一种优化的达比加群酯关键中间体合成工艺
文献发布时间:2024-04-18 19:59:31
技术领域
本发明涉及医药中间体的制备相关技术领域,具体为一种优化的达比加群酯关键中间体合成工艺。
背景技术
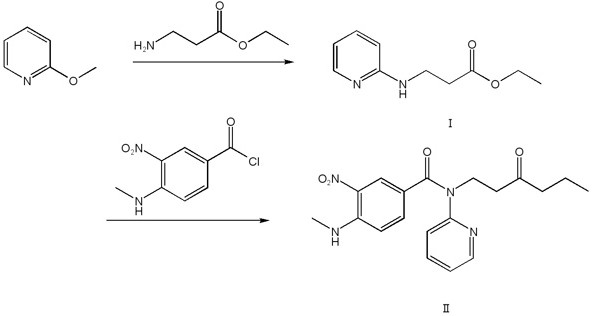
达比加群酯(英文名称Dabigatranetexilate,)是一种新型的直接凝血酶抑制剂,此药品是由德国勃林格殷格翰公司开发,于2008年4月在德国和英国率先上市,这是继华法林之后50年来上市的首个新类别口服抗凝血药物,而3-[(3-氨基-4-甲胺基苯甲酰基)(吡啶-2-基)氨基]丙酸乙酯(化合物II)是合成达比加群酯的一个关键中间体,而3-(吡啶-2-基氨基)丙酸乙酯(化合物I)是合成化合物II的关键原料。
目前的合成工艺合成化合物Ⅱ时必须合成化合物Ⅰ,即3-(吡啶-2-基氨基)丙酸乙酯,现有合成方法有:
路线一
林国强[CN1861596]等人报道了合成路线一,用2-氨基吡啶为起始原料,与丙烯酸乙酯发生迈克尔加成反应,一步合成得到3-(吡啶-2-基氨基)丙酸乙酯。尽管反应只需一步,但由于2-氨基吡啶的溶解性差,丙烯酸乙酯易聚合,3-(吡啶-2-基氨基)丙酸乙酯的活性及溶解性优于2-氨基吡啶,易进一步反应,得到化合物3,增加了分离纯化的难度,最终分离收率低,不到50%,产品纯度低。
路线二
(2) 邢松松 【中国医药杂志,41(5) ,321-325;2010】 等报道了合成路线二,该方法是先合成2-氯吡啶N-氧化物和β-氨基丙酸乙酯盐酸盐;再由2-氯吡啶N-氧化物与β-氨基丙酸乙酯盐酸盐反应生成3-(吡啶-2-基氨基)丙酸乙酯N-氧化物;最后经Pd/C催化氢化生成3-(吡啶-2-基氨基)丙酸乙酯,收率低,耗时长。且由于该方法使用了3-氨基丙酸乙酯盐酸盐,它在碱性条件下解离成不稳定的游离3-氨基丙酸乙酯,在加热的条件下易自身缩合,这必然会降低收率,增加分离难度,文献报道要用柱层析分离就已经证明了这一点;同时还要用到髙沸点的叔戊醇,不利于溶剂回收,
发明内容
针对上述合成达比加群酯关键中间体化合物的原料合成工艺的不足,本发明提供一种优化的达比加群酯关键中间体合成工艺,通过2-甲氧基吡啶的醚键与胺基化合物3-氨基丙酸乙酯之间的反应的,高收率,高纯度的得到化合物Ⅰ。再通过化合物Ⅰ合成达比加群酯关键中间体,使得产物收率得到有效提升,同时降低反应产物提纯处理的难度。
本发明反应路线如下所示:
本发明技术方案如下所示:
一种优化的达比加群酯关键中间体合成工艺,包括以下步骤:
第一步,氮气保护下,低温将催化剂溶于有机溶剂中,搅拌混合后,室温下加入2-甲氧基吡啶以及3-氨基丙酸乙酯,加热反应,反应结束后,分离产物,得到化合物Ⅰ;
第二步,在氮气保护的条件下,将上一步所得化合物Ⅰ加入到250mL有机溶剂中,加入反应物化合物3-硝基-4-甲胺基-苯甲酰氯,加入碱催化剂,加入干燥剂,加热回流反应,得到化合物Ⅱ。
进一步的,所述第一步反应所用溶剂为四氢呋喃。
进一步的,所述第二步反应所用溶剂为二氯甲烷和氯仿中的一种。
进一步的,所述第一步反应所用催化剂为正丁基锂,所述正丁基锂存在形式为2.5mol/L的己烷溶液。
进一步的,所述第一步反应反应温度为60~65℃,优选为60℃。
进一步的,所述第一步反应所用时间为30~50min,优选为30min。
进一步的,所述第二步反应所用碱催化剂为吡啶或三乙胺。
进一步的,所述第一步反应各物质的摩尔量之比为2-甲氧基吡啶:3-氨基丙酸乙酯:催化剂=1:1.6:2.0~2.2,优选为1:1.6:2.1。
进一步的,所述第二步反应各物质的摩尔比为化合物Ⅰ:3-硝基-4-甲胺基-苯甲酰氯:碱催化剂=1:1:2~4,优选为1:3。
本发明的有益效果是:1.通过2-甲氧基吡啶的醚键与胺基化合物3-氨基丙酸乙酯之间的反应的,高收率,高纯度的得到关键中间体化合物Ⅱ的关键原料化合物Ⅰ,有效提高反应的收率;2.同时,本发明工艺步骤少,可以简化合成工艺,减少反应步骤,使其更加适应但大规模工业化生产的需求;3.本发明的反应原料易得,反应产物分离简单,副产物便于分离,也可以有效的提高化合物Ⅰ以及最终产物化合物Ⅱ的纯度。
附图说明
图1为本发明的工艺路线流程示意图;
图2为本发明的化合物Ⅰ与化合物Ⅱ的化学结构示意图;
图3为本发明的路线一工艺路线流程示意图;
图4为本发明路线二工艺路线流程示意图。
具体实施方式
下面将结合本发明实施例及附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
第一步,氮气保护下,-78℃下将0.21mol(2.5mol/L己烷溶液)正丁基锂加入200ml四氢呋喃中,搅拌15min后升温至室温,加入0.1mol(10.9g)2-甲氧基吡啶,继续加入0.16mol的3-氨基丙酸乙酯,逐渐加热至60℃,60℃下加热反应30min,反应结束后,缓慢加入无水丙醇淬灭反应,然后蒸发溶剂,加入100ml水与100ml二氯甲烷溶解,溶液用二氯甲烷萃取2~3次,合并有机相,有机相用饱和食盐水洗涤2~3次,蒸发溶剂,产物用50ml甲苯重结晶,得到18.8g化合物Ⅰ,收率96.9%,纯度98.5%;
第二步,在氮气保护的条件下,将上一步所得0.1mol(19.4g)化合物Ⅰ加入到250mL有机溶剂二氯甲烷中,加入0.3mol碱催化剂三乙胺,加入0.1mol化合物3-硝基-4-甲胺基-苯甲酰氯,加入无水硫酸钠0.2mol,搅拌回流反应4~6h,待反应完成后,趁热过滤,滤渣用二氯甲烷洗涤2~3次,向滤液中缓慢加入100ml蒸馏水,加入稀盐酸中和至中性,分离有机相,有机相用饱和食盐水洗涤2~3次,无水硫酸钠干燥,减压蒸发浓缩,产物继续用60ml甲苯重结晶,得到32.8g化合物Ⅱ,收率96.1%,纯度98.6%。
第一步,氮气保护下,-78℃下将0.20mol(2.5mol/L己烷溶液)正丁基锂200ml加入四氢呋喃中,搅拌15min后升温至室温,加入0.1mol(10.9g)2-甲氧基吡啶,继续加入0.16mol的3-氨基丙酸乙酯,逐渐加热至60℃,60℃下加热反应30min,反应结束后,缓慢加入无水丙醇淬灭反应,然后蒸发溶剂,加入100ml水与100ml二氯甲烷溶解,溶液用二氯甲烷萃取2~3次,合并有机相,有机相用饱和食盐水洗涤2~3次,蒸发溶剂,产物用50ml甲苯重结晶,得到18.5g化合物Ⅰ,收率95.3%,纯度98.3%。
第一步,氮气保护下,-78℃下将0.22mol(2.5mol/L己烷溶液)正丁基锂加入200ml四氢呋喃中,搅拌15min后升温至室温,加入0.1mol(10.9g)2-甲氧基吡啶,继续加入0.16mol的3-氨基丙酸乙酯,逐渐加热至60℃,60℃下加热反应30min,反应结束后,缓慢加入无水丙醇淬灭反应,然后蒸发溶剂,加入100ml水与100ml二氯甲烷溶解,溶液用二氯甲烷萃取2~3次,合并有机相,有机相用饱和食盐水洗涤2~3次,蒸发溶剂,产物用50ml甲苯重结晶,得到18.7g化合物Ⅰ,收率96.3%,纯度98.6%.
第一步,氮气保护下,-78℃下将0.21mol(2.5mol/L己烷溶液)正丁基锂加入200ml四氢呋喃中,搅拌15min后升温至室温,加入0.1mol(10.9g)2-甲氧基吡啶,继续加入0.18mol的3-氨基丙酸乙酯,逐渐加热至60℃,60℃下加热反应30min,反应结束后,缓慢加入无水丙醇淬灭反应,然后蒸发溶剂,加入100ml水与100ml二氯甲烷溶解,溶液用二氯甲烷萃取2~3次,合并有机相,有机相用饱和食盐水洗涤2~3次,蒸发溶剂,产物用50ml甲苯重结晶,得到18.8g化合物Ⅰ,收率96.9%,纯度98.7%。
第二步,在氮气保护的条件下,将上一步所得0.1mol(19.4g)化合物Ⅰ加入到250mL有机溶剂二氯甲烷中,加入0.4mol碱催化剂三乙胺,加入0.1mol化合物3-硝基-4-甲胺基-苯甲酰氯,加入无水硫酸钠0.2mol,搅拌回流反应4~6h,待反应完成后,趁热过滤,滤渣用二氯甲烷洗涤2~3次,向滤液中缓慢加入100ml蒸馏水,加入稀盐酸中和至中性,分离有机相,有机相用饱和食盐水洗涤2~3次,无水硫酸钠干燥,减压蒸发浓缩,产物继续用60ml甲苯重结晶,得到32.9g化合物Ⅱ,收率96.7%,纯度98.7%。
第二步,在氮气保护的条件下,将上一步所得0.1mol(19.4g)化合物Ⅰ加入到250mL有机溶剂氯仿中,加入0.3mol碱催化剂三乙胺,加入0.1mol化合物3-硝基-4-甲胺基-苯甲酰氯,加入无水硫酸钠0.2mol,搅拌回流反应4~6h,待反应完成后,趁热过滤,滤渣用二氯甲烷洗涤2~3次,向滤液中缓慢加入100ml蒸馏水,加入稀盐酸中和至中性,分离有机相,有机相用饱和食盐水洗涤2~3次,无水硫酸钠干燥,减压蒸发浓缩,产物继续用60ml甲苯重结晶,得到32..2g化合物Ⅱ,收率94.6%,纯度97.8%。
第二步,在氮气保护的条件下,将上一步所得0.1mol(19.4g)化合物Ⅰ加入到250mL有机溶剂二氯甲烷中,加入0.3mol碱催化剂吡啶,加入0.1mol化合物3-硝基-4-甲胺基-苯甲酰氯,加入无水硫酸钠0.2mol,搅拌回流反应4~6h,待反应完成后,趁热过滤,滤渣用二氯甲烷洗涤2~3次,向滤液中缓慢加入100ml蒸馏水,加入稀盐酸中和至中性,分离有机相,有机相用饱和食盐水洗涤2~3次,无水硫酸钠干燥,减压浓缩,产物继续用60ml甲苯重结晶,得到30.9g化合物Ⅱ,收率90.9%,纯度98.3%。
对于本领域技术人员而言,显然本发明不限于上述示范性实施例的细节,而且在不背离本发明的精神或基本特征的情况下,能够以其他的具体形式实现本发明。因此,无论从哪一点来看,均应将实施例看作是示范性的,而且是非限制性的,本发明的范围由所附权利要求而不是上述说明限定,因此旨在将落在权利要求的等同要件的含义和范围内的所有变化囊括在本发明内。不应将权利要求中的任何附图标记视为限制所涉及的权利要求。
此外,应当理解,虽然本说明书按照实施方式加以描述,但并非每个实施方式仅包含一个独立的技术方案,说明书的这种叙述方式仅仅是为清楚起见,本领域技术人员应当将说明书作为一个整体,各实施例中的技术方案也可以经适当组合,形成本领域技术人员可以理解的其他实施方式。