固态氧化水凝胶及其制备方法和应用、衍生化试剂盒、生物组织氧化前处理和衍生化方法
文献发布时间:2024-04-18 19:58:26
技术领域
本发明属于生物分析技术领域,具体涉及固态氧化水凝胶及其制备方法和应 用、衍生化试剂盒、生物组织氧化前处理和衍生化方法。
背景技术
质谱成像技术是生物组织中代谢物原位表征的重要手段,通过对空间位置的 逐点扫描,获取质谱信息和位置的对应矩阵,通过数据软件处理获得代谢物的空 间位置可视化。为了实现质谱成像技术对生物组织中低丰度、难电离代谢物的原 位表征通常会对目标组织进行衍生化。组织衍生化是通过用化学反应将高质谱响 应的官能团引入待分析的代谢物结构中,然后用质谱成像平台检测和可视化衍生 化之后高质谱响应的离子,从而实现低丰度、难电离的代谢物的原位表征。
目前利用组织原位衍生化方法已经实现了几类代谢物的质谱成像分析,根据 目标代谢物所含官能团的不同,可以分为以下几类:含有氨基的氨基酸、含有羧 基的脂肪酸、含有碳碳双键的不饱和脂肪酸、含有羰基的脂肪醛和氧化脂肪酸、 含有酚羟基的神经递质和含有醇羟基的胆固醇类等。其中醇羟基类代谢物的衍生 化试剂和方法很少且具有局限性,比如甜菜碱醛只能够针对极少数个别代谢物进 行衍生化;此外,利用胆固醇氧化酶对胆固醇类物质先进行组织原位氧化,然后 再衍生化能够提高胆固醇类物质的质谱响应,但是该方法只针对胆固醇类物质, 覆盖度低。
含一、二级醇羟基的代谢物在生物体内分布广泛,比如羟基脂肪酸、脂肪醇、 胆固醇及其衍生物(胆汁酸类和维生素类代谢物)等,它们具有重要的生物学功 能,了解其在生物样本中的空间分布及相对含量对解析生理病理机制有重要意义。 目前还没有合适的组织原位衍生化方法能够同时对上述所有含一、二级醇羟基的 代谢物进行衍生化。
发明内容
有鉴于此,本发明提供了固态氧化水凝胶及其制备方法和应用、衍生化试剂 盒、生物组织氧化前处理和衍生化方法,本发明提供的固态氧化水凝胶能够将生 物组织中一级和二级醇羟基代谢物中的羟基氧化为醛基和/或酮基,然后用衍生化 试剂对氧化形成的醛基和/或酮基进行衍生化,提高了生物组织原位衍生化对羟基 代谢物的覆盖度。
为了解决上述技术问题,本发明提供了一种固态氧化水凝胶,包括氧化剂、 水凝胶和水;所述氧化剂包括三氧化铬水溶液、Jones试剂、Sarrett试剂、PCC试 剂和三氧化铬冰醋酸水溶液中的一种或多种;
优选的,所述氧化剂在氧化水凝胶中的摩尔浓度为19~145mmol/L。
本发明还提供了上述技术方案所述固态氧化水凝胶的制备方法,包括以下步 骤:
将氧化剂、水凝胶和水混合,得到含有氧化剂的水凝胶溶液;
将所述含有氧化剂的水凝胶溶液静置,得到所述固态氧化水凝胶。
优选的,所述含有氧化剂的水凝胶溶液中水凝胶的质量浓度为5~45%。
本发明还提供了上述技术方案所述固态氧化水凝胶或上述技术方案所述制备 方法制备得到的固态氧化水凝胶在生物组织氧化前处理中的应用。
本发明还提供了一种衍生化试剂盒,包括独立包装的固态水凝胶、固态氧化 水凝胶和固态衍生化水凝胶;
所述固态氧化水凝胶为上述技术方案所述固态氧化水凝胶或上述技术方案所 述制备方法制备得到的固态氧化水凝胶。
本发明还提供了一种生物组织氧化前处理方法,包括以下步骤:
将固态氧化水凝胶贴附于生物组织切片表面进行氧化反应;所述固态氧化水 凝胶为上述技术方案所述固态氧化水凝胶或上述技术方案所述制备方法制备得到 的固态氧化水凝胶。
优选的,所述氧化反应的温度为2~25℃,所述氧化反应的时间为10min~1.5h。
优选的,所述生物组织切片中的代谢物包括羟基代谢物;
所述羟基代谢物包括羟基脂肪酸类代谢物、脂肪醇类代谢物、胆固醇和胆固 醇的衍生物中的一种或多种。
本发明还提供了一种生物组织原位衍生化方法,包括以下步骤:
将固态衍生化水凝胶贴附于前处理生物组织切片表面进行衍生化反应;
所述前处理生物组织切片为按照上述技术方案所述氧化前处理方法处理得到 的前处理生物组织切片。
有益技术效果:本发明提供了一种固态氧化水凝胶,包括氧化剂、水凝胶和 水;所述氧化剂包括三氧化铬水溶液、Jones试剂、Sarrett试剂、PCC试剂和三氧 化铬冰醋酸水溶液中的一种或多种。本发明提供的固态氧化水凝胶包含三氧化铬, 其中的铬为六价态,具有强氧化性,固态氧化水凝胶能够将生物组织中一级和二 级醇羟基代谢物的羟基氧化为醛基和/或酮基,氧化形成的醛基和/或酮基经过进一 步衍生化后进行质谱成像分析有效提高了羟基代谢物的质谱响应,提高了对羟基 代谢物的检测覆盖度;同时本发明提供的固态氧化水凝胶具有较好的氧化选择性, 将生物组织切片中的一级和二级醇羟基代谢物氧化为醛和酮,几乎不会过氧化成 羧酸,也不会影响羟基代谢物中的双键。本发明提供的固态氧化水凝胶提高了羟 基代谢物的质谱响应,有效解决了羟基类物质原位衍生化的困难,从而能够实现 对羟基代谢物进行原位表征。
附图说明
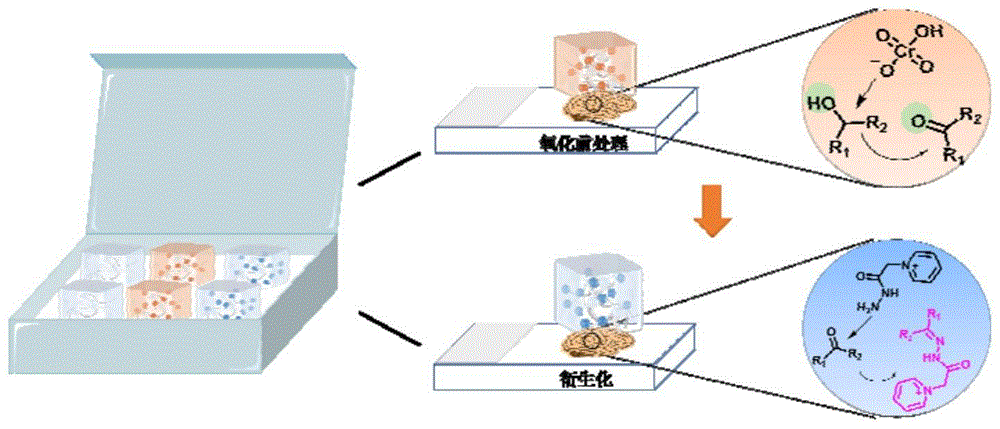
图1为固态水凝胶、固态氧化水凝胶和固态衍生化水凝胶试剂盒的构成及工作 原理示意图;
图2为固态水凝胶、固态氧化水凝胶和固态衍生化水凝胶处理和干扰扣除流程图;
图3为应用例1中脑组织切片氧化和衍生化前后脂肪醇(13:3)、羟基脂肪酸 (14:2)和胆固醇的质谱成像结果对比图;
图4为应用例2肾组织切片氧化和衍生化前后脂肪醇(17:0)、脂肪醇(16:2) 和羟基脂肪酸(15:2)的质谱成像结果对比图;
图5为应用例1、2的羟基脂肪酸、脂肪醇、胆固醇及其衍生物代表性的质谱 成像图;
图6为应用例1~4每种组织中鉴定到代谢物数量的柱状图;
图7为应用例1~4每种组织中3类代谢物数量的韦恩图;
图8为应用例5加标大鼠肝组织匀浆中1-十二醇、1-十二醛、2-十二醇和2-十 二酮离子强度的柱状图及氧化产率;
图9为应用例6中ZDF(fa/fa)糖尿病大鼠和ZDF(fa/+)正常对照大鼠脑组 织中的羟基脂肪酸(13:2)、羟基脂肪酸(14:2)、羟基脂肪酸(16:3)、脂肪醇(10:3)、 脂肪醛(14:4)和羰基脂肪酸(18:2;O)的质谱成像结果对比图;
图10为应用例6中ZDF(fa/fa)糖尿病大鼠和ZDF(fa/+)正常对照大鼠脑 组织中羟基脂肪酸、脂肪醇分析热图;其中“糖”代表糖尿病组,“正”代表正常 对照组;
图11为应用例6中ZDF(fa/fa)糖尿病大鼠和ZDF(fa/+)正常对照大鼠脑 组织中脂肪醛和羰基脂肪酸分析热图;其中“糖”代表糖尿病组,“正”代表正常 对照组;
图12为应用例7中ZDF(fa/fa)糖尿病大鼠和ZDF(fa/+)正常对照大鼠肾 组织中的羟基脂肪酸(8:2)、羟基脂肪酸(11:2)、羟基脂肪酸(15:2)、羟基脂肪 酸(15:3)脂肪醛(11:3)和羰基脂肪酸(18:2;O)的质谱成像结果对比图;
图13为应用例7中ZDF(fa/fa)糖尿病大鼠和ZDF(fa/+)正常对照大鼠肾 组织的羟基脂肪酸、脂肪醇分析热图;其中“糖”代表糖尿病组,“正”代表正常 对照组。
图14为应用例7中ZDF(fa/fa)糖尿病大鼠和ZDF(fa/+)正常对照大鼠肾 组织的羰基脂肪酸和脂肪醛分析热图;其中“糖”代表糖尿病组,“正”代表正常 对照组。
具体实施方式
本发明提供了一种固态氧化水凝胶,包括氧化剂、水凝胶和水;
所述氧化剂包括三氧化铬水溶液、Jones试剂、Sarrett试剂、PCC试剂和三氧化铬冰醋酸水溶液中的一种或多种,优选为三氧化铬水溶液、Jones试剂或三氧化铬冰醋酸 水溶液,更优选为Jones试剂。在本发明中,所述水凝胶优选包括琼脂、琼脂糖或明胶 中的一种或多种,更优选为明胶;在本发明中,当氧化剂包括两种以上上述具体物质时, 本发明对具体物质的配比无特殊限定,采用任意配比即可。
在本发明中,所述三氧化铬水溶液氧化剂优选按照下述方法制备得到:将三氧化铬 溶于水,得到三氧化铬水溶液。在本发明中,所述三氧化铬水溶液中三氧化铬的摩尔浓度优选为1.3~2.7mol/L,更优选为2.67mol/L。
在本发明中,所述Jones试剂优选按照下述方法制备得到:将三氧化铬和浓硫酸混合,得到第一溶液,将所述第一溶液加水稀释,得所述Jones试剂。在本发明中,所述 Jones试剂中三氧化铬的摩尔浓度优选为1.3~2.7mol/L,更优选为2.67mol/L。在本发明 中,所述浓硫酸和水的体积比优选为22~24:77,更优选为为23:77。本发明对所述混合 无特殊限定,只要能够混合均匀即可。
在本发明中,所述Sarrett试剂优选按照下述方法制备得到:将三氧化铬和吡啶混合, 得到第一溶液,利用二氯甲烷稀释所述第一溶液,得到Sarrett试剂。在本发明中,所述Sarrett试剂中三氧化铬的摩尔浓度优选为1.3~2.7mol/L,更优选为2.67mol/L。在本发明中,所述吡啶和二氯甲烷的体积比优选为22~24:77,更优选为23:77。本发明对所述混 合无特殊限定,只要能够混合均匀即可。
在本发明中,所述PCC试剂优选按照下述方法制备得到:将氯铬酸吡啶盐(PCC) 溶于水,得到PCC试剂。在本发明中,所述PCC试剂中PCC的摩尔浓度优选为 1.3~2.7mol/L,更优选为2.67mol/L。本发明对所述溶解无特殊限定,只要能够溶解完全 即可。
在本发明中,所述三氧化铬冰醋酸水溶液优选按照下述方法制备得到:将三氧化铬 和冰醋酸第一混合,得到第一溶液;将所述第一溶液和水第二混合,得到三氧化铬冰醋酸水溶液。在本发明中,所述氧化铬冰醋酸水溶液中三氧化铬的摩尔浓度优选为 1.3~2.7mol/L,更优选为2.67mol/L。在本发明中,所述冰醋酸和水的体积比优选为 22~24:77,更优选为为23:77。本发明对所述溶解无特殊限定,只要能够溶解完全即可。
在本发明中,所述氧化剂中活性组分(三氧化铬或PCC)在氧化水凝胶中的摩尔浓度优选为19~145mmol/L,更优选48~96mmol/L,最优选为48mmol/L。在本发明中,所 述水凝胶占氧化水凝胶的质量百分含量优选为5~45%,更优选为14~16%。
本发明还提供了上述技术方案所述固态氧化水凝胶的制备方法,包括以下步骤:
将氧化剂、水凝胶和水混合,得到含有氧化剂的水凝胶溶液;
将所述含有氧化剂的水凝胶溶液静置,得到所述固态氧化水凝胶。
本发明将氧化剂、水凝胶和水混合,得到含有氧化剂的水凝胶溶液。在本发明中,所述氧化剂中的活性组分(三氧化铬或PCC)在含有氧化剂的水凝胶溶液中摩尔浓度优 选为19~145mmol/L,更优选48~96mmol/L,进一步优选为48mmol/L。在本发明中,所 述水凝胶占含有氧化剂的水凝胶溶液的质量百分含量优选为5~45%,更优选为14~16%。 在本发明中,所述混合的温度优选为65~75℃,更优选为68~70℃。本发明对所述混合 的方式无特殊要求只要能够混合均匀即可。
得到含有氧化剂的水凝胶溶液后,本发明将所述含有氧化剂的水凝胶溶液静置,得 到所述固态氧化水凝胶。在本发明中,所述静置前优选还包括冷却。在本发明中,所述冷却后溶液的温度优选为室温,所述室温的温度优选为20~35℃,更优选为25~30℃; 所述冷却的时间优选为0.5~2h,更优选为1~1.5h。本发明对所述冷却的方式无特殊限定,只要能够冷却得到所需的温度即可。
在本发明中,所述静置的温度优选为-20~4℃,更优选为-15~2℃;所述静置的时间 优选4~10h,更优选为5~8h,最优选为6h。本发明优选将含有氧化剂的水凝胶溶液置于冰箱中得到静置的温度。在本发明中,所述静置优选在塑形模具中进行,本发明对所 述塑形模具的形状和结构没有特殊要求。在本发明中,所述氧化水凝胶的厚度优选为 0.8~1.5cm,更优选为1~1.2cm。本发明通过控制静置的温度和时间,使所述含有氧化剂 的水凝胶溶液更有效的凝固成具有一定弹性和机械强度的固态氧化水凝胶。
本发明还提供了上述技术方案所述固态氧化水凝胶或上述技术方案所述制备方法 制备得到的固态氧化水凝胶在生物组织氧化前处理中的应用。
本发明还提供了一种衍生化试剂盒,包括独立包装的固态水凝胶、固态氧化水凝胶 和固态衍生化水凝胶。图1为固态水凝胶、固态氧化水凝胶和固态衍生化水凝胶试剂盒的构成及工作原理示意图。
在本发明中,所述固态水凝胶优选包括水凝胶和水;在本发明中,所述水凝胶包括琼脂、琼脂糖或明胶中的一种或多种,优选为明胶。在本发明中,所述水凝胶占固态水 凝胶的质量百分含量优选为5~45%,更优选为14~16%;
所述固态水凝胶的制备方法,优选包括以下步骤:
将水凝胶和水混合,得到混合溶液;
将所述混合溶液静置,得到固态水凝胶;
本发明将水凝胶和水混合,得到混合溶液。在本发明中,所述混合溶液中水凝胶的质量百分含量优选为5~45%,更优选为14~16%。在本发明中,所述混合的温度优选为 65~75℃,更优选为68~70℃。本发明对所述混合无特殊要求,只要能够混合均匀即可。
得到混合溶液后,本发明将所述混合溶液静置,得到固态水凝胶。在本发明中,所述静置的温度优选为-20~4℃,更优选为-15~2℃;所述静置的时间优选4~10h,更优选为5~8h,最优选为6h;在本发明中,所述静置优选在塑形模具中进行,本发明对所述 塑形模具的形状和结构没有特殊要求。本发明通过控制静置的温度和时间使所述混合溶 液更有效的凝固成具有一定弹性和机械强度的固态水凝胶。
在本发明中,所述固态氧化水凝胶为上述技术方案所述固态氧化水凝胶或上述技术 方案所述制备方法制备得到的固态氧化水凝胶。
在本发明中,所述固态衍生化水凝胶优选包括衍生化试剂、水凝胶、甲酸和水;所述衍生化试剂优选包括吉拉德试剂P(Girard's reagent P,GP)和/或吉拉德试剂T(Girard's reagent T,GT),更优选为GP。在本发明中,当所述衍生化试剂为GP和GT时,本发明 对所述GP和GT的用量配比无特殊限定,采用任意配比即可。在本发明中,所述水凝 胶优选包括明胶或琼脂,更优选为明胶。
在本发明中,所述衍生化试剂在包含水凝胶、衍生化试剂、甲酸和水的混合溶液中的摩尔浓度优选为0.66~1320mmol/L,优选为66~990mmol/L,最优选为165~660mmol/L。在本发明中,所述水凝胶在包含水凝胶、衍生化试剂、甲酸和水的混合溶液中的质量百 分含量优选为5~45%,更优选为14~16%,所述甲酸在包含水凝胶、衍生化试剂、甲酸 和水的混合溶液中的质量浓度优选为0.5~3%,更优选为1~2%。在本发明中,所述衍生 化水凝胶的制备方法,优选包括以下步骤:
将水凝胶、衍生化试剂、甲酸和水混合,得到混合溶液;
将所述混合溶液静置,得到固态衍生化水凝胶;
本发明将水凝胶、衍生化试剂、甲酸和水混合,得到混合溶液。在本发明中,所述混合溶液中水凝胶的质量百分含量优选为5~45%,更优选为14~16%。在本发明中,所 述混合溶液中衍生化试剂的摩尔浓度优选为0.66~1320mmol/L,更优选为 66~990mmol/L,最优选为165~660mmol/L。本发明对所述混合无特殊要求,只要能够混 合均匀即可。
得到混合溶液后,本发明将所述混合溶液静置,得到固态衍生化水凝胶。在本发明中,所述静置的温度优选为-20~4℃,更优选为-15~2℃;所述静置的时间优选4~10h, 更优选为5~8h,最优选为6h;在本发明中,所述静置优选在塑形模具中进行,本发明 对所述塑形模具的形状和结构没有特殊要求。本发明通过控制静置的温度和时间使所述 混合溶液更有效的凝固成具有一定弹性和机械强度的固态衍生化水凝胶。
本发明还提供了一种生物组织氧化前处理方法,包括以下步骤:
将固态氧化水凝胶贴附于生物组织切片表面进行氧化反应;所述固态氧化水凝胶为 上述技术方案所述固态氧化水凝胶或上述技术方案所述制备方法制备得到的固态氧化水凝胶。
在本发明中,所述生物组织切片优选包括脑矢状面组织切片、脑冠状面组织切片、肝组织切片、生殖腺组织切片、肺组织切片、肾组织切片、心组织切片、肌肉组织切片、 脾组织切片、肾上腺组织切片或肿瘤组织切片,更优选为脑矢状面组织切片、肝组织切 片、肾组织切片或心组织切片。在本发明中,所述生物组织切片中的代谢物优选包括羟 基代谢物;所述羟基代谢物优选包括一级和二级醇羟基代谢物;所述羟基代谢物优选包 括羟基脂肪酸类代谢物、脂肪醇类代谢物、胆固醇和胆固醇的衍生物中的一种或多种, 更优选为羟基脂肪酸类代谢物或脂肪醇类代谢物。在本发明中,所述胆固醇的衍生物优 选包括胆汁酸类代谢物或维生素类代谢物。在本发明中,所述羟基代谢物中的羟基优选 包括一级醇和/或二级醇。
本发明对所述生物组织切片的制备方法无特殊要求,采用本领域常规的方式即可。 在本发明中,制备所述组织切片的方法优选包括以下步骤:
将大鼠乙醚麻醉,断头处死后迅速解剖获取生物组织;
将所述生物组织依次进行包埋和切片,得到所述生物组织切片。
本发明将大鼠乙醚麻醉,断头处死后迅速解剖获取生物组织。在本发明中,所述大鼠优选为Sprague-Dawley(SD)正常雄性大鼠。本发明对所述获取组织的方式无特殊要 求,采用本领域常规的方式即可。本发明优选将得到的生物组织冷冻保存。在本发明中, 所述冷冻保存的温度优选为-80℃。
得到生物组织后,本发明将所述生物组织依次进行包埋和切片,得到所述生物组织 切片。在本发明中,所述包埋前优选将冷冻的生物组织进行平衡处理。在本发明中,所述平衡处理的温度优选为-22~-18℃,更优选为-20℃,时间优选为10~14h,更优选为12h。在本发明中,所述包埋用包埋剂优选为莱卡冷冻切片包埋剂(Leica Cryo-Gel),本发明 对所述包埋的具体实施过程没有特殊要求。在本发明中,所述包埋剂可以将生物组织固 定于底托上,防止切片组织松动或者掉落。在本发明中,所述切片优选在切片机中进行, 本发明对所述切片的具体实施过程没有特殊要求,在本发明中,所述生物组织切片的厚 度优选为10~14μm,更优选为12μm。本发明优选将相邻两张组织切片贴附于同一张载 玻片的左右两侧,左侧为空白对照组织切片,右侧为实验用组织切片。
在本发明中,所述固态氧化水凝胶的尺寸优选和所述同一张载玻片右侧的生物组织 切片的尺寸一致。在本发明中,所述氧化反应的温度优选为2~25℃,更优选为18~23℃; 所述氧化反应的时间优选为10min~1.5h,更优选为10~30min,最优选为10min。
在本发明中,生物组织切片中羟基代谢物中的羟基在所述氧化反应过程中被氧化为 醛基和/或酮基,当所述羟基为一级醇时被氧化为醛,当所述羟基为二级醇时被氧化为酮。 本发明提供的固态氧化水凝胶能够将生物组织切片中的羟基代谢物氧化为醛和/或酮,几 乎不会过氧化成羧酸;当羟基代谢物为不饱和的羟基类物质,氧化时双键也不会受到影 响。本发明提供的氧化水凝胶针对性较强,提高了组织中羟基代谢物的灵敏度和覆盖度。
在本发明中,所述固态氧化水凝胶处理后优选还包括:将固态氧化水凝胶从生物组 织切片表面除去。
本发明还提供了一种生物组织原位衍生化方法,包括以下步骤:
将固态衍生化水凝胶贴附于前处理生物组织切片表面进行衍生化反应;
所述前处理生物组织切片为按照上述技术方案所述氧化前处理方法处理得到的前 处理生物组织切片。
在本发明中,所述衍生化水凝胶优选包括衍生化试剂、水凝胶、甲酸和水;所述衍生化试剂优选包括吉拉德试剂P(Girard's reagent P,GP)和/或吉拉德试剂T(Girard'sreagent T,GT),更优选为GP。在本发明中,当所述衍生化试剂为GP和GT时,本发明 对所述GP和GT的用量配比无特殊限定,采用任意配比即可。在本发明中,所述水凝 胶优选包括明胶或琼脂,更优选为明胶。
在本发明中,所述衍生化试剂在包含水凝胶、衍生化试剂、甲酸和水的混合溶液中的摩尔浓度优选为0.66~1320mmol/L,优选为66~990mmol/L,最优选为165~660mmol/L。在本发明中,所述水凝胶在包含水凝胶、衍生化试剂、甲酸和水的混合溶液中的质量百 分含量优选为5~45%,更优选为14~16%,所述甲酸在包含水凝胶、衍生化试剂、甲酸 和水的混合溶液中的质量浓度优选为0.5~3%,更优选为1~2%。在本发明中,所述衍生 化水凝胶的制备方法,优选包括以下步骤:
将水凝胶、衍生化试剂、甲酸和水混合,得到混合溶液;
将所述混合溶液静置,得到固态衍生化水凝胶;
本发明将水凝胶、衍生化试剂、甲酸和水混合,得到混合溶液。在本发明中,所述混合溶液中水凝胶的质量百分含量优选为5~45%,更优选为14~16%。在本发明中,所 述混合溶液中衍生化试剂的摩尔浓度优选为0.66~1320mmol/L,更优选为 66~990mmol/L,最优选为165~660mmol/L。本发明对所述混合无特殊要求,只要能够混 合均匀即可。
得到混合溶液后,本发明将所述混合溶液静置,得到固态衍生化水凝胶。在本发明中,所述静置的温度优选为-20~4℃,更优选为-15~2℃;所述静置的时间优选4~10h, 更优选为5~8h,最优选为6h;在本发明中,所述静置优选在塑形模具中进行,本发明 对所述塑形模具的形状和结构没有特殊要求。本发明通过控制静置的温度和时间使所述 混合溶液更有效的凝固成具有一定弹性和机械强度的衍生化水凝胶。
在本发明中,所述衍生化反应的温度优选为15~25℃,更优选为18~23℃;所述衍生化反应的时间优选为15min~5h,更优选为30min~3h,进一步优选为1h~2h。
在本发明中,所述衍生化反应后优选还包括:
将固态衍生化水凝胶从生物组织切片的表面移除后进行干燥。
在本发明中,所述干燥优选为真空烘干,所述干燥的温度优选为15~25℃;所述干燥的时间优选为1~4h,更优选为2~3h。
本发明提供的将生物组织切片中羟基氧化为醛基和/或酮基后再进行衍生化,提高了 待检测代谢物的检测灵敏度。本发明给出了将不易衍生化的物质针对性的进行官能团转 化,得到易衍生化物质后用已知成熟的第二步反应引入高质谱响应的基团,从而实现原 位组织质谱成像分析。此外也可以针对同一类物质,通过转化为不同官能团的物质实现衍生化。具体为,针对羟基类物质还可以寻找其他氧化剂针对性的将羟基氧化成羧基, 然后对羧基官能团利用已知的成熟衍生化试剂进行质谱成像检测,同样也可以检测该类 物质。本发明提供的原位衍生化方法能够实现生物组织内低丰度、难电离、缺乏合适衍 生化试剂的代谢物的质谱成像检测和原位分析。
本发明还提供了一种生物组织代谢物非诊断和治疗为目的的检测方法,包括以下步 骤:
采用质谱成像技术对所述原位衍生化生物组织切片中的代谢物进行表征;所述原位 衍生化生物组织切片为上述技术方案所述原位衍生化方法得到的原位衍生化生物组织切片。
在本发明中,所述表征的方法优选包括以下步骤:
采用质谱成像技术,获得衍生化生物组织切片中衍生化代谢物离子的质荷比与离子 强度、位置关系的多维数据阵;
设定质荷比容差为5mDa,由扣除干扰后的组织成像结果中可以得到羟基代谢物经氧化、衍生化后产物的空间分布及质荷比与离子强度、位置关系的多维数据阵;
根据得到的羟基代谢物经氧化和衍生化后产物的质荷比计算出氧化和衍生化之前 的羟基代谢物的精确分子量,然后进行代谢物鉴定。
本发明采用质谱成像技术,获得衍生化生物组织切片中衍生化代谢物离子的质荷比 与离子强度、位置关系的多维数据阵。本发明对所述质谱成像技术没有特殊要求,采用本领域技术熟知的质谱成像技术即可;在本发明的实施例中,所述质谱成像技术为本发 明人自主研发的空气动力辅助解吸电喷雾离子化质谱成像技术(Air flow assisteddesorption electrospray ionization mass spectrometry imaging,AFADESI-MSI)。
在本发明中,所述质谱成像技术的参数为:以体积比为8:2的乙腈和水的混合溶液作为喷雾溶液,设定采集模式为正离子模式,设定电压为7kV,喷雾流速为5μL/min, 喷雾气压力为0.7MPa,X轴扫描速度为0.2mm/s,Y轴步进距离为0.2mm/s。本发明对 衍生化生物组织切片中的代谢物按空间位置逐点进行扫描检测,获得衍生化生物组织切 片中衍生化代谢物离子的质荷比与离子强度、位置关系的多维数据阵。
获得衍生化生物组织切片中衍生化代谢物离子的质荷比与离子强度、位置关系的多 维数据阵后,考虑到生物组织内原本存在的含醛(酮)基类物质会对该方法的氧化产物造成干扰,本发明设定质荷比容差为5mDa,扣除同一张载玻片左侧生物组织切片的干 扰,得到羟基代谢物经氧化、衍生化后产物的质荷比与离子强度、位置关系的多维数据 阵。在本发明中,所述左边侧生物组织切片的制备方法优选按照制备右侧衍生化生物组 织切片的方法制备得到,不同之处在于,将固态氧化水凝胶替换为固态水凝胶;固态水 凝胶中不含有氧化剂,其它成分与氧化水凝胶一致。左侧生物组织切片用生物组织切片 和右侧衍生化生物组织切片用生物组织切片为同一生物组织的相邻切片。本发明对扣除 左侧生物组织切片的背景的方式无特殊要求,采用本领域常规的扣除背景的方式即可, 下述所有的衍生化质谱成像结果都是扣除干扰的结果。
根据得到的羟基代谢物经氧化和衍生化后产物的质荷比计算出氧化和衍生化之前 的羟基代谢物的精确分子量。本发明对所述计算的方法无特殊要求,采用本领域常规的计算方法即可。
得到氧化和衍生化后羟基代谢物的质荷比(m/z)与离子强度后,本发明优选还包括计算得到氧化和衍生化之前羟基代谢物的精确分子量M。由于氧化过程会丢失两个 氢,GP衍生过程会得到一个GP分子然后丢失一分子水,所以M=m/z-132.0556
本发明通过对羟基代谢物的[M+H]
在本发明中,所述衍生化加标生物组织匀浆条的制备方法包括以下步骤:
将生物组织匀浆和标准品储备液混合,得到加标生物组织匀浆;
将所述加标生物组织匀浆固化,得到加标生物组织匀浆条;
将所述加标生物组织匀浆条按照上述技术方案所述氧化前处理和衍生化方法进行 衍生化,得到衍生化加标生物组织匀浆条。
本发明将生物组织匀浆和含有代谢物标准品的标准储备液混合,得到加标生物组织 匀浆。在本发明中,所述生物组织匀浆优选为生物组织和生理盐水的混合溶液,在本发明中,所述生物组织的质量和生理盐水的体积比优选为3:40;在本发明中,所述含有代 谢物标准品的标准储备液中的代谢物标准品优选包括羟基脂肪酸代谢物、脂肪醇类代谢 物、胆固醇和胆固醇的衍生物中的一种或多种。在本发明中,所述加标生物组织匀浆中 代谢物标准品的摩尔浓度优选为0.5~3.3mmol/L。
得到加标生物组织匀浆后,本发明将所述加标生物组织匀浆固化,得到加标生物组 织匀浆条。本发明对所述固化的具体实施方案没有特殊要求,在本发明的具体实施例中, 所述固化为:用打孔器于PVC不干胶上制作连续的2×5mm矩形孔;将打孔后的PVC 不干胶粘附于正电荷防脱载玻片上;利用微量移液器吸取5μL加标生物组织匀浆于载玻 片的矩形孔中;将载玻片置于室温干燥4h。
得到加标生物组织匀浆条后,本发明将所述加标生物组织匀浆条按照上述技术方案 所述衍生化方法进行氧化和衍生化,得到衍生化加标生物组织匀浆条。
在本发明中,通过实验测量衍生化加标生物组织匀浆条获得数据的方法与所述衍生 化生物组织切片的获得方法相同,在此不再赘述。
为了进一步说明本发明,下面结合实施例对本发明提供的技术方案进行详细 地描述,但不能将它们理解为对本发明保护范围的限定。
实施例1
将Jones试剂、明胶和水70℃混合,得到混合溶液,混合溶液中Jones试剂 的摩尔浓度为48mmol/L,明胶的质量百分含量为15%;将混合溶液转移至塑形容 器冷却1h至25℃后置于4℃冰箱置6h,得到厚度为1cm的氧化水凝胶。
应用例1
将明胶和水70℃混合,得到质量浓度为15%的明胶水溶液,转移至塑形容器 冷却1h至25℃后置于4℃冰箱静置6h,得到厚度为1cm的固态水凝胶;
将明胶、摩尔浓度为2.67mol/L的三氧化铬水溶液和水70℃混合,得到三氧 化铬质量浓度为48mmol/L,明胶质量浓度为15%的含有氧化剂的水凝胶溶液;将 含有氧化剂的水凝胶溶液转移至塑形容器冷却1h至25℃后置于4℃冰箱静置6h, 得到厚度为1cm的固态氧化水凝胶;
将吉拉德试剂P、明胶、甲酸和水70℃混合,得到混合溶液,混合溶液中吉 拉德试剂P的摩尔浓度为330mmol/L,甲酸的质量百分含量为1%,明胶的质量 百分含量为15%;将混合溶液转移至塑形容器冷却1h至25℃后置于4℃冰箱置 6h,得到厚度为1cm的固态衍生化水凝胶;
将雄性SD大鼠乙醚麻醉,然后断头处死,在冰上剖取脑组织,置于-80℃保 存;将冷冻保存的脑组织转移到-20℃平衡12h后用Lecia Cryo-Gel包埋剂固定于 底托上,然后对需要研究位置进行厚度为12μm的连续切片,选取两片大鼠脑组 织矢状面相邻切片,分别相邻贴附于同一张载玻片的左右两侧。
将固态水凝胶和固态氧化水凝胶切制成30mm×15mm×10mm的尺寸,再按照 脑组织切片的大小进行微调,将固态水凝胶贴附于左侧脑组织切片,将固态氧化 水凝胶贴附于右侧脑组织切片并在25℃放置10min,移除固态水凝胶和氧化水凝 胶;然后切制两块30mm×15mm×10mm尺寸的衍生化水凝胶,贴附于上述脑组织 切片并于20℃衍生化反应2h,将衍生化反应后的组织切片置于真空干燥器20℃ 干燥3h,得到氧化和衍生化脑组织切片。
配制体积比为8:2的乙腈和水的混合液作为喷雾溶剂,利用AFADESI-MSI 对原位衍生化脑组织切片进行质谱成像检测,得到原位衍生化脑组织切片中离子 的质荷比与离子强度、位置关系的多维矩阵;检测参数按照如下方式进行限定: 测量脑组织的长(X mm)宽(Y mm),根据X轴设定扫描速度为0.2mm/s,Y轴 步进距离为0.2mm/次,计算并设定每张载玻片上切片的扫描每行的时间(X/0.2s) 和扫描的行数(Y/0.2),设置电压为7kV,喷雾针流速为5μL/min,喷雾气压力 为0.7MPa,采集模式为正离子模式;得到衍生化脑组织切片中离子的质荷比与离 子强度、位置关系的多维矩阵。按照上述干扰扣除的方式将左侧的干扰扣除得到 氧化和衍生化的质谱成像结果。
图2为固态水凝胶、固态氧化水凝胶和固态衍生化水凝胶处理和干扰扣除流 程示意图。
考虑到羟基类物质主要在负离子模式下检测,选取与上述脑组织切片相邻的 切片不经过处理直接利用AFADESI-MSI进行质谱成像检测。检测参数按照如下 方式进行限定:测量脑组织的长(X mm)宽(Y mm),根据X轴设定扫描速度 为0.2mm/s,Y轴步进距离为0.2mm/次,计算并设定每张载玻片上切片的扫描每 行的时间(X/0.2s)和扫描的行数(Y/0.2),设置电压为-7KV,喷雾针流速为 5μL/min,喷雾气压力为0.7MPa,采集模式为负离子模式,得到未经衍生化的脑 组织切片的质谱成像结果。
图3左侧无处理栏检测结果为未经处理直接进行质谱成像检测的结果,右侧 衍生化栏的检测结果为氧化和衍生化脑组织切片的质谱成像检测结果。由图3可 以看出,大鼠脑组织切片的脂肪醇(13:3)、羟基脂肪酸(14:2)和胆固醇未经衍 生化处理时检测不到,而经过氧化和衍生化后强度明显提高,质谱成像结果清晰 可见,这说明本发明提供的检测方法针对羟基类物质具有很好的检测能力,经过 代谢物注释在脑组织切片中共检测到四类(羟基脂肪酸、脂肪醇、胆固醇及其衍 生物)共202个代谢产物(图6)。
应用例2
按照应用例1的方法对原位氧化和衍生化肾组织切片进行质谱成像检测,不 同之处在于,生物组织切片为大鼠的肾组织切片,肾组织切片的尺寸为 20mm×20mm×10mm;
按照应用例1中关于对未经处理的脑组织切片进行质谱成像检测的方法对未 经处理的肾组织切片进行质谱成像检测。
得到的未经处理肾组织切片和原位氧化和衍生化肾组织切片的质谱成像结果 对比图,如图4所示。图4左侧无处理栏检测结果为未经处理直接对肾组织进行 质谱成像检测的结果,右侧衍生化检测结果为原位氧化和衍生化肾组织切片的质 谱成像检测结果。由图4可以看出,大鼠肾组织切片的脂肪醇(17:0)、脂肪醇(16:2) 和羟基脂肪酸(15:2)不经氧化和衍生化处理不能检测到,而经过氧化和衍生化 后强度明显提高,质谱成像结果清晰可见,这说明本发明提供的试剂盒针对肾组 织内的羟基类物质具有很好的检测能力。经过代谢物注释在脑组织切片中共检测 到三类(羟基脂肪酸、脂肪醇、胆固醇及其衍生物)共133个代谢产物(图6)。
应用例3
按照应用例1的方法制备得到原位氧化和衍生化肝组织切片,按照应用例1 的检测方法对原位氧化和衍生化肝组织切片进行质谱成像检测,检测得到三类(羟 基脂肪酸、脂肪醇、胆固醇及其衍生物)共164个代谢产物(图6)。
应用例4
按照应用例1的方法制备得到原位氧化和衍生化心组织切片,不同之处在于, 心组织切片的尺寸为15mm×15mm×10mm;按照应用例1的检测方法对原位氧化 和衍生化肝组织切片进行质谱成像检测,检测得到三类(羟基脂肪酸、脂肪醇、 胆固醇及其衍生物)共110个代谢产物(图6)。
图5为应用例1、2中检测到的代表性羟基脂肪酸类、脂肪醇类、胆固醇及其 衍生物的质谱成像空间分布图,由图5可以看出脑组织和肾组织中的微区清晰可 见,可以明显看出代谢物的微区分布。
将应用例1~4得到的大鼠脑、肾、肝和心的质谱数据进行Metlin、HMDB、 LipidMaps数据库比对,并通过脂质碳链数增加及不饱和度增加规则,共得到至 少126个羟基脂肪酸类代谢物,80个脂肪醇类代谢物,44个胆固醇及其衍生物; 羟基脂肪酸类代谢物如表1所示,脂肪醇类代谢物如表2所示,胆固醇及其衍生 物如表3所示。其中代谢物一栏中括号中的数字分别代表碳原子数和饱和度,例 如羟基脂肪酸(24:6)中24代表碳原子数,6代表不饱和度。
根据表1~3绘制应用例1~4组织切片中鉴定得到的代谢物数量柱状图,如图 6所示。由图6可以看出脑、肾、肝和心组织切片中共检测到三类(羟基脂肪酸、 脂肪醇、胆固醇及其衍生物)分别为202、133、164和110个代谢物。
根据表1~3用R语言绘制应用例1~4组织切片中3类代谢物分别在四种组织 中的数量韦恩图,如图7所示。由图7可以看出胆固醇及其衍生物主要分布在脑, 羟基脂肪酸和脂肪醇类代谢物在四种器官中数量和种类差别不大,但也是脑中的 羟基代谢物数量最多,代谢物在组织中的分布具有差异性。
表1应用例1~4检测到羟基脂肪酸类代谢物
/>
/>
/>
/>
表2应用例1~4检测到的脂肪醇类代谢物
/>
/>
表3应用例1~4检测到的胆固醇及其衍生物
/>
/>
应用例5
将0.4mL生理盐水与0.3g大鼠肝组织混合得到匀浆液,稀释十倍得到组织匀 浆,取组织匀浆95μL;取含有1-十二醇、2-十二醇、1-十二醛和2-十二酮的标准 储备液5μL,分别配制成含上述标准代谢物浓度为0.5mmol/L的加标大鼠肝组织 匀浆;
取打孔器于PVC不干胶上制作连续的2mm×5mm矩形孔,再将PVC不干胶 粘附于正电荷防脱载玻片上;利用微量移液器准确吸取5μL非加标肝组织匀浆 和四种标准品肝组织匀浆于载玻片的类矩形孔中,每种匀浆平行3个,并置于室 温干燥4h,制备大鼠肝组织匀浆条。
将实施例1制备得到固态氧化水凝胶切制成2mm×5mm×10mm的尺寸,再按 照组织匀浆条大小进行微调,将固态氧化水凝胶贴附于不同加标组织匀浆条,于 室温放置10min,完成大鼠匀浆条中代谢物的氧化反应;移除固态氧化水凝胶, 然后切制同样大小的固态衍生化水凝胶,同时贴附于该组织匀浆条,于室温放置 2h,完成氧化后衍生化的匀浆条制作,然后置于真空干燥器干燥1h;
利用AFADESI-MSI对氧化和衍生化的匀浆条进行质谱成像检测,首先配制 体积比为8:2的乙腈和水的混合液作为喷雾溶剂,然后测量匀浆条的长宽,根据X轴扫描速度为0.2mm/s,Y轴步进距离为0.2mm/次,计算每张载玻片上切片的 扫描每行的时间和扫描的行数,设置电压为7kV,喷雾针流速为5μL/min,喷雾 气压力为0.7MPa,采集模式为正离子模式,获得氧化后衍生化处理的匀浆条中离 子的质荷比与离子强度、位置关系的多维矩阵;
由于1-十二醇、2-十二醇、1-十二醛和2-十二酮的氧化和衍生化的m/z都是318.2534,所以先用MassImager数据处理软件得到氧化和衍生化的非加标肝组织 匀浆条中m/z 318.2534的离子强度;然后得到加标肝组织匀浆条中的1-十二醇、 2-十二醇、2-十二醛和2-十二酮的离子强度;最后4种加标匀浆条的离子强度同 时扣除非加标肝组织匀浆条中m/z 318.2534的离子强度,得到标准品经氧化和衍 生化后产物的质荷比与离子强度的二维数据。
由于1-十二醛是1-十二醇的氧化产物,2-十二酮是2-十二醇的氧化产物,所 以用1-十二醇的离子强度除以1-十二醛的离子强度作为羟基氧化为醛的产率,用 2-十二醇的离子强度除以2-十二酮的离子强度作为羟基氧化为酮的产率。其中羟 基氧化为醛的产率为18.12%,羟基氧化为酮的产率为38.58%(图8)。
该结果也进一步说明了一、二级醇在衍生化试剂盒中氧化水凝胶的处理下实 现一级醇转化为醛,二级醇转化为酮,进一步的衍生化水凝胶可以提高代谢物的 质谱响应,该试剂盒的衍生化结果是准确的。
应用例6
以雄性自发性2型ZDF(fa/fa)糖尿病大鼠为检测对象,按照应用例1制备 原位氧化和衍生化脑组织矢状面切片;以雄性ZDF(fa/+)正常对照大鼠为检测 对象,按照应用例1制备原位氧化和衍生化脑组织矢状面切片,并进行质谱成像 分析。代表性代谢物的质谱成像图如图9所示,由图9可看出,糖尿病大鼠和正 常大鼠的脑组织切片成像清晰,ZDF(fa/fa)糖尿病大鼠中羟基脂肪酸(13:2)、 羟基脂肪酸(14:2)、羟基脂肪酸(16:3)、脂肪醇(10:3)离子强度明显低于正常 对照ZDF(fa/+)大鼠,降低的微区主要是嗅球、海马和小脑;ZDF(fa/fa)糖尿 病大鼠中脂肪醛(14:4)和羰基脂肪酸(18:2;O)离子强度明显高于正常对照ZDF(fa/+)大鼠,升高的微区主要是嗅球、海马、基底核和丘脑。
由质谱成像数据分别得到正常对照组和糖尿病组大鼠脑组织中有显著差异的 羟基脂肪酸和脂肪醇及其离子强度,随后用R语言进行热图分析(图10),得到 糖尿病组和正常对照组大鼠脑组织切片中羟基脂肪酸和脂肪醇分析热图。从图10 中可以发现,这些代谢物在糖尿病大鼠脑组织中发生了明显变化,于正常对照组 相比,糖尿病大鼠脑组织中羟基脂肪酸和脂肪醇的离子强度明显降低。
进一步由同一张载玻片中左边(固态水凝胶和衍生化水凝胶处理)的成像结 果得到对照组和糖尿病组有显著差异的羰基脂肪酸和脂肪醛及其离子强度,然后 用R语言进行热图分析(图11),得到糖尿病组和正常对照组组织切片中羰基脂 肪酸和脂肪醛分析热图。由图11可看出,糖尿病大鼠和正常对照大鼠的脑组织切 片成像清晰,代谢物离子强度具有明显差异,其中糖尿病大鼠的脑组织中羰基脂 肪酸和脂肪醛的离子强度明显高于正常对照大鼠。
应用例7
按照应用6的方法对ZDF(fa/fa)糖尿病大鼠和ZDF(fa/+)正常对照大鼠 的肾组织切片进行质谱成像分析,代表性代谢物质谱成像结果如图12所示。由图 12可看出,糖尿病大鼠和正常大鼠的肾组织切片成像清晰,代谢物的离子强度具 有明显差别。ZDF(fa/fa)糖尿病大鼠肾组织中羟基脂肪酸(8:2)、羟基脂肪酸 (11:2)、羟基脂肪酸(15:2)、羟基脂肪酸(15:3)的离子强度显著高于正常对照 组;ZDF(fa/fa)糖尿病大鼠肾组织中脂肪醛(11:3)和羰基脂肪酸(18:2;O) 的离子强度也明显高于正常对照组,升高的微区主要是肾皮质。
由质谱成像分别得到糖尿病组和正常对照组具有显著差异的羟基脂肪酸和脂 肪醇及其离子强度,然后用R语言进行热图分析(图13),得出糖尿病组和对照 组组织切片中羟基脂肪酸、脂肪醇分析热图。由图13可看出,糖尿病大鼠的肾组 织中羟基脂肪酸和脂肪醇离子强度明显高于正常对照大鼠。
进一步由同一张载玻片中左边(固态水凝胶和衍生化水凝胶处理)的成像结 果分别得到糖尿病组和对照组具有显著差异的羰基脂肪酸和脂肪醛及其离子强 度,然后用R语言进行热图分析(图14),得到糖尿病组和正常对照组大鼠肾组 织切片中羰基脂肪酸和脂肪醛分析热图。由图14可看出,糖尿病大鼠肾组织中羰 基脂肪酸和脂肪醛离子强度明显高于对照大鼠。
尽管上述实施例对本发明做出了详尽的描述,但它仅仅是本发明一部分实施 例,而不是全部实施例,人们还可以根据本实施例在不经创造性前提下获得其他 实施例,这些实施例都属于本发明保护范围。
- 氧化铝载体及其制备方法、乙烯环氧化反应用银催化剂及乙烯环氧化制备环氧乙烷的方法
- 一种谷胱甘肽过氧化物酶测定试剂盒及其制备方法和应用
- 蛇床子素单端甲基氧化衍生物的制备方法
- 固态衍生化水凝胶及其制备方法和应用
- 一种羧乙基壳聚糖衍生物/氧化魔芋甘露聚糖可注射水凝胶及其制备方法和应用