一种低温水解制备草甘膦的方法及装置
文献发布时间:2024-04-18 20:00:25
技术领域
本发明属于草甘膦农药技术领域,尤其涉及一种低温水解制备草甘膦的方法及装置。
背景技术
草甘膦,别名N-(膦酸甲基)甘氨酸(简写为PMG),是孟山都开发的一种低毒的内吸传导型广谱灭生性除草剂。当前,国内外主流的生产路线有两种,一种是以亚氨基二乙酸(IDA)为原料,该工艺过程简单,工艺条件较缓和,产品收率较高,但由于对设备要求高,原料的限制性,国外居多;一种是以亚磷酸二烷基酯、甘氨酸为原料,也称甘氨酸法,是我国主流的生产工艺,占比约70%。该法生产出的草甘膦纯度高且工艺稳定,符合我国产业结构与国情。
近年来,随着环保核查与清洁生产政策制度的不断提高,为巩固行业的龙头地位,大中小行业针对该法从母液减量化与资源化、C、N、P 3种主要元素的利用率、装置的自动化与连续化等方面进行了大量研究。然而原子利用率低、副产物多、能耗高的问题,仍然客观存在。为解决此问题,本发明小组在先前的研究中通过明确反应机理,分别从原料配比优化、合成脱水、各阶段温度与PH精准调控、以及母液科学套用等方面,提高甘氨酸的转换率、有效避免酯化阶段的其他含有甲氧基的副产水解,从而提升草甘膦的定向生成,实现草甘膦原粉收率突破77%。但是没有从源头解决母液量大问题。此外,由于使用该方法合成草甘膦需要使用大量的甲醇和三乙胺,后续回收处理过程复杂,费用较高,这也增加了草甘膦生产成本,且环保压力较大。
专利CN 116041388 A提供了一种草甘膦水解的酸解脱醇方法,通过梯级反应段靠压力差汽提,同时解决传统间歇式水解生产效率低,综合能耗高的问题。但是由于草甘膦酸解反应的特点,冷凝液与原母液三乙胺回收过程仍然需要加入大量的碱中和,导致酸碱的浪费。且脱溶水解仍然在较高的温度进行,副产物生成量并没有降低。
发明内容
有鉴于此,本发明的目的在于提供一种低温水解制备草甘膦的方法及装置,该方法采用鼓入空气除甲醛,优于传统氧化工艺,温度调控大幅缩短了酸解脱溶时间,较传统工艺缩短20~35%;减少酸碱消耗,降低母液废水与废盐的生成,同时实现无水体系溶剂回收。
本发明提供了一种低温水解制备草甘膦的方法,包括以下步骤:
1)将甘氨酸、烷基酯和多聚甲醛在甲醇和叔胺的存在下,反应后得到合成液S1;
2)将所述合成液S1进行真空脱溶,得到缩合液S2;
3)将所述缩合液S2和盐酸混合,进行酸化水解反应,酸化过程鼓入空气,升温脱酸,再调节pH和降温,结晶,水洗并干燥后得到草甘膦。
优选地,所述步骤1)中多聚甲醛、甲醇、三乙胺、甘氨酸和烷基酯的摩尔比为(1.5~1.9):(5-10):(0.7~1.1):1:(0.8~1.05)。
优选地,步骤2)脱溶过程中,甲醇和叔胺的脱除率均为40~70wt%;
脱溶的温度为80~110℃,脱溶的时间小于等于100s。
优选地,步骤2)中真空脱溶采用的真空度为0.015~0.04MPa。
优选地,所述缩合液S2补加甲醇后和酸混合;
补加的甲醇和甘氨酸的摩尔比为(1.5~3.3):1。
优选地,酸为盐酸;
甘氨酸和盐酸的摩尔比为1:(1.8~2.3);
所述盐酸的质量浓度为30~32%。
优选地,所述缩合液S2以滴加的方式和酸混合;
酸化水解反应的时间为35~45min,温度为50~70℃。
优选地,升温脱酸的温度控制在90~113℃区间内;
结晶的温度≤30℃。
优选地,水洗得到原母液和洗涤用水,部分洗涤用水作为结晶后离心一次水洗水套用。
本发明提供了一种用于生产上述技术方案所述方法制备的草甘膦的装置,包括依次设置的合成反应釜、缩合液旋转单元、缩合液储罐、酸化水解反应釜和结晶反应釜;
所述酸化水解反应釜还连接酸回收单元。
本发明提供了一种低温水解制备草甘膦的方法,包括以下步骤:1)将甘氨酸、烷基酯和多聚甲醛在甲醇和叔胺的存在下,反应后得到合成液S1;2)将所述合成液S1进行真空脱溶,得到缩合液S2;3)将所述缩合液S2和盐酸混合,进行酸化水解反应,酸化过程鼓入空气,升温脱酸,再调节pH和降温,结晶,水洗并干燥后得到草甘膦。该方法与传统方法相比,具有以下有益效果:一、降低反应终点,提高元素利用率,使草甘膦收率与品质得到极大地提升;二、大幅度减少了精馏设备占地面积,降低脱酸与溶剂回收能耗,实现稀酸回用,促进绿色循环;三、缩减盐酸和碱的消耗以及废水排放量,在草甘膦行业节能降耗减碳方面具有深远意义;具体盐酸与液碱单耗分别降低28~35%与55~60%,水解脱酸时间减少20~35%。
附图说明
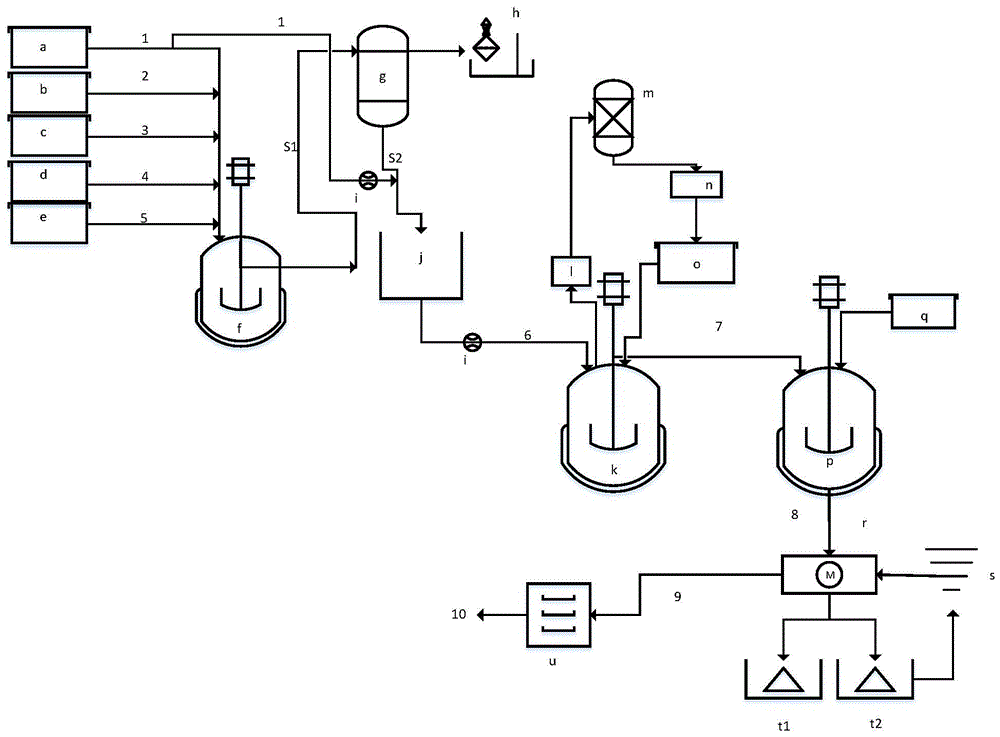
图1为本发明生产草甘膦的装置示意图。
具体实施方式
本发明提供了一种低温水解制备草甘膦的方法,包括以下步骤:
1)将甘氨酸、烷基酯和多聚甲醛在甲醇和叔胺的存在下,反应后得到合成液S1;
2)将所述合成液S1进行常压或真空脱溶,得到缩合液S2;
3)将所述缩合液S2和盐酸混合,进行酸化水解反应,酸化过程鼓入空气,升温脱酸,再调节pH和降温,结晶,水洗并干燥后得到草甘膦。
本发明通过降低酸化水解反应终点,提高元素利用率,使草甘膦收率与品质得到极大提升;大幅度减少精馏设备占地面积,降低脱酸与溶剂回收能耗,实现稀酸回用,促进绿色循环;缩减盐酸和碱的消耗以及废水排放量,在草甘膦行业节能降耗减碳等方面具有深远意义。本申请中盐酸和液碱单耗分别降低28~35%与55~60%;水解脱酸的时间减少20~35%。
本发明将甘氨酸、烷基酯和多聚甲醛在甲醇和叔胺的存在下,反应后得到合成液S1。
在本发明中,所述烷基酯为亚磷酸二甲酯;叔胺为三乙胺。本发明以甘氨酸、烷基酯和多聚甲醛为原料,甲醇和叔胺分别为溶剂和催化剂,在一定温度和pH值下经解聚、加成与缩合得到合成液S1。
在本发明中,所述步骤1)中多聚甲醛、甲醇、三乙胺、甘氨酸和烷基酯的摩尔比为(1.5~1.9):(5-10):(0.7~1.1):1:(0.8~1.05)。
本发明优选将甲醇和三乙胺加入至反应釜中,再将多聚甲醛先加入至反应釜中,在32~42℃下进行解聚反应,待多聚甲醛完全溶解后,再将甘氨酸加入至反应釜中,在温度37~52℃下反应30~55min;待反应结束后加入烷基酯,在47~60℃下反应,保温8~10min后补加三乙胺和烷基酯,调节体系pH值为7.5~7.8,反应50~100min,得到合成液S1。为了调控亚磷酸二甲酯的选择性,减少水解副反应,同时控制体系酸碱度,加速甘氨酸成盐,促进正向反应过程;本发明中三乙胺和烷基酯分批次加入,缩合过程中补加量分别占三乙胺和烷基酯所需总量的4~6%和2~4%。
得到合成液S1后,本发明将所述合成液S1进行常压或真空脱溶,得到缩合液S2。本发明中脱溶将甲醇和叔胺进行脱除;优选采用旋蒸、闪蒸和分子蒸馏等类似方式,实现快速脱除,优选采用旋蒸方式。脱溶可以在常压下进行,也可以在真空条件下进行;本发明优选在真空或者微真空状态下进行脱溶;真空脱溶采用的真空度为0.015~0.04MPa。所述脱溶的温度为80~110℃,脱溶的时间小于等于100s;所述脱溶的温度优选为85~95℃,脱溶的时间优选为70~100s。本发明优选脱溶过程中,甲醇和叔胺的脱除率均为40~70wt%;优选的,脱溶过程中,甲醇的脱除率为52~60%,叔胺的脱除率为40~50%。具体实施例中,脱溶的温度为110℃或90℃;脱溶的时间为90s,脱溶的真空度为0.035MPa、或0.025MPa。
得到缩合液S2后,本发明将所述缩合液S2和酸混合,进行酸化水解反应,酸化过程鼓入空气,升温脱酸,再调节pH和降温,结晶,水洗并干燥后得到草甘膦。
本发明优选将所述缩合液S2补加甲醇后和酸混合;所述补加的甲醇和甘氨酸的摩尔比为(1.5~3.3):1;具体实施例中,补加的甲醇和甘氨酸的摩尔比为2.8:1、2.2:1。本发明补加少量甲醇,升温过程随着甲醇的脱除,甲醛夹带除体系。
本发明采用的酸优选为盐酸;所述甘氨酸和盐酸的摩尔比为1:(1.8~2.3);所述盐酸的质量浓度为30~32%,优选为30.5~31.5%。具体实施例中,盐酸的质量浓度为30.3%、31.2%、31.3%。
所述缩合液S2无需经过冷冻盐水降温直接与酸混合酸化;本发明中缩合液S2优选以滴加的方式和酸混合,滴加与酸化同时进行;酸化水解反应的时间为35~45min,酸化水解反应的温度为50~70℃。
本发明在酸化过程中需要辅助鼓入空气进行氧化,待缩合液S2与盐酸混合完全后停止鼓入空气。本发明优选采用补加甲醇夹带和空气氧化共同除甲醛的方式,优于单一的空气氧化,能够提高草甘膦的收率,还能改善浆料的流动性。
酸化水解反应结束后,本发明优选升温脱酸;升温的时间为45~60min;升温脱酸的温度控制在90~113℃区间内;具体实施例中,升温的时间为5min,升温脱酸的终点温度为110℃。本发明中酸化水解的时间和脱溶脱酸的总时间为80~100min。具体实施例中,酸化水解的时间为40min,脱酸的时间为50min。
本发明优选将升温脱酸过程中回收的酸性气体经冷凝回收至酸吸收塔作为补水回用,循环至需要的浓度,直接作为产品回用至体系。
升温脱酸结束后,将计量好的液碱加入至反应釜中调节pH,中和后打入结晶釜进行结晶;结晶的温度≤30℃,优选低于20℃;结晶的时间为3~7小时。结晶得到的结晶液进行离心和水洗,得到的原母液和洗涤用水分开收集,部分洗涤用水作为离心一次水洗水套用。
水洗后进行干燥,得到草甘膦。本发明对制备得到的草甘膦采用GB/T12686-2017规定的方法进行分析,纯度达到96.5%以上,草甘膦原粉收率达到78.0%以上。
本发明提供的上述工艺突破了现有工艺,先进行溶剂脱溶再进行酸性水解,同时酸性水解过程回收酸,同步实现提浓套用,不但可以显著降低水解与溶剂精馏回收过程能耗,抑制副产物生成,大幅度提升了原子利用率,同时可以降低酸碱消耗,降低母液量。
本发明还提供了一种用于生产上述技术方案所述方法制备的草甘膦的装置,包括依次设置的合成反应釜、缩合液旋转单元、缩合液储罐、酸化水解反应釜和结晶反应釜;
所述酸化水解反应釜还连接盐酸回收单元。
图1为本发明甘氨酸-烷基酯法生产草甘膦的装置示意图,其中,a-甲醇计量槽、b-三乙胺计量槽、c-亚磷酸二甲酯计量槽、d-甘氨酸计量槽、e-多聚甲醛计量槽、f-合成反应釜、g-缩合液旋转设备、h-醇胺储罐、i-流量调节阀、j-缩合液储罐、k-酸化水解反应釜、l-稀酸冷凝罐、m-盐酸吸收塔、n-盐酸大槽、o-盐酸计量槽、p-结晶反应釜、q-液碱计量槽、r-离心机、s-工业水槽、t1-原母液槽、t2-洗涤水槽、u-干燥机;
1-甲醇管、2-三乙胺管、3-亚磷酸二甲酯管、4-甘氨酸管、5-多聚甲醛管、S1-缩合液管、S2-浓缩缩合液管、6-缩合液S2与甲醇混合管路、7-水解液、8-结晶液、8-草甘膦湿粉、9-草甘膦成品。
在本发明中,合成反应釜的底部通过管道与缩合液旋转单元相联;缩合液旋转单元的底部与缩合液储罐相联;所述缩合液储罐的底部与酸化水解反应釜的顶部相联;酸化水解反应釜的底部与结晶反应釜的顶部相联。
酸化水解反应釜还连接有酸回收单元。所述酸回收单元包括依次串联的稀酸冷凝罐、酸吸收塔和酸循环吸收槽,酸循环吸收槽连接至酸计量槽的顶部。脱酸气体经冷凝回收至酸吸收塔作为补水回用。
在本发明中,所述合成反应釜的顶部分别与甲醇、三乙胺、多聚甲醛、甘氨酸和烷基酯的计量槽相联;甲醇计量槽的底部还与缩合液储罐相联;流量调节阀分别串联在甲醇计量槽至缩合液储罐、缩合液储罐至水解反应釜的管道上。本发明通过流速调节,控制反应选择性。
结晶反应釜的顶部还连接液碱计量槽,底部通过管道连接至离心机的顶部;离心机的顶部还连接工业水槽;离心机的底部连接原母液槽、洗涤水槽和草甘膦洗料装置。所述洗涤水槽的底部连接至工业水槽,作为部分工业水套用。
采用上述装置采用甘氨酸-烷基酯法生产草甘膦,通过先脱溶,再酸化水解,使得反应终点温度降低,提高了元素利用率,草甘膦的品质和收率均得到提升;大幅度减少了精馏设备占地面积,降低脱酸与溶剂回收能耗,实现稀酸回用,促进绿色循环。
为了进一步说明本发明,下面结合实施例对本发明提供的一种低温水解制备草甘膦的方法及装置进行详细地描述,但不能将它们理解为对本发明保护范围的限定。
装置实施例
合成反应釜底部通过管道与浓缩液旋蒸设备相联,旋蒸设备底部联接浓缩液储罐;浓缩液储罐底部通过管道与水解反应釜顶部相联;
合成反应釜顶部与甲醇、三乙胺、多聚甲醛、甘氨酸和亚磷酸二甲酯的计量槽相联;同时甲醇计量槽底部还与浓缩液储罐相连,流量调节阀分别串联在甲醇计量槽至缩合液储罐,缩合液储罐至水解反应釜的管道上。通过流速调节,控制反应选择性;
水解反应釜底部与结晶反应釜顶部相联,同时联接盐酸回收装置;结晶反应釜顶部联接液碱计量槽,底部通过管道联接至离心机顶部,顶部还联接有工业水槽;离心机底部同时联接母液槽、洗涤水槽与草甘膦洗料装置,湿料进一步烘干得到草甘膦成品;
稀酸回收装置由稀酸冷凝罐、盐酸吸收塔,盐酸循环吸收大槽组成,依次串联,盐酸循环吸收大槽联接至盐酸计量槽顶部;
洗涤水槽底部还联接至工业水槽,作为部分工业水套用。
对比例1
甲醇和三乙胺计量槽中分别备料22.0mol和2.15mol,打入合成反应釜,开启搅拌。然后将备料好的多聚甲醛(4.15mol)打入反应釜,控温38℃开始解聚反应。多聚甲醛完全溶解之后,将备料好的2.31mol甘氨酸打入,控温42℃反应53分钟。待反应结束,将计量槽备好的2.68mol亚磷酸二甲酯分批次快速加入合成反应釜,控温53℃,保温8min后分别补加三乙胺0.095mol与亚磷酸二甲酯0.062mol,调解体系pH为7.65,反应80分钟,反应结束即可得到合成液S1。然后将合成液S1打入水解反应釜,60℃加入计量好的盐酸7.30mol开始水解反应,然后升温开加热套脱去甲醇和其他副产物,控制升温脱溶时间100分钟,反应终点125℃。最后加入计量好的液碱中和后打入结晶反应釜,降温至30℃进行结晶反应,后经水洗,干燥,得到草甘膦成品。
固体草甘膦收率达到75.27%,母液与草甘膦原粉的总收率达到79.82%,草甘膦成品含量95.71%。
对比例2
甲醇、三乙胺与多聚甲醛计量槽中分别备料22.0mol、2.15mol和4.15mol,打入合成反应釜,开启搅拌,控温38℃开始解聚反应。多聚甲醛完全溶解之后,将备料好的2.31mol甘氨酸打入,控温42℃反应53分钟。待反应结束,将计量槽备好的2.38mol亚磷酸二甲酯分批次快速加入合成反应釜,控温53℃,保温8min后分别补加三乙胺0.095mol与亚磷酸二甲酯0.062mol,调解体系pH为7.65,反应80分钟,反应结束即可得到合成液S1。将合成液S1打入缩合液旋蒸分离装置进行常压脱醇、脱胺,控制脱溶时间80s,温度145℃,脱溶结束后得到S2。于水解反应釜加入4.80mol,含量为30.3%的盐酸,控制60℃开始酸化反应,酸化滴加反应时间40分钟,混合结束开始升温脱酸,升温反应时间50分钟,终点温度110℃。最后加入计量好的液碱中和后打入结晶反应釜,降温至30℃进行结晶反应4h,后经水洗,干燥,得到草甘膦成品。
固体草甘膦收率达到74.35%,母液与草甘膦原粉的总收率为79.35%,草甘膦成品含量94.15%。
对比例3
甲醇、三乙胺与多聚甲醛计量槽中分别备料22.0mol、2.15mol和4.15mol,打入合成反应釜,开启搅拌,控温38℃开始解聚反应。多聚甲醛完全溶解之后,将备料好的2.31mol甘氨酸打入,控温42℃反应53分钟。待反应结束,将计量槽备好的2.38mol亚磷酸二甲酯分批次快速加入合成反应釜,控温53℃,保温8min后分别补加三乙胺0.095mol与亚磷酸二甲酯0.062mol,调解体系pH为7.65,反应80分钟,反应结束即可得到合成液S1。将合成液S1打入缩合液旋蒸分离装置进行微负压脱醇、脱胺,控制脱溶时间300s,温度110℃,真空度0.035MPa,脱溶结束后得到缩合液S2。于水解反应釜加入4.80mol,含量为30.3%的盐酸,控制60℃开始酸化反应,酸化滴加反应时间40分钟,混合结束开始升温脱酸,升温反应时间50分钟,终点温度110℃。最后加入计量好的液碱中和后打入结晶反应釜,降温至30℃进行结晶反应4h,后经水洗,干燥即可得到草甘膦成品。
固体草甘膦收率为73.8%,母液与草甘膦原粉的总收率为78.13%,草甘膦成品含量93.97%。
实施例1
甲醇、三乙胺与多聚甲醛计量槽中分别备料22.0mol、2.15mol和4.15mol,打入合成反应釜,开启搅拌,控温38℃开始解聚反应。多聚甲醛完全溶解之后,将备料好的2.31mol甘氨酸打入,控温42℃反应53分钟。待反应结束,将计量槽备好的2.38mol亚磷酸二甲酯分批次快速加入合成反应釜,控温53℃,保温8min后分别补加三乙胺0.095mol与亚磷酸二甲酯0.062mol,调解体系pH为7.65,反应80分钟,反应结束即可得到合成液S1。将合成液S1打入缩合液旋蒸分离装置进行微负压脱醇、脱胺,控制脱溶时间90s,温度90℃,真空度0.025MPa,脱溶结束后得到缩合液S2。于水解反应釜加入4.80mol,含量为30.3%的盐酸,控制60℃开始酸化反应,酸化滴加反应时间40分钟,混合结束开始升温脱酸,升温反应时间50分钟,终点温度110℃。最后加入计量好的液碱中和后打入结晶反应釜,降温至30℃进行结晶反应4h,后经水洗,干燥即可得到草甘膦成品。
固体草甘膦收率可以达到74.83%,母液与草甘膦原粉的总收率为80.03%,草甘膦成品含量94.53%。
实施例2
甲醇、三乙胺与多聚甲醛计量槽中分别备料22.0mol、2.15mol和4.15mol,打入合成反应釜,开启搅拌,控温38℃开始解聚反应。多聚甲醛完全溶解之后,将备料好的2.31mol甘氨酸打入,控温42℃反应53分钟。待反应结束,将计量槽备好的2.38mol亚磷酸二甲酯分批次快速加入合成反应釜,控温43℃,保温8min后分别补加三乙胺0.095mol与亚磷酸二甲酯0.062mol,调解体系pH为7.65,反应80分钟,反应结束即可得到合成液S1。将合成液S1打入缩合液旋蒸分离装置进行微负压脱醇、脱胺,控制脱溶时间90s,温度90℃,真空度0.025MPa,脱溶结束后得到S2。于水解反应釜加入4.80mol,含量为30.3%的盐酸,控制60℃开始酸化反应,酸化过程中采取鼓空气氧化,促进甲醛转化,酸化滴加反应时间40分钟,混合结束开始升温脱酸,升温反应时间50分钟,终点温度110℃。最后加入计量好的液碱中和后打入结晶反应釜,降温至30℃进行结晶反应4h,后经水洗,干燥即可得到草甘膦成品。
固体草甘膦收率达到75.35%,母液与草甘膦原粉的总收率为80.82%,草甘膦成品含量95.21%。
实施例3
本实施例与实施例2均采取辅助强化方式除酸化液体系中甲醛,其他条件不变,不同的是,采取补加甲醇的方式,甲醇:甘氨酸的摩尔比=2.8,在高温脱溶过程中随着甲醇的脱除,甲醛夹带出体系。固体草甘膦收率达到75.83%,母液与草甘膦原粉的总收率达到81.25%,草甘膦成品含量95.9%。
实施例4
本实施例与实施例3不同的是,采取补加甲醇配合空气氧化的方式共同除甲醛,甲醇:甘氨酸的摩尔比=2.2。固体草甘膦收率达到76.47%,母液与草甘膦原粉的总收率为81.7%,草甘膦成品含量96.52%。
实施例5
本实施例与实施例4相比,其他条件不变,不同的是,盐酸投加量由4.80mol降至3.8mol。固体草甘膦收率仅为74.35%,母液与草甘膦原粉的总收率78.99%,草甘膦成品含量95.08%。
实施例6
本实施例与实施例4相比,其他条件不变,不同的是,酸化盐酸控制含量31.2-31.3%。固体草甘膦收率达到76.75%,母液与草甘膦原粉的总收率达到81.88%,草甘膦成品含量96.83%。
实施例7
本实施例与实施例6相比,其他条件不变,不同的是,降温结晶过程控制终点析晶温度改为18℃,提升结晶率。固体草甘膦收率达到77.31%,母液与草甘膦原粉的总收率达到82.33%,草甘膦成品含量96.75%。
实施例8
本实施例与实施例7相比,其他条件不变,不同的是,草甘膦洗涤过程采用的工业水,50%替换为实施例7的工业洗涤用水的洗涤水套用。固体草甘膦收率达到78.04%,母液与草甘膦原粉的总收率达到83.54%,草甘膦成品含量96.58%。
表1实施例和对比例的草甘膦收率和纯度
由表1可知,对比例1采用现有的传统工艺,先水解酸化,在脱溶过程中进行溶剂回收,对比例2~3、实施例1~实施例8,均采取先缩合液脱溶,再中低温酸化工艺。从上表1以及实施案例数据可以显著看出,改进先脱溶后水解工艺可以降低酸消耗与能耗;实施案例2至实施例8进一步表明改进工艺辅助酸化过程强化除甲醛,可以达到甚至远高于传统工艺的收率,最优组别原粉收率与总收率分别高于现有工艺(对比例1)2.77%与3.72%。进一步,对比例1与对比例3和实施例1相比较,说明微负压缩合液脱溶相较于常压高温脱醇、脱胺更合适,且缩合液旋蒸时间不易过长,否则料液可能变质,粘性加强,影响流动性与收率。综合对比实施例2与实施例3和实施例4,实验组(先脱溶后水解),更优选必须强化辅助除甲醛手段,否则在酸解后期,甲醛作用下,目标草甘膦转变为增甘膦或者甲基草甘膦,影响含量和收率。实施例4与实施5对比说明,虽然三乙胺提前脱除,盐酸单耗可以下降,但是必须保证盐酸加入量促进草甘膦前体完全水解。实施例6~实施例8,分别对盐酸含量、结晶温度以及科学套用过程进一步优化,进一步提升结晶率与收率。
表2实施例和对比例的节能数据归纳如下
由以上实施例可知,本发明提供了一种低温水解制备草甘膦的方法,包括以下步骤:1)将甘氨酸、烷基酯和多聚甲醛在甲醇和叔胺的存在下,反应后得到合成液S1;2)将所述合成液S1进行常压或真空脱溶,得到缩合液S2;3)将所述缩合液S2和盐酸混合,进行酸化水解反应,酸化过程鼓入空气,升温脱酸,再调节pH和降温,结晶,水洗并干燥后得到草甘膦。该方法与传统方法相比,具有以下有益效果:一、降低反应终点,提高元素利用率,使草甘膦收率与品质得到极大地提升;二、大幅度减少了精馏设备占地面积,降低脱酸与溶剂回收能耗,实现稀酸回用,促进绿色循环;三、缩减盐酸和碱的消耗以及废水排放量,在草甘膦行业节能降耗减碳方面具有深远意义;具体盐酸与液碱单耗分别降低28~35%与55~60%,水解脱酸时间减少20~35%。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。