)家族的成员,是一种特征在于N末端的两个溴结构域(BD结构域)和C末端的额外末端结构域(ET结构域)的蛋白质。两个BD结构域识别组蛋白的N末端尾部的乙酰化赖氨酸残基并与之相互作用。ET结构域被认为在募集多种转录调节因子中起支架功能,但尚未完全表征。BRD4已被证实位于超级增强子区域,该区域通常位于重要致癌基因如c-MYC、Bcl-xL和BCL-6的上游,并且在调节它们的表达中起关键作用。基于其通过将相关转录调节剂募集到特定基因组位点来调节基因表达的作用,BRD4是用于治疗和/或预防多种人类癌症如中线癌、急性髓性白血病(AML)、多发性骨髓瘤(MM)、伯基特淋巴瘤(BL)和前列腺癌的候选药物靶标。
已开发若干种小分子BET溴结构域抑制剂,例如JQ1、iBET和OTX15,它们在包括BL在内的各种癌症的某些临床前模型中显示出治疗潜力。几乎所有BL病例都含有c-myc基因易位,使其处于位于IgH上游的超级增强子的控制下,从而驱动异常高水平的c-MYC表达、肿瘤发展和维持。BRD4抑制剂的临床前研究证实了其抑制BL细胞系中c-MYC和增殖的能力;然而,这些抑制剂的IC
材料和方法
实验设计和工序的细节如下:
根据所公布的方法合成抑制剂JQ1、OTX-15和泊马度胺。
1.K
表面等离子体共振(SPR)实验在Biacore3000(GE Healthcare)上进行。将Myc标记的人小脑蛋白固定在与抗Myc抗体偶联的羧甲基化葡聚糖表面(CM5)胺上,以识别Myc标记。利用NTA/Ni
所有化合物均在100%DMSO原种板中以3倍连续稀释制备,最高浓度为5mM。将化合物从原种板转移至测定板并稀释到不含DMSO的运行缓冲液中。将所有化合物以六浓度系列运行,最终测定最高浓度为100μM。
数据分析在Scrubber 2(BioLogic软件,Campbell,Australia)中进行。减去空白并针对标准DMSO曲线校正DMSO的数据。所有报告的KD值表示至少N=2的平均值,并且通过使用1:1拟合算法拟合至最少五个浓度而获得。数据示于下表1中,其中“a”是<1μM的K
表1.示例性CLM的表面等离子体共振数据
/>
/>
/>
/>
/>
/>
/>
2.细胞和试剂
NAMALWA、Ramos、CA-46和DAUDI细胞购自ATCC并按照说明进行保存。针对BRD4(#E2A7X)、c-MYC(#D84C12)、PARP(#46D11)的抗体购自Cell Signaling Technology。肌动蛋白(#A5441)抗体购自SigmaAldrich。二抗(#7074、#7076)购自Cell SignalingTechnology。MG132(#M7449)购自SigmaAldrich。Carfizomib(#S2853)购自Selleck。
2.蛋白质印迹分析
将培养的细胞收集在含有40mM HEPES(pH 7.4)、140mM NaCl、2.5mM EDTA、1%NP-40、0.1%SDS和蛋白酶抑制剂混合物的裂解缓冲液中。离心10分钟(14000rpm)后,收集上清液以通过BCA方法测定蛋白质浓度,并通过标准方案进行免疫印迹。使用Bio-Rad ClarityECL Western Blotting Substrate在Bio-Rad ChemiDocTM MP成像系统上显现蛋白质印迹结果。
3.RT-PCR
用得自Bio-Rad的Aurum
4.增殖测定
/>
为了评估抑制剂对增殖的影响,将细胞(50,000/100μl)接种在96孔组织培养板中,然后添加指定浓度的化合物。72小时后,添加100μL/孔的重构CellTiter-Glo(CTG)试剂(得自Promega的#G7572),并在得自BioTek的Cytation 3成像读取仪上读数。通过将经处理细胞的测定读数与对照的经DMSO处理的细胞比较来确定相对细胞生长。
表2.本公开的示例性PROTAC
/>
/>
/>
/>
/>
/>
/>
/>
/>
表3.示例性雄激素受体PROTAC的特征
/>
/>
/>
/>
/>
*DC50(nM)和IC50(nM):A<1
1<=B<10
10<=C<100
D>=100
**Dmax(降解%)
A>75
50 C<=50 表4.示例性BDR4 PROTAC的特征 *DC50(nM)和IC50(nM): A<1 1<=B<10 10<=C<100 D>=100 **Dmax(降解%) A>75 50 C<=50 表5.示例性雌激素受体PROTAC的特征 /> /> /> /> /> + *DC50(nM)和IC50(nM): A<1 1<=B<10 10<=C<100 D>=100 **Dmax(降解%) A>75 50 C<=50 表6.示例性Tau PROTAC的特征 /> **Dmax(降解%) A>75 50 C<=50 5.工业适用性 描述了一种新的双官能分子,其通过PROTAC技术含有BRD4或雄激素受体募集部分和E3连接酶人小脑蛋白募集部分。本公开的双官能分子主动降解BRD4,导致显著且持久的下游MYC抑制以及强大的细胞增殖抑制和凋亡诱导。PROTAC介导的蛋白质降解为通过传统方法靶向“不可摧毁的”病理蛋白提供了有前景的策略。 本申请中通篇引用的所有参考文献、专利、待决专利申请和已公布专利的内容以引用方式明确并入本文。 所属领域的技术人员仅使用常规实验就认识到或能够确定本文所述的特定实施方案的许多等效物。此类等效物旨在被所附权利要求书涵盖。应当理解,本文所述的详细实例和实施方案仅为了举例说明目的而给出,且决不视为对本发明的限制。根据其的各种修改或变化将被所属领域的技术人员考虑到,并且包含在本申请的实质和范围内且被视为在所附权利要求书的范围内。例如,可以改变成分的相对数量以优化所期望的效应,可以添加其他成分,且/或可以用类似成分取代一种或多种所述成分。与本公开的系统、方法和工艺相关的其他有利特点和功能从所附权利要求书将显而易见。此外,所属领域的技术人员仅使用常规实验就认识到或能够确定本文所述的本发明的特定实施方案的许多等效物。此类等效物旨在被所附权利要求书涵盖。