一种三苯基膦乙酰丙酮羰基铑的合成方法
文献发布时间:2024-04-18 19:58:53
技术领域
本发明涉及化学催化领域,具体涉及一种三苯基膦乙酰丙酮羰基铑的合成方法。
背景技术
三苯基膦乙酰丙酮羰基铑,是丙烯氢甲酰化合成丁辛醇的核心催化剂,产品丁醇主要作为树脂、油漆和粘接剂的溶剂和增塑剂的原料。辛醇可直接作为有机溶剂用于油漆涂料、造纸和纺织等行业,但其最重要的用途是制成邻苯二甲酸二辛酯(DOP)。由于我国经济的发展,丁辛醇的市场需求量不断加大,产品一直处于供不应求的状态。
具体来说,三苯基膦乙酰丙酮羰基铑的合成方法有:
(1)按照Bonati等[J.Chem.Soc.,3156,1964公开的制备方法],将研细的水合氯化铑(RhCl3·nH2O)粉末与一氧化碳反应生成四羰基二氯二铑[Rh(CO)2Cl]2,四羰基二氯二铑再与乙酰丙酮在碳酸钡的作用下于石油醚(沸程60-80℃)中回流一星期制得乙酰丙酮二羰基铑[Rh(acac)(CO)2],在苯做溶剂下乙酰丙酮二羰基铑和三苯基膦反应,制备三苯基膦乙酰丙酮羰基铑。
(2)按照Ю.C.BapIIIaвсκий,Τ.Г.ЧерKисова.Ж.Неорг[M].Xим:1966.1709-1711.的制备方法,先合成乙酰丙酮二羰基铑,在无氧的条件下,合成三苯基膦乙酰丙酮羰基铑。
(3)CN103709205A提出的合成方法为:(1)在沉淀反应的条件下,将三氯化铑水溶液与沉淀剂接触,以使铑元素沉淀,得到含铑元素的沉淀;(2)将所述含铑元素的沉淀进行洗涤,洗涤至所述含铑元素的沉淀中氯离子含量低于1000ppm;(3)在合成乙酰丙酮二羰基铑的反应的条件下,将步骤(2)得到的所述含铑元素的沉淀溶解后与乙酰丙酮进行接触,得到含有乙酰丙酮二羰基铑的反应液;(4)在合成乙酰丙酮三苯基膦羰基铑的反应的条件下,将所述含有乙酰丙酮二羰基铑的反应液中的乙酰丙酮二羰基铑与三苯基膦接触。
(4)CN105669769A公开了三水合三氯化铑与三苯基膦溶液回流反应制得三苯基膦氯化铑;在氮气保护下,将制备的三苯基膦氯化铑与N,N-二甲基甲酰胺混合,再加入乙酰丙酮,回流加热反应,冷却至室温后,浓缩溶液,加入冰水,放置,析出晶体,过滤,滤饼水洗,真空干燥得乙酰丙酮三苯基膦羰基铑。
(5)CN105503959A公开了将以含有铑膦络合物的氢甲酰化催化剂废液为起始原料,包括:步骤a,在酸性条件下对所述废液进行氧化处理;步骤b,以经步骤a处理后的废液为原料合成乙酰丙酮三苯基膦羰基铑。
(6)CN106674285A公开的制备方法包括:(1)配制一定浓度的三氯化铑的水溶液,与甲苯混合,然后通入一氧化碳气体加热后进行反应;(2)向溶液中加入乙酰丙酮钠盐,继续反应;(3)向反应液中加入一定量的三苯基膦,加热反应;(4)反应液过滤,先后用去离子水、乙醇及正己烷洗涤,真空干燥得到乙酰丙酮三苯基膦羰基铑产品。
(7)CN107759640A公开的制备方法包括:(1)先用冰醋酸洗涤纯化三碘化铑废泥,再在氮气保护下,用混酸与纯化的三碘化铑废泥在一定温度下反应;(2)将混酸蒸馏至剩余1%~10%,再加入碳酸盐至无气泡产生,滤出不溶物;(3)加入DMF升温至一定温度后加入乙酰丙酮反应,加入去离子水,过滤干燥得到乙酰丙酮二羰基铑;(4)用环己烷溶解乙酰丙酮二羰基铑后升温至一定温度,加入三苯基膦反应,滤出析出晶体,真空干燥得到乙酰丙酮三苯基膦羰基铑。
(8)CN107759640A公开的制备方法包括:将氧化铑与乙酰丙酮搅拌混合,加入引发剂,加热至回流温度下反应,当体系从黑色变成棕黄色后,加入三苯基膦的脂肪醛继续在回流温度下进行反应,反应结束冷却后加入去离子水经过滤、去离子水洗涤滤饼,滤饼真空干燥后可得乙酰丙酮三苯基膦羰基铑。
(9)CN115651030A公开的制备方法包括:将水合三氯化铑与DMF混合,在油浴中加热至100-130℃,再加入乙酰丙酮,在140-160℃下回流;将反应液冷却至室温,向体系中加入水和有机溶剂,搅拌2-5min,将用有机溶剂溶解的三苯基膦缓慢加入体系中并搅拌,再向体系中加入水,搅拌2-5min,加入碱水溶液搅拌后停止反应;除去低沸点的有机溶剂,静置、过滤得到黄色固体,将黄色固体洗涤、真空干燥后即得到乙酰丙酮三苯基膦羰基铑
(10)文献《贵金属》Nov 2005Vol26 No.1,称取一定量的RhCl3·nH2O,分别加入适量的N,N-二甲基甲酰胺、乙酰丙酮,升温至140~160℃,充分反应1h,溶液逐渐由红棕色变为橙黄色。冷却,加入适量去离子水,即有大量紫红色沉淀生成,过滤,水洗,干燥得乙酰丙酮二羰基铑(I)。在氮气气氛和搅拌下,缓缓向乙酰丙酮二羰基铑(I)的甲苯溶液中加入适量三苯基膦的甲苯溶液,反应1h。蒸去大部分溶剂,冷却,即有金黄色晶体析出,过滤,真空干燥即得到乙酰丙酮三苯基膦羰基铑(I),2步的单程总收率为91%。
(11)文献《贵金属》Nov 2016Vol37 No.2,作者以在N,N-二甲基甲酰胺介质中,将水合三氯化铑与乙酰丙酮加热回流,一步合成了乙酰丙酮·二羰基铑([Rh(CO)2(acac)]2),产率为93%;以正丁烷为介质,[Rh(CO)2(acac)]2与三苯基磷(PPh3)定量反应,得到乙酰丙酮·三苯基膦·羰基铑(Rh(acac)(PPh3)(CO)),产率为99%。
以上的制备方法普遍存在着下列不足中的一方面或者多方面:
(1)不是无水无氧条件下合成,催化剂活性普遍偏低;
(2)反应时间长,操作流程多,产物纯度低,产物收率不高;
(3)大多数方法采用N,N-二甲基甲酰胺做溶剂,再用大量的水将反应液进行稀释,N,N-二甲基甲酰胺进入废液,造成废液里的铑回收难度大,铑回收率低,铑损耗大;
(4)废液达标处理困难,废液处理花费代价大;
(5)用N,N-二甲基甲酰胺作为溶剂,反应后的N-N二甲基甲酰胺废液存在较大的异味,据国外的资料,使用N-N二甲基甲酰胺的场所禁止育龄妇女进入,说明此试剂及废液对操作人员的身体十分不友好。
发明内容
为了克服了上述方法的不足,本发明提供了一种三苯基膦乙酰丙酮羰基铑的制备方法,包括以下步骤:
步骤1在尾气排气口有液封装置的反应釜中将氯铑酸铵用一定量的无水醇溶解,开启搅拌,先通入氮气不少于10分钟,赶走溶液和反应釜里的空气,设定温度,再通入CO,反应完毕,过滤,滤去沉淀得到滤液。
步骤2滤液再抽入反应釜中,开启搅拌,加入一定量的乙酰丙酮继续反应一段时间,得到乙酰丙酮二羰基铑溶液。
步骤3向步骤2所得的反应液中加入一定量的三苯基膦,立即有亮黄色沉淀产生,加完三苯基膦后持续反应一段时间。
步骤4过滤沉淀,先用水洗涤易溶于水的杂质,最后用有机溶剂洗去有机杂质,得到三苯基膦乙酰丙酮羰基铑纯品。
进一步地,所述的无水醇,选自无水甲醇、无水乙醇、无水异丙醇中的一种或几种;从经济性和安全性等考虑,优选无水乙醇,所述氯铑酸铵的醇溶液的浓度为0.1~0.3摩尔/升,且要求氯铑酸铵的醇溶液PH值为6-7。
进一步地,要求通气管插入到溶液底部,通气的顺序的先通入氮气不少于10分钟以便赶走溶液和反应釜里的空气,再通入CO。
进一步地,要求步骤一的反应温度控制在40℃-45℃,反应是吸热反应,温度过低,反应速度慢,温度过高,CO很快从醇溶液中逸出,通过多次试验,最优温度为45℃。
进一步地,鼓泡速度优选每秒钟3-5个气泡,反应终点的判断是溶液变为透明的淡黄色。CO的鼓泡速度慢,反应需要的时间长,鼓泡速度过快,的确会加快反应速度,部分CO来不及反应就逸出,造成CO的浪费,根据作者多年的生产经验,每秒钟鼓3-5个气泡是最适合的。
进一步地,所述乙酰丙酮的加入量为乙酰丙酮与铑的摩尔比为2.4~2.6;乙酰丙酮与铑摩尔比小于2.4,反应速度慢,亦会导致乙酰丙酮配位不完全,产品收率低,乙酰丙酮与铑摩尔比大于2.6,造成试剂浪费。
进一步地,步骤三所述三苯基膦的加入量为三苯基膦与铑的摩尔比为0.95~1。三苯基膦以固体形式加入反应釜中,反应温度为45℃~50℃。
进一步地,步骤4所述的有机溶剂为丙酮。
本发明的反应机理及反应过程为:
1)氯铑酸铵与一氧化碳的配位反应;(NH
2)二氯四羰基铑与乙酰丙酮的配位反应;Rh
3)乙酰丙酮二羰基铑与三苯基膦的取代反应:
Rh(CO)
本发明的有益效果包括:
(1)本发明提供的方法具有反应条件温和、产物收率高、产物纯度高、成本低等优点,以氯铑酸铵为原料,与一氧化碳气体和乙酰丙酮反应,得乙酰丙酮二羰基铑,乙酰丙酮二羰基铑加入三苯基膦,生成三苯基膦乙酰丙酮羰基铑。过滤沉淀,先用水洗,再用有机试剂洗涤,实施例得到三苯基膦乙酰丙酮羰基铑产物,收率可达94%以上、纯度99%;
(2)本发明采用氯铑酸铵作为起始原料,相较于氯化铑等其它铑盐,醇溶液酸度适中,无需加碱中和,不会引入杂质,从而在高产率的前提下,还能够保证产品的高纯度;
(3)此外本发明提供的方法还具有对操作人员友好,母液处理简单,产生废液少,废液中的铑容易回收,生产成本低等优点,非常适于工业化大规模生产。
附图说明
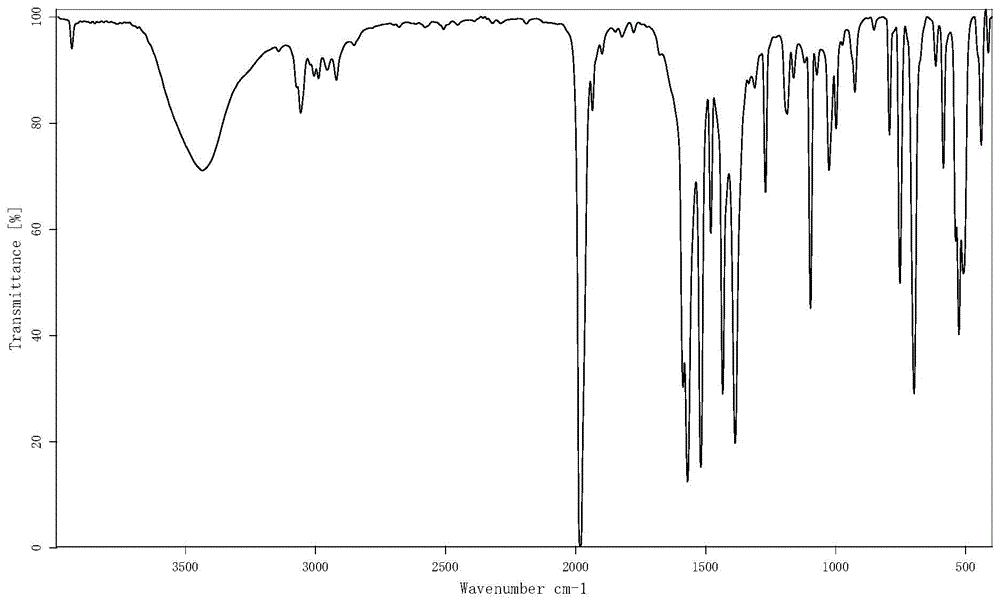
图1为三苯基膦乙酰丙酮羰基铑的红外图谱图。
图2为三苯基膦乙酰丙酮羰基铑(I)的1H-NMR谱图。
图3为三苯基膦乙酰丙酮羰基铑(I)的13C-NMR谱图。
具体实施方式
实施例1
取1000克氯铑酸铵,铑含量27.8%,用27升无水乙醇溶解,置于反应釜中,将CO通气管插入到溶液底部,开启搅拌,先通氮气10分钟,赶走溶液和反应釜里的空气,再通入一氧化碳,通气速度以每秒钟鼓5个气泡,开启加热设定温度40℃,待观察到反应溶液变成透明的淡黄色,关闭一氧化碳,关闭加热,冷却至室温,过滤,滤液再抽入反应釜中,开启搅拌,加入650克乙酰丙酮,继续反应60分钟,缓慢加入固体三苯基膦673克,加完三苯基膦后在45℃反应2小时,过滤,先用水洗,洗至洗水用5%浓度的硝酸银检验后无白色沉淀,再用丙酮洗涤除去有机杂质,抽干,置于防爆真空干燥箱内干燥至恒重,得三苯基膦乙酰丙酮羰基铑1264克,产率95%。
实施例2
取3000克氯铑酸铵,铑含量27.8%,用27升无水乙醇溶解,置于反应釜中,将CO通气管插入到溶液底部,开启搅拌,先通氮气15分钟,赶走溶液和反应釜里的空气,再通入一氧化碳,通气速度为每秒钟鼓5个气泡。开启加热,设定温度45℃,待观察到反应溶液变成透明的淡黄色,关闭一氧化碳,关闭加热,冷却至室温,过滤,滤液再抽入反应釜中,开启搅拌,加入2110克乙酰丙酮,继续反应60分钟,缓慢加入固体三苯基膦2125克,加完三苯基膦后50℃反应2小时,过滤,先用水洗,洗至洗水用5%浓度的硝酸银检验后无白色沉淀,再用丙酮洗涤除去有机杂质,抽干,置于防爆真空干燥箱内干燥至恒重,得三苯基膦乙酰丙酮羰基铑3751克,产率94%。
实施例1-2制得的产品的铑含量及杂质测定
(1)根据GB/T34609.1-2017铑化合物化学分析方法第1部分,测定了样品铑含量为20.9%与计算值铑20.9%非常相近。
(2)根据GB/T34609.2-2017铑化合物化学分析方法第2部分,测定了样品杂质(见表1),表1显示产品具有有很高(99%)的纯度。
表1实施例1-2制得的三苯基膦乙酰丙酮羰基铑的ICP杂质测定结果
实施例1-2制得的产品的结构表征
(1)红外光谱分析
实施例1-2制得的三苯基膦乙酰丙酮羰基铑(I)的红外谱图见图1,从图谱的峰来看,表示样品中主要成分为三苯基膦乙酰丙酮羰基铑,与文献报道完全一致。
(2)核磁共振分析
如图2所示的三苯基膦乙酰丙酮羰基铑(I)的1H-NMR谱图和图3所示的三苯基膦乙酰丙酮羰基铑(I)的13C-NMR谱图显示,实施例1-2制得的三苯基膦乙酰丙酮铑(I)的核磁共振氢谱与碳谱均与文献报道一致。
从红外光谱和核磁共振氢谱来看,没有明显的杂峰,表明三苯基膦乙酰丙酮羰基铑纯度很高。
对比例1
取1000克水合三氯化铑(氯化铑水合物),铑含量38%,用37升无水乙醇溶解,置于反应釜中,将CO通气管插入到溶液底部,开启搅拌,先通氮气10分钟,赶走溶液和反应釜里的空气,再通入一氧化碳,通气速度以每秒钟鼓5个气泡,开启加热40℃,待观察到反应溶液变成透明的淡黄色,关闭一氧化碳,关闭加热,冷却至室温,过滤,滤液再抽入反应釜中,开启搅拌,加入961克乙酰丙酮,继续反应60分钟,缓慢加入固体三苯基膦961克,加完三苯基膦后在45℃反应2小时,过滤,先用水洗,洗至洗水用5%浓度的硝酸银检验后无白色沉淀,再用丙酮洗涤除去有机杂质,抽干,置于防爆真空干燥箱内干燥至恒重,得三苯基膦乙酰丙酮羰基铑1291克,产率71%。采用高效液相色谱分析纯度,纯度为93%。
对比例2
取1000克水合三氯化铑(氯化铑水合物),铑含量38%,用37升无水乙醇溶解,置于反应釜中,将CO通气管插入到溶液底部,开启搅拌,先通氮气10分钟,赶走溶液和反应釜里的空气,再通入一氧化碳,通气速度以每秒钟鼓5个气泡。,开启加热40℃,待观察到反应溶液变成透明的淡黄色,关闭一氧化碳,关闭加热,冷却至室温,过滤,滤液再抽入反应釜中,开启搅拌,加入961克乙酰丙酮,用碱将反应釜中溶液的PH值调到7左右,继续反应60分钟,缓慢加入固体三苯基膦968克,加完三苯基膦后在45℃反应2小时,过滤,先用水洗,洗至洗水用5%浓度的硝酸银检验后无白色沉淀,再用丙酮洗涤除去有机杂质,抽干,置于防爆真空干燥箱内干燥至恒重,得三苯基膦乙酰丙酮羰基铑1618克,产率89%。采用高效液相色谱分析纯度,纯度为91%。
实施例1-2与对比例1-2的产品产率及纯度对比如表2所示。
表2实施例1-2与对比例1-2的产品产率及纯度对比
由表1可知,本发明采用氯铑酸铵作为起始原料,相较于氯化铑等其它铑盐,醇溶液酸度适中,无需加碱中和,不会引入杂质,从而在高产率的前提下,还能够保证产品的高纯度,这对于合成工艺及产品量产来讲都至关重要。