偶氮化合物及其盐的前药
文献发布时间:2024-04-18 20:00:50
技术领域
本发明涉及通式I的化合物及其治疗用途。
本公开还涉及通式I的化合物的药物组合物以及使用其进行治疗的方法。
背景技术
疼痛是一个复杂的多维概念,它有助于启动信号级联以响应任何有害刺激。在痛觉中,许多类型的受体被激活,它们的信号传导通路各不相同。这些信号传导通路可被视为通过靶向疼痛转导分子来调节疼痛的位点,从而在多种疼痛综合症中产生镇痛效果。
间质性膀胱炎/膀胱疼痛综合征(IC/BPS)、膀胱过度活动症(OAB)通常具有其它慢性盆腔疼痛病症的合并症,如IBS和子宫内膜异位症,以及更多全身性疼痛病症。外周炎症会产生多种炎性介质,如缓激肽、前列腺素(PGE2)、嘌呤(ATP)、蛋白酶和NGF,其作用于伤害性感觉神经元中表达的它们的同源受体以激活细胞内信号转导通路。
因此,本发明提供了可用于治疗间质性膀胱炎/膀胱疼痛综合征(IC/BPS)、膀胱过度活动症(OAB)、慢性咳嗽、子宫内膜异位症、神经性疼痛、偏头痛、瘙痒症、带状疱疹后遗神经痛、尿路感染相关疼痛、骨关节炎、关节炎和尿失禁的化合物。
附图说明
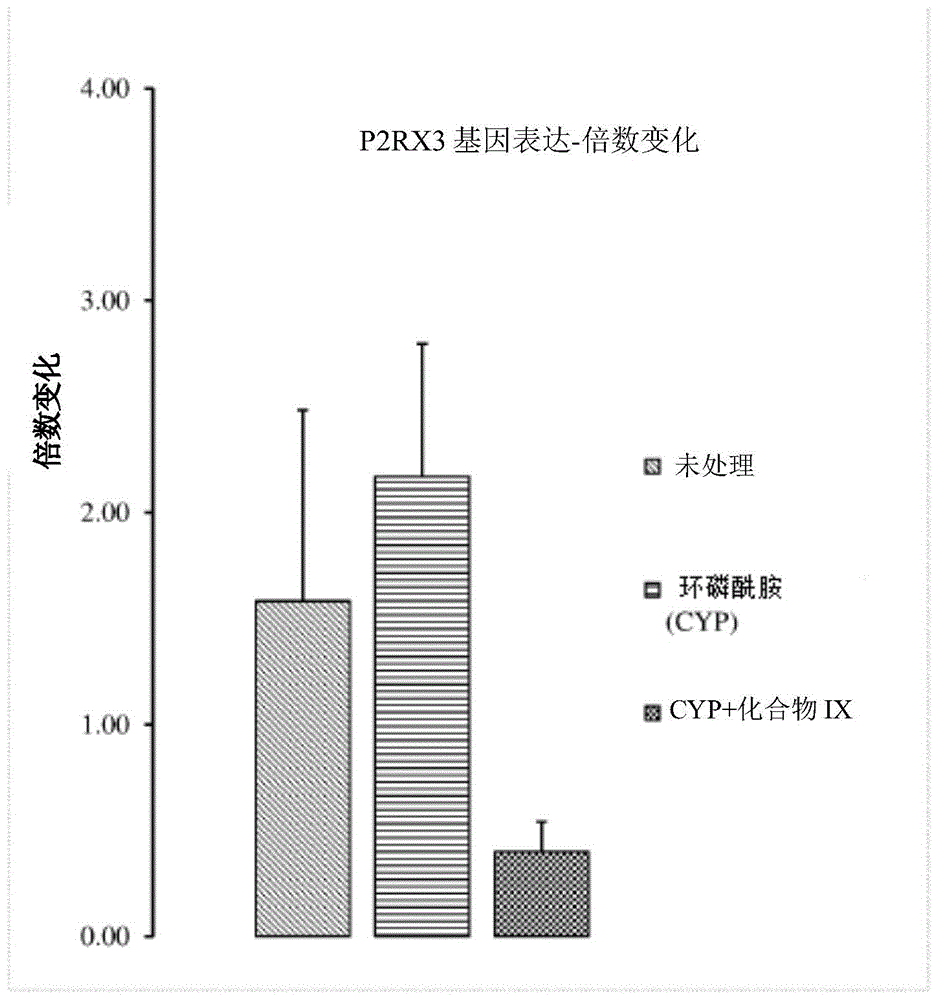
图1-使用从用测试药物化合物与环磷酰胺组合处理的RT4细胞中分离的RNA(随后的cDNA合成),通过定量RT-PCR进行基因表达分析。条形图表示P2RX3基因表达的倍数变化。所有值均以平均值±SEM表示图2-化合物XI对雄性SD大鼠中CFA诱导的机械性痛觉超敏的效果(使用上下法)%MPE;CFA注射后第2天。数据以%MPE(平均值±SEM)表示,****p<0.0001与CFA对照组相比,在双因素ANOVA然后是Bonferroni多重比较检验中,n=08。
图3.化合物XI对机械性痛觉超敏中平均缩爪阈值的效果。数据以平均值±SEM表示,n=8,单因素ANOVA然后进行Dunnett多重比较检验*P<0.05、***P<0.0001与溶媒对照相比。
图4:化合物No.449和化合物No.54对急性的环磷酰胺(CYP)诱导的膀胱炎模型的效果。数据以AUC%疼痛评分(平均值±SEM)表示。
图5:化合物XI对慢性的环磷酰胺(CYP)诱导的膀胱炎模型的效果。数据以AUC%疼痛评分(平均值±SEM)表示。
图6:化合物No.43对急性的环磷酰胺(CYP)诱导的膀胱炎模型的效果。数据以AUC%疼痛评分(平均值±SE)表示。
图7:化合物XIII对急性的环磷酰胺(CYP)诱导的膀胱炎模型的效果。数据以AUC%疼痛评分(平均值±SEM)表示。
图8:在Sprague Dawley大鼠中单次口服给予化合物No.12后,化合物No.12和化合物XI的平均值±SD浓度-时间曲线。
图9:在Sprague Dawley大鼠中单次口服给予化合物No.43后,化合物No.43和化合物XI的平均值±SD浓度-时间曲线。
图10:在Sprague Dawley大鼠中单次口服给予化合物No.446后,化合物No.446和化合物XI的平均值±SD浓度-时间曲线。
图11:在Sprague Dawley大鼠中单次口服给予化合物No.449后,化合物No.449和化合物XI的平均值±SD浓度-时间曲线。
图12:在Sprague Dawley大鼠中单次口服给予化合物XIII后,化合物XIII和化合物XI的平均值±SD浓度-时间曲线。
图13:在Sprague Dawley大鼠中单次口服给予化合物No.54后,化合物No.54和化合物XI的平均值±SD浓度-时间曲线。
图14:化合物XI对急性的环磷酰胺(CYP)诱导的膀胱炎模型的效果。数据以膀胱压力(mm hg)表示(平均值±SE)。
发明内容
本发明提供了有价值的前药,其克服了现有活性物质的缺点,即活性物质被生物膜的不充分吸收或母体化合物的不利代谢。此外,与母体化合物相比,这些新型前药具有改善的药代动力学特征。
在一个实施方式中,本发明提供了通式(I)的化合物、其药学上可接受的盐或溶剂化物:
其中,
R
R
R
在一个实施方式中,本发明提供了通式I-A的化合物、其药学上可接受的盐或溶剂化物;
其中,R
R
R
R
R
R
另一方面,本发明提供了用于治疗间质性膀胱炎/膀胱疼痛综合征(IC/BPS)、膀胱过度活动症(OAB)、慢性咳嗽、子宫内膜异位症、神经性疼痛、偏头痛、瘙痒症、带状疱疹后遗神经痛、尿路感染相关疼痛、骨关节炎、关节炎和尿失禁的方法,该方法包括给予选自通式(I)的化合物、其药学上可接受的盐或溶剂化物。
另一方面,本发明提供了用于治疗间质性膀胱炎/膀胱疼痛综合征(IC/BPS)、膀胱过度活动症(OAB)、慢性咳嗽、子宫内膜异位症、神经性疼痛、偏头痛、瘙痒症、带状疱疹后遗神经痛、尿路感染相关疼痛、骨关节炎、关节炎和尿失禁的方法,该方法包括给予选自通式(I-A)的化合物、其药学上可接受的盐或溶剂化物。
另一方面,本发明提供了用于间质性膀胱炎/膀胱疼痛综合征(IC/BPS)、膀胱过度活动症(OAB)、慢性咳嗽、子宫内膜异位症、神经性疼痛、偏头痛、瘙痒症、带状疱疹后遗神经痛、尿路感染相关疼痛、骨关节炎、关节炎和尿失禁的方法,该方法包括给予选自通式(I)的化合物、其药学上可接受的盐或其溶剂化物。
另一方面,本发明提供了用于间质性膀胱炎/膀胱疼痛综合征(IC/BPS)、膀胱过度活动症(OAB)、慢性咳嗽、子宫内膜异位症、神经性疼痛、偏头痛、瘙痒症、带状疱疹后遗神经痛、尿路感染相关疼痛、骨关节炎、关节炎和尿失禁的用途的方法,该方法包括给予选自通式(I-A)的化合物、其药学上可接受的盐或溶剂化物。
另一方面,本发明提供了用于由感染、创伤、手术、内窥镜程序、或声音或导管的穿过引起的下尿路粘膜刺激所导致的疼痛、烧灼感、尿急、尿频和其它不适的症状缓解的方法,该方法包括给予选自通式(I)的化合物、其药学上可接受的盐或其溶剂化物。
另一方面,本发明提供了用于由感染、创伤、手术、内窥镜程序或声音或导管的穿过引起的下尿路粘膜刺激所导致的疼痛、烧灼感、尿急、尿频和其它不适的症状缓解的方法,该方法包括给予选自通式(I-A)的化合物、其药学上可接受的盐或溶剂化物。
在一个实施方式中,本发明涉及药物组合物,其包含至少一种选自以下的化合物:通式(I)的化合物、其药学上可接受的盐、溶剂化物;以及药学上可接受的赋形剂。
在一个实施方式中,本发明涉及药物组合物,其包含至少一种选自以下的化合物:通式(I-A)的化合物、其药学上可接受的盐、溶剂化物;以及药学上可接受的赋形剂。
具体实施方式
本文提供的详细描述和实例是示例性的,并且落入本发明范围内的任何修改或变化对于本领域技术人员来说将是显而易见的。此外,除非另有定义,本文中使用的所有技术和科学术语应具有本领域普通技术人员所理解的含义。
如本文所用,术语“前药”是指在给予后通过化学或生理过程在体内释放生物活性化合物的前体化合物。在一个实施方式中,前药具有所需的生物活性。在另一个实施方式中,前药转化为具有所需生物活性的治疗活性化合物。
本公开的药物组合物可以是本领域技术人员已知的任何形式。本发明的药物组合物可以口服、肠胃外、通过吸入喷雾、局部、直肠、鼻腔、口腔或通过植入的储库给予,优选口服给予,或通过注射给予。例如,在一些实施方式中,药物组合物是用于口服递送的产品形式,所述产品形式选自于由片剂、浓缩物、干粉、液体、胶囊、小丸剂(pellet)和丸剂(pill)组成的组。本文公开的药物组合物还可以进一步包含载体、粘合剂、稀释剂和赋形剂。
术语“一个(a/an)”并不表示数量的限制,而是表示存在至少一个所提及的项目。
本文所用的术语“约”,当指可测量值时,意在涵盖与指定值±10%、优选±5%、更优选±1%并且还更优选±0.1%的变化。
本文所用的术语“药物组合物”用于本发明的目的,是指处于药物制剂形式的组合物,其中所述组合物可以通过选自于由口服、肠胃外、鼻腔、局部、直肠、口腔、眼、阴道、耳或植入的储库组成的组的途径给予。
如本文所用,术语“药学上可接受的”是指与组合物的所有其它药物成分相容并且对用组合物治疗的个体无害的盐、载体、赋形剂和其它组合物成分。
术语“药学上可接受的载体”是指可以与本发明的化合物一起向患者给予且不破坏其药理学活性的无毒载体。
本文所用的术语“药学上可接受的赋形剂”包括溶媒、佐剂或稀释剂或其它辅助物质,例如公众容易获得的本领域常规的那些物质。例如,药学上可接受的赋形剂包括pH调节和缓冲剂、张力调节剂、稳定剂、润湿剂等。
如本文所用,术语“溶剂化物”是式(I)化合物或其药学上可接受的盐与化学计量或非化学计量量的溶剂的物理形式。
如本文所用,术语“盐”是指本发明化合物的酸式盐或碱式盐。碱性化合物的盐是与无机酸、有机羧酸、有机磺酸等形成的盐。合适的酸的实例包括盐酸、氢溴酸、硫酸、硝酸、高氯酸、富马酸、马来酸、磷酸、乙醇酸、乳酸、水杨酸、琥珀酸、对甲苯磺酸、酒石酸、乙酸、柠檬酸、甲磺酸、甲酸、苯甲酸、丙二酸、萘-2-磺酸、三氟乙酸和苯磺酸。酸性化合物的盐是与碱(即,阳离子物质)形成的,例如碱金属和碱土金属阳离子,如钠、锂、钾、钙和镁离子,以及铵阳离子,例如铵、三甲基铵和二乙基铵。
如本文所用,术语“烷基”是指具有1至约12个碳原子的直链或支链饱和脂肪族基团,例如甲基、乙基、丙基、异丙基、正丁基、异丁基、叔丁基等。烷基还指具有1至约2个碳原子、具有1至约3个碳原子、具有1至约4个碳原子、具有1至约5个碳原子、具有1至约6个碳原子、具有1至约7个碳原子、具有1至约8个碳原子、具有1至约9个碳原子、具有1至约10个碳原子。
如本文所用,术语“亚烷基(alkylene)”是指衍生自具有1至约6个碳原子的烷基的直链或支链饱和二价脂肪族基团,例如-CH
术语“烯基”是指具有至少2个碳原子和至少一个碳-碳双键的直链或支链烃。烯基可具有任何合适数量的双键,包括但不限于1、2、3、4、5或更多数量。优选的烯基包括乙烯基(-CH=CH
术语“炔基”表示具有2至12个碳原子并具有至少1-2个炔基不饱和位点的炔基,炔基包括乙炔基(-C≡CH)、炔丙基(-CH
术语“环烷基”表示具有单环(例如环己基)或多个稠环(例如降冰片基)的3至12个碳原子的饱和碳环基团。环烷基包括环丙基、环丁基、环戊基、环己基、环庚基、降冰片基等。
术语“杂环”表示含有C3-C12原子的环,其中至多4个碳原子被选自于由O、S或N组成的组的杂原子取代。杂环进一步含有“杂环烷基”或“杂芳基”。
术语“杂环烷基”表示根据上述定义的C3-C12环烷基,其中至多4个碳原子被选自于由O、S或N组成的组的杂原子取代。杂环烷基包括吡咯烷、哌啶、哌嗪、吗啉、四氢呋喃、四氢噻吩、3-甲基-3,9-二氮杂螺[5.5]十一烷、2-甲基-2,6-二氮杂螺[3.3]庚烷、间二氧杂环戊烯等。杂环烷基可以是单环、双环或螺环。
术语“芳基”表示由含有6-14个碳原子的一个或多个稠合环组成的环状芳烃基团,其中至少一个环本质上是芳香性的,例如苯基、萘基、1,2,3,4-四氢萘基、茚满基等。
术语“杂芳基”表示由含有5-14个环原子、优选含有5-10个环原子的一个或多个稠合环组成的环状芳烃基团,其中至少一个环本质上是芳香性的,并且其含有至少一个选自N、O或S的杂原子,例如吡啶基、吡咯基、呋喃基、噻吩基、咪唑基、噁唑基、异噁唑基、噻唑基、异噻唑基、吡唑基、1,2,3-三唑基、1,2,4-三唑基、苯并噻吩基、苯并三唑基、异苯并噻吩基、吲哚基、异吲哚基、苯并咪唑基、咪唑并[1,2-a]吡啶基、苯并噻唑基、苯并噁唑基、喹嗪基、喹唑啉基、苯并喹啉基等。
如本文所用的术语氨基酸是指两种立体异构形式,称为“D”和“L”。任何氨基酸的D型和L型具有相同的物理性质和化学反应活性,但令平面偏振光的平面旋转相等但方向相反,并且以不同的速率与不对称试剂反应。蛋白质中所有天然存在的氨基酸均为L型。氨基酸包括赖氨酸、缬氨酸、色氨酸、苯丙氨酸、甲硫氨酸、亮氨酸、苏氨酸、异亮氨酸、精氨酸、组氨酸、酪氨酸、肉碱、丝氨酸、谷氨酰胺、天冬氨酸、脯氨酸、甘氨酸、半胱氨酸、丙氨酸、谷氨酸。氨基酸可以以“D”或“L”对映异构体存在。
术语“卤素”表示氯、碘、氟和溴。
如本文所用,术语“离去基团”或“L”或“L
术语“氨基”是指基团-NH
术语“烷基氨基”是指基团-NH烷基。
术语“二烷基氨基”是指基团-N(烷基)
术语“烷氧基”是指基团-O-烷基。
术语“芳氧基”是指基团-O-芳基。
术语“环烷氧基”是指基团-O-环烷基。
术语“杂环烷氧基”是指基团-O-杂环烷基。
术语“杂芳氧基”是指基团-O-杂芳基。
术语“烷基酯”是指基团-亚烷基-O-(CO)-R。
术语“氨基芳基”是指基团-亚芳基-氨基。
术语“烷基醇”或“羟烷基”是指被一个或多个-OH基团取代的烷基。
术语“氨基酸残基”或“氨基酸侧链”是指片段-C(O)-C(H/R)
术语“任选”是指随后描述的事件或情况可以发生或可以不发生,并且该描述包括事件或情况发生的情况和不发生的情况。
出于本发明的目的,术语“取代的”应理解为包括用一个或多个不同原子添加或替换官能团或化合物中包含的一个或多个原子。
除非个别取代基的定义另有限制,上述基团,如“烷基”、“烯基”、“炔基”、“环烷基”、“杂环”、“杂环烷基”、“取代的杂环烷基”、“芳基”和“杂芳基”等基团可以任选地被一个或多个取代基取代,该取代基选自于由以下组成的组:“C1-C8烷基”、“C2-C8烯基”、“C2-C8炔基”、“环烷基”、“杂环烷基”、“芳基”、“杂芳基”、“氨基”、“烷基氨基”、“二烷基氨基”、“酰基”、“酰氧基”、“酰氨基”、“氨基羰基”、“烷氧基羰基”、“烷氧基烷氧基”、“烷氧基羰氧基”、“氨基甲酸酯”、“亚磺酰基”、“磺酰基”、“烷氧基”、“硫醇基”、“卤素”、“羧基”、“三卤甲基”、“氰基”、“羟基”、“巯基”、“硝基”、“磷酸酯”、“烷基磷酸酯”等。优选的一个或多个取代基的数量选自1至10;更优选数量为1至5。
在一个实施方式中,前药本身可能缺乏或具有所需的生物活性。在本发明的一个实施方式中,说明书中公开的化合物本身可以具有所需的生物活性。
在一个实施方式中,本发明的化合物含有一个或多个不对称碳原子,并因此作为外消旋体和外消旋混合物、单一对映异构体、非对映异构体混合物和单独的非对映异构体存在。这些化合物的所有此类异构体形式明确地包括在本发明中。每个立体异构源的碳可以是R或S构型。在一个实施方式中,化合物的对映异构体、非对映异构体、外消旋体明确地包括在本发明中。
在一个实施方式中,本发明提供了通式I-A的化合物、其药学上可接受的盐或溶剂化物:
其中,R
R
R
R
R
R
在一个实施方式中,本发明提供了通式I-A的化合物、其药学上可接受的盐或溶剂化物;其中,R
在一个实施方式中,本发明提供了通式I-A的化合物、其药学上可接受的盐或溶剂化物;其中,R
在一个实施方式中,本发明提供了通式I-A的化合物、其药学上可接受的盐或溶剂化物;其中,R
在一个实施方式中,本发明提供了通式I-A的化合物、其药学上可接受的盐或溶剂化物;其中,R
在一个实施方式中,本发明提供了通式I-A的化合物、其药学上可接受的盐或溶剂化物;其中,R
在一个实施方式中,本发明提供了通式I-A的化合物、其药学上可接受的盐或溶剂化物;其中,R
在一个实施方式中,本发明提供了通式I的化合物、其药学上可接受的盐或溶剂化物:
其中,
R
R
R
在一个实施方式中,本发明提供了通式I的化合物、其药学上可接受的盐或溶剂化物;其中,R
在一个实施方式中,本发明提供了通式I的化合物、其药学上可接受的盐或溶剂化物;其中,R
在一个实施方式中,本发明提供了通式I的化合物、其药学上可接受的盐或溶剂化物;其中,R
在一个实施方式中,本发明提供了通式I的化合物、其药学上可接受的盐或溶剂化物;其中,R
在一个实施方式中,本发明提供了通式I的化合物、其药学上可接受的盐或溶剂化物;其中,R
在一个实施方式中,本发明提供了通式I的化合物、其药学上可接受的盐或溶剂化物;其中,R
在一个实施方式中,本发明涉及通式(II)的化合物、其药学上可接受的盐或溶剂化物:
其中,
R
R
条件是R
在一个实施方式中,本发明提供了通式II的化合物、其药学上可接受的盐或溶剂化物;其中,R
在一个实施方式中,本发明提供了通式II的化合物、其药学上可接受的盐或溶剂化物;其中,R
在一个实施方式中,本发明提供了通式II的化合物、其药学上可接受的盐或溶剂化物;其中,R
在一个实施方式中,本发明提供了通式II的化合物、其药学上可接受的盐或溶剂化物;其中,R
在一个实施方式中,本发明提供了通式II的化合物、其药学上可接受的盐或溶剂化物;
其中,R
在一个实施方式中,本发明提供了通式II的化合物、其药学上可接受的盐或溶剂化物;
其中,R
在一方面,本发明涉及通式(III)的化合物、其药学上可接受的盐或溶剂化物:
R
R
在一个实施方式中,本发明提供了通式III的化合物、其药学上可接受的盐或溶剂化物;
其中,R
在一个实施方式中,本发明提供了通式III的化合物、其药学上可接受的盐或溶剂化物;
其中,R
在一个实施方式中,本发明提供了通式III的化合物、其药学上可接受的盐或溶剂化物;
其中,R
在一个实施方式中,本发明提供了选自于由以下组成的组的化合物:
其药学上可接受的盐或溶剂化物。
在另一方面,通式(III-A)的化合物、其药学上可接受的盐或溶剂化物:
其中,
R
R
在一个实施方式中,本发明提供了选自于由以下组成的组的化合物:
/>
其药学上可接受的盐或溶剂化物。
在另一方面,通式(III-B)的化合物、其药学上可接受的盐或溶剂化物:
其中,R
在一个实施方式中,本发明提供了选自于由以下组成的组的化合物:
其药学上可接受的盐或溶剂化物。
在另一方面,通式(III-C)的化合物、其药学上可接受的盐或溶剂化物:
/>
其中,
R
R
在一个实施方式中,本发明提供了选自于由以下组成的组的化合物:
其药学上可接受的盐或溶剂化物。
在一个实施方式中,本发明涉及通式(IV)的化合物、其药学上可接受的盐或溶剂化物;
其中,
R
R
R
条件是R
在一个实施方式中,本发明涉及通式(IV-A)的化合物、其药学上可接受的盐或溶剂化物;
其中,
R
R
R
在一个实施方式中,本发明提供了选自于由以下组成的组的化合物:
其药学上可接受的盐或溶剂化物。
在一个实施方式中,本发明涉及通式(IV-B)的化合物、其药学上可接受的盐或溶剂化物;
其中,
R
R
R
在一个实施方式中,本发明涉及通式(IV-C)的化合物、其药学上可接受的盐或溶剂化物;
其中,
R
R
条件是R
在一个实施方式中,本发明提供了选自于由以下组成的组的化合物:
其药学上可接受的盐或溶剂化物。
在一方面,本发明涉及通式(V)的化合物、其药学上可接受的盐或溶剂化物:
其中,
R
R
R’
条件是R
在一个实施方式中,本发明涉及通式(V-A)的化合物、其药学上可接受的盐或溶剂化物;
其中;
R
R
R’
在一个实施方式中,本发明涉及通式(V-B)的化合物、其药学上可接受的盐或溶剂化物;
其中,
R
R
在一个实施方式中,本发明涉及通式(V-C)的化合物、其药学上可接受的盐或溶剂化物;
其中,
R
R’
一方面,本发明涉及通式(VI)的化合物、其药学上可接受的盐或溶剂化物:
其中,
R
R
条件是R
在一个实施方式中,本发明涉及通式(VI-A)的化合物、其药学上可接受的盐或溶剂化物;
其中,
R
R
在一个实施方式中,本发明涉及通式(VI-B)的化合物、其药学上可接受的盐或溶剂化物;
其中,
R
R
在一个实施方式中,本发明涉及通式(VI-C)的化合物、其药学上可接受的盐或溶剂化物:
其中,
R
在一方面,本发明涉及通式(VII)的化合物、其药学上可接受的盐或溶剂化物;
其中,
R
R
条件是R
在一个实施方式中,本发明涉及通式(VII-A)的化合物、其药学上可接受的盐或溶剂化物;
其中,
R
R
在一个实施方式中,本发明提供了选自于由以下组成的组的化合物:
其药学上可接受的盐或溶剂化物。
在一个实施方式中,本发明涉及通式(VII-B)的化合物、其药学上可接受的盐或溶剂化物;
其中,
R
R
在一个实施方式中,本发明涉及通式(VII-C)的化合物、其药学上可接受的盐或溶剂化物;
其中R
在一个实施方式中,本发明提供了选自于由以下组成的组的化合物:
其药学上可接受的盐或溶剂化物。
在一方面,本发明涉及通式(VIII)的化合物、其药学上可接受的盐或溶剂化物:
其中,
R
R’
条件是R
在一个实施方式中,本发明提供了选自于由以下组成的组的化合物:
其药学上可接受的盐或溶剂化物。
在一个实施方式中,本发明涉及通式(VIII-A)的化合物、其药学上可接受的盐或溶剂化物;
其中,
R
R’
在一个实施方式中,本发明提供了选自于由以下组成的组的化合物:
/>
其药学上可接受的盐或溶剂化物。
在一个实施方式中,本发明涉及式(VIII-B)的化合物、其药学上可接受的盐或溶剂化物;
其中,
R
R’
在一个实施方式中,本发明提供了选自于由以下组成的组的化合物:
其药学上可接受的盐或溶剂化物。
在一个实施方式中,本发明涉及通式(VIII-C)的化合物、其药学上可接受的盐或溶剂化物:
其中,R’
在一个实施方式中,本发明提供了选自于由以下组成的组的化合物:
/>
/>
/>
其药学上可接受的盐。
在一个实施方式中,本发明提供了选自于由表1所列化合物及其药学上可接受的盐或溶剂化物组成的组的化合物。
在一个实施方式中,本发明提供了用于治疗至少一种选自以下的疾病病症的方法:间质性膀胱炎/膀胱疼痛综合征(IC/BPS)、膀胱过度活动症(OAB)、慢性咳嗽、子宫内膜异位症、神经性疼痛、偏头痛、瘙痒症、带状疱疹后遗神经痛、尿路感染相关疼痛、骨关节炎、关节炎和尿失禁,所述方法包括给予至少一种选自于由表1所列化合物及其药学上可接受的盐或溶剂化物组成的组的化合物。
在一个实施方式中,本发明提供了使用至少一种选自于由表1所列化合物、其药学上可接受的盐或溶剂化物组成的组的化合物用于治疗至少一种选自以下的疾病病症的方法:间质性膀胱炎/膀胱疼痛综合征(IC/BPS)、膀胱过度活动症(OAB)、慢性咳嗽、子宫内膜异位症、神经性疼痛、偏头痛、瘙痒症、带状疱疹后遗神经痛、尿路感染相关疼痛、骨关节炎、关节炎和尿失禁。
在一个实施方式中,表1和表2中给出的编号被视为供说明书中参考的化合物号。
表1:化合物
/>
/>
/>
/>
/>
/>
/>
/>
/>
在一个实施方式中,本发明涉及通式(IX)的化合物、其药学上可接受的盐或溶剂化物:
其中,R
R
R
R
在一个实施方式中,本发明涉及通式(IX-A)的化合物、其药学上可接受的盐或溶剂化物:
其中,R
其中,R
R
在一个实施方式中,本发明提供了至少一种选自于由以下组成的组的化合物:
/>
/>
其药学上可接受的盐或溶剂化物。
在一个实施方式中,本发明涉及通式(IX-B)的化合物、其药学上可接受的盐或溶剂化物:
其中,R
在一个实施方式中,本发明提供了至少一种选自于由以下组成的组的化合物:
/>
/>
其药学上可接受的盐或溶剂化物。
在一个实施方式中,本发明涉及通式(IX-C)的化合物、其药学上可接受的盐或溶剂化物:
其中,R
R
在一个实施方式中,本发明提供了至少一种选自于由以下组成的组的化合物:
/>
/>
/>
/>
/>
/>
/>
/>
/>
其药学上可接受的盐或溶剂化物。
在一个实施方式中,本发明涉及通式(IX-D)的化合物、其药学上可接受的盐或溶剂化物:
其中,R
R
在一个实施方式中,本发明提供了化合物
其药学上可接受的盐或溶剂化物。
在一个实施方式中,本发明涉及通式(X)的化合物、其药学上可接受的盐或溶剂化物:
其中,
R
R’
R
R
R
R
在一个实施方式中,本发明提供了至少一种选自于由以下组成的组的化合物:
/>
其药学上可接受的盐或溶剂化物。
在一个实施方式中,本发明提供了至少一种选自于由表2所列化合物及其药学上可接受的盐或溶剂化物组成的组的化合物。
在一个实施方式中,本发明提供了用于治疗至少一种选自以下的疾病病症的方法:间质性膀胱炎/膀胱疼痛综合征(IC/BPS)、膀胱过度活动症(OAB)、慢性咳嗽、子宫内膜异位症、神经性疼痛、偏头痛、瘙痒症、带状疱疹后遗神经痛、尿路感染相关疼痛、骨关节炎、关节炎和尿失禁,所述方法包括给予至少一种选自于由表2所列化合物、其药学上可接受的盐或溶剂化物组成的组的化合物。
在一个实施方式中,本发明提供了使用至少一种选自于由表2所列化合物、其药学上可接受的盐或溶剂化物组成的组的化合物用于治疗至少一种选自以下的疾病病症的方法:间质性膀胱炎/膀胱疼痛综合征(IC/BPS)、膀胱过度活动症(OAB)、慢性咳嗽、子宫内膜异位症、神经性疼痛、偏头痛、瘙痒症、带状疱疹后遗神经痛、尿路感染相关疼痛、骨关节炎、关节炎和尿失禁。
表2:化合物
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
/>
另一方面,本发明提供了用于治疗至少一种选自以下的疾病病症的方法:间质性膀胱炎/膀胱疼痛综合征(IC/BPS)、膀胱过度活动症(OAB)、慢性咳嗽、子宫内膜异位症、神经性疼痛、偏头痛、瘙痒症、带状疱疹后遗神经痛、尿路感染相关疼痛、骨关节炎、关节炎和尿失禁,所述方法包括给予选自通式(I)或(I-A)的化合物、其药学上可接受的盐或溶剂化物。
另一方面,本发明提供了用于治疗至少一种选自以下的疾病病症的方法:间质性膀胱炎/膀胱疼痛综合征(IC/BPS)、膀胱过度活动症(OAB)、慢性咳嗽、子宫内膜异位症、神经性疼痛、偏头痛、瘙痒症、带状疱疹后遗神经痛、尿路感染相关疼痛、骨关节炎、关节炎和尿失禁,所述方法包括给予选自以下的化合物:通式(II)、式(III)、式(III-A)、式(III-B)、式(III-C)、式(IV)、式(IV-A)、式(IV-B)、式(IV-C)、式(V)、式(V-A)、式(V-B)、式(V-C)、式(VI)、式(VI-A)、式(VI-B)、式(VI-C)、式(VII)、式(VII-A)、式(VII-B)、式(VII-C)、式(VIII)、式(VIII-A)、式(VIII-B)、式(VIII-C)、式(IX)、式(IX-A)、式(IX-B)、式(IX-C)、式(IX-D)、式(X)的化合物、其药学上可接受的盐或溶剂化物。
另一方面,本发明提供了一种用于由感染、创伤、手术、内窥镜程序、或声音或导管的穿过引起的下尿路粘膜刺激所导致的疼痛、烧灼感、尿急、尿频和其它不适的症状缓解的方法,所述方法包括给予治疗有效量的式(I)或式(I-A)的化合物、其药学上可接受的盐或溶剂化物。
另一方面,本发明提供了一种用于由感染、创伤、手术、内窥镜程序、或声音或导管的穿过引起的下尿路粘膜刺激所导致的疼痛、烧灼感、尿急、尿频和其它不适的症状缓解的方法,所述方法包括给予选自以下的化合物:通式(II)、式(III)、式(III-A)、式(III-B)、式(III-C)、式(IV)、式(IV-A)、式(IV-B)、式(IV-C)、式(V)、式(V-A)、式(V-B)、式(V-C)、式(VI)、式(VI-A)、式(VI-B)、式(VI-C)、式(VII)、式(VII-A)、式(VII-B)、式(VII-C)、式(VIII)、式(VIII-A)、式(VIII-B)、式(VIII-C)、式(IX)、式(IX-A)、式(IX-B)、式(IX-C)、式(IX-D)、式(X)的化合物、其药学上可接受的盐或溶剂化物。
在又一方面,本发明涉及药物组合物,其包含至少一种选自以下的化合物:通式(I-A)、式(I)、式(II)、式(III)、式(III-A)、式(III-B)、式(III-C)、式(IV)、式(IV-A)、式(IV-B)、式(IV-C)、式(V)、式(V-A)、式(V-B)、式(V-C)、式(VI)、式(VI-A)、式(VI-B)、式(VI-C)、式(VII)、式(VII-A)、式(VII-B)、式(VII-C)、式(VIII)、式(VIII-A)、式(VIII-B)、式(VIII-C)、式(IX)、式(IX-A)、式(IX-B)、式(IX-C)、式(IX-D)、式(X)的化合物、其药学上可接受的盐或溶剂化物,以及药学上可接受的赋形剂。
在另一方面,本发明提供了用于治疗至少一种选自以下的疾病病症的用途的药物组合物:间质性膀胱炎/膀胱疼痛综合征(IC/BPS)、膀胱过度活动症(OAB)、慢性咳嗽、子宫内膜异位症、神经性疼痛、偏头痛、瘙痒症、带状疱疹后遗神经痛、尿路感染相关疼痛、骨关节炎、关节炎和尿失禁,包括给予选自以下的化合物:通式(I-A)、式(I)、式(II)、式(III)、式(III-A)、式(III-B)、式(III-C)、式(IV)、式(IV-A)、式(IV-B)、式(IV-C)、式(V)、式(V-A)、式(V-B)、式(V-C)、式(VI)、式(VI-A)、式(VI-B)、式(VI-C)、式(VII)、式(VII-A)、式(VII-B)、式(VII-C)、式(VIII)、式(VIII-A)、式(VIII-B)、式(VIII-C)、式(IX)、式(IX-A)、式(IX-B)、式(IX-C)、式(IX-D)、式(X)的化合物、其药学上可接受的盐或溶剂化物。
在另一方面,本发明提供了用于治疗至少一种选自选自以下的疾病病症的方法:间质性膀胱炎/膀胱疼痛综合征(IC/BPS)、膀胱过度活动症(OAB)、慢性咳嗽、子宫内膜异位症、神经性疼痛、偏头痛、瘙痒症、带状疱疹后遗神经痛、尿路感染相关疼痛、骨关节炎、关节炎和尿失禁,其包括给予选自以下的化合物:
其药学上可接受的盐或溶剂化物。
在另一方面,本发明提供了用于治疗至少一种选自以下的疾病病症的方法:膀胱过度活动症(OAB)、慢性咳嗽、子宫内膜异位症、神经性疼痛、偏头痛、瘙痒症、带状疱疹后遗神经痛、尿路感染相关疼痛、骨关节炎、关节炎和尿失禁;所述方法包括给予选自以下的化合物:
(化合物XIII);其药学上可接受的盐或溶剂化物。
以下实施例是出于说明本发明的目的而给出的,并且不应被视为限制本发明的范围。
实施例-01:1-甲基-L-色氨酸(E)-2-((2,6-二氨基吡啶-3-基)二氮烯基)苯基酯的制备
步骤-01:N-(叔丁氧羰基)-1-甲基-L-色氨酸的制备
在0℃温度下,向处于1,4-二噁烷(40mL)中的1-甲基-L-色氨酸(2.0g)的搅拌溶液中添加氢氧化钠(364mg/20mL水),然后在相同温度下添加二碳酸二叔丁酯(2.7mL)。将反应混合物在0℃下搅拌4h,然后在室温下继续搅拌另外16h。反应完成后(通过TLC监测:在二氯甲烷中的20%甲醇),减压蒸发过量的溶剂,随后使用1N盐酸酸化反应混合物。用乙酸乙酯(3×1500mL)对该混合物进行萃取,随后用水(3×500mL)洗涤。将合并的有机层用无水硫酸钠干燥并减压蒸发以获得粗品。将粗品用正戊烷洗涤并减压干燥,以得到N-(叔丁氧羰基)-1-甲基-L-色氨酸(1.4g,48%收率),为灰白色固体。
LCMS:96.83%(m/z:317.39),[M-1]
1H-NMR(400MHz,DMSO-d
步骤-02:(S)-3-(112-吲哚-3-基)-2-((叔丁氧羰基)氨基)丙酸(E)-2-((2,6-二氨基吡啶-3-基)二氮烯基)苯基酯的制备
向处于二氯甲烷(6.0mL)中的(E)-2-((2,6-二氨基吡啶-3-基)二氮烯基)苯酚(0.500g)的搅拌溶液中添加二环己基碳二亚胺(1.16g)、4-(二甲基氨基)吡啶(0.12g)和N-(叔丁氧羰基)-1-甲基-L-色氨酸(1.2g)。将反应混合物在室温下搅拌16h。通过TLC(流动相:在庚烷中的50%乙酸乙酯)监测反应的完成。反应完成后,将反应混合物用去矿化水淬灭并用处于二氯甲烷中的5%甲醇(3×100mL)萃取。将合并的有机层用无水硫酸钠干燥并减压蒸发以获得粗品。将粗品用正戊烷洗涤并减压干燥,以得到(S)-3-(112-吲哚-3-基)-2-((叔丁氧羰基)氨基)丙酸(E)-2-((2,6-二氨基吡啶-3-基)二氮烯基)苯基酯(0.1g,50%纯度),为黄色固体。
LCMS:50.45%(m/z:530.14),[M+1]
步骤-03:1-甲基-L-色氨酸(E)-2-((2,6-二氨基吡啶-3-基)二氮烯基)苯基酯的制备:
在0℃温度下,向处于二氯甲烷(20mL)中的(S)-3-(1H-吲哚-3-基)-2-((叔丁氧羰基)氨基)丙酸(E)-2-((2,6-二氨基吡啶-3-基)二氮烯基)苯基酯(1g)的搅拌溶液中添加处于1,4-二噁烷(2mL)中的4M盐酸,然后将反应混合物在室温下另外搅拌16h。反应完成后(通过TLC监测:流动相:处于庚烷中的50%乙酸乙酯),减压蒸发过量的溶剂,以得到粗物质。通过prep-HPLC(T3柱,三氟乙酸:乙腈,1:1)纯化粗品,以得到1-甲基-L-色氨酸(E)-2-((2,6-二氨基吡啶-3-基)二氮烯基)苯基酯(0.09g,11%),为黄色固体。
LCMS:95.22%(m/z:430.15[M+1]
1
按照实施例-01的步骤-02反应条件(DCC、DMAP、DCM),已经合成了以下酯前药,其中相应的酸是商业获得的。
/>
/>
/>
实施例-18:丙酸(E)-2-((2,6-二氨基吡啶-3-基)二氮烯基)苯基酯的制备
在0℃温度下,向处于二氯甲烷(8mL)中的(E)-2-((2,6-二氨基吡啶-3-基)二氮烯基)苯酚盐酸盐(0.2g)的溶液中添加三乙胺(0.25mL)和丙酰氯(0.1g),并将反应混合物在室温下搅拌3h。反应完成后(通过TLC监测),用冷水(10mL)淬灭反应物质并用二氯甲烷(3×30mL)进行萃取。将合并的有机层用水(2×100mL)洗涤,经无水硫酸钠干燥,并减压浓缩,以获得粗物质,将其通过Prep HPLC(60%乙腈:甲酸)纯化,以提供丙酸(E)-2-((2,6-二氨基吡啶-3-基)二氮烯基)苯基酯。(0.042g,19.5%)
LCMS:99.52%(m/z:286.2,[M+1]
1
按照实施例-18的反应条件(三乙胺,DCM);已经合成了以下酯前药,其中相应的酰氯是商业获得的。
实施例-20:1-甲基哌啶-2-甲酸(E)-2-((2,6-二氨基吡啶-3-基)二氮烯基)苯基酯的制备
向处于二氯甲烷(3mL)中的(E)-2-((2,6-二氨基吡啶-3-基)二氮烯基)苯酚(0.3g)的搅拌溶液中添加吡啶(3mL)、1,1’-羰基二咪唑(0.68g)和4-(二甲基氨基)吡啶(0.067g)。将所得反应混合物在0℃下搅拌30min。向上述搅拌溶液中添加1-甲基哌啶-2-甲酸(0.592g)并将所得溶液在室温下搅拌16h。通过TLC(流动相:处于己烷中的70%乙酸乙酯)监测反应混合物。反应完成后,将反应混合物用水淬灭,用二氯甲烷(3×100mL)萃取,经无水硫酸钠干燥,减压蒸发,以获得粗物质。通过prep HPLC[方法:(A)0.5%TFA水溶液(B)100%乙腈]纯化粗品,并将产物级分冻干,以得到1-甲基哌啶-2-甲酸(E)-2-((2,6-二氨基吡啶-3-基)二氮烯基)苯基酯(0.037g,9%),为黄色固体。
LCMS:95.00%(m/z:355.25,[M+1]
1
实施例-21:二乙基氨基甲酸(E)-2-((2,6-二氨基吡啶-3-基)二氮烯基)苯基酯的制备
向处于二氯甲烷(10mL)中的(E)-2-((2,6-二氨基吡啶-3-基)二氮烯基)苯酚盐酸盐(0.200g)的溶液中添加二环己基碳二亚胺(0.466g)和4-(二甲基氨基)吡啶(0.046g)并在室温下搅拌15min。之后,加入二乙基氨基甲酸(0.114g)并将反应混合物在室温下搅拌24h。反应完成后(通过TLC监测),将反应物质用冷水(10mL)淬灭并用二氯甲烷(3×30mL)萃取,用水(2×100mL)洗涤,经无水硫酸钠干燥,并在真空下浓缩以获得粗物质。通过prepHPLC[70%乙腈/30%TFA]纯化粗物质,以提供二乙基氨基甲酸(E)-2-((2,6-二氨基吡啶-3-基)二氮烯基)苯基酯(0.025g,10.1%),为黄色固体。
LCMS:99.70%(m/z:329.20,[M+1]
1
实施例-22:4-甲基-1,4-二氮杂环庚烷-1-甲酸(E)-2-((2,6-二氨基吡啶-3-基)二氮烯基)苯基酯的制备
在0℃和氮气气氛下,向处于10mL二氯甲烷中的(E)-2-((2,6-二氨基吡啶-3-基)二氮烯基)苯酚氯化氢(0.500g)的搅拌溶液中添加N,N-二异丙基乙胺(2.43g)和1,1’-羰基二咪唑(0.918g),然后是4-(二甲基氨基)吡啶(0.116g)。将反应混合物在0℃温度下搅拌10min,然后添加1-甲基1,4-二氮杂环庚烷(0.861g)。将反应混合物在室温下搅拌26h。反应完成后,用水淬灭混合物并用二氯甲烷(3×50mL)进行萃取。将合并的有机层经无水硫酸钠干燥并减压蒸发以获得粗物质。通过快速色谱法(Flash chromatography)富集粗品,然后通过prep HPLC[方法:TFA缓冲液]纯化,并将产物级分冻干,以得到0.080g的4-甲基-1,4-二氮杂环庚烷-1-甲酸(E)-2-((2,6-二氨基吡啶-3-基)二氮烯基)苯基酯HCl盐,为红色固体。
LCMS:97.21%(m/z:370.22[M+1]+,4.39min(运行10min),214nm)。
1
实施例-23:吗啉-4-甲酸(E)-2-((2,6-二氨基吡啶-3-基)二氮烯基)苯基酯的制备:
向处于二氯甲烷:吡啶(1:1.5mL)中的(E)-2-((2,6-二氨基吡啶-3-基)二氮烯基)苯酚盐酸盐(0.2g)的溶液中添加吗啉4-碳酰氯(0.279g)并在室温下搅拌24h。反应完成后(通过TLC监测),用冷水(10mL)淬灭反应物质并用二氯甲烷(3×30mL)进行萃取。将有机层用水(2×100mL)进行洗涤,用无水硫酸钠干燥,并减压浓缩以获得粗物质。用处于二氯甲烷中的9%甲醇作为洗脱剂,通过使用快速色谱法纯化粗品,以提供4-甲基哌嗪-1-甲酸(E)-2-((2,6-二氨基吡啶-3-基)二氮烯基)苯基酯(0.035g,13.5%),为黄色固体。
LCMS:98.18%(m/z:341.19[M+1]
1
实施例-24:4-甲基哌嗪-1-甲酸(E)-2-((2,6-二氨基吡啶-3-基)二氮烯基)苯基酯的制备
向处于二氯甲烷(10mL)中的(E)-2-((2,6-二氨基吡啶-3-基)二氮烯基)苯酚盐酸盐(0.2g)的溶液中添加4-甲基哌嗪-1-碳酰氯(0.305g)和4-(二甲基氨基)吡啶(0.045g)并在室温下搅拌24h。反应完成后(通过TLC监测),用冷水(10mL)淬灭反应物质并用二氯甲烷(3×30mL)进行萃取。将有机层用水(2×100mL)洗涤,用盐水洗涤,经无水硫酸钠干燥,并减压浓缩以获得粗物质。用10%甲醇:二氯甲烷作为洗脱剂,通过使用快速色谱法纯化粗品,以提供4-甲基哌嗪-1-甲酸(E)-2-((2,6-二氨基吡啶-3-基)二氮烯基)苯基酯(0.051g,19.1%),为黄色固体。
LCMS:96.78%(m/z:356.20,[M+1]
1
按照实施例-24的反应条件(酰氯、DMAP、DCM);已经合成了以下氨基甲酸酯前药,其中相应的酰氯是商业获得的。
实施例-26:(2-(二甲基氨基)乙基)(甲基)氨基甲酸(E)-2-((2,6-二氨基吡啶-3-基)二氮烯基)苯基酯盐酸盐的制备
在0℃下,向处于二氯甲烷(2.5mL)和吡啶(2.5mL)中的(E)-2-((2,6-二氨基吡啶-3-基)二氮烯基)苯酚盐酸盐(0.25g)的搅拌溶液中添加1,1’-羰基二咪唑(0.456g)和4-(二甲基氨基)吡啶(0.057g),并将所得混合物在0℃下搅拌30min,然后添加N
LCMS:98.96%(m/z:358.25[M+1]
1
按照实施例-26的反应条件(CDI、吡啶、DMAP、DCM);已经合成了以下氨基甲酸酯前药,其中相应的仲胺是商业获得的。
/>
/>
实施例35:4-吗啉代哌啶-1-羧酸(E)-2-((2,6-二氨基吡啶-3-基)二氮烯基)苯基酯的制备
在0℃下,向处于二氯甲烷(10mL)中的(E)-2-((2,6-二氨基吡啶-3-基)二氮烯基)苯酚盐酸盐(0.2g)的溶液中添加吡啶(0.288g)、1,1’-羰基二咪唑(0.6g)和4-(二甲基氨基)吡啶(0.046g),并在室温下搅拌30min。之后,加入4-(哌啶-4-基)吗啉(0.32g)并将反应混合物在室温下搅拌48h。反应完成后(通过TLC监测),用冷水(10mL)淬灭反应物质并用二氯甲烷(3×30mL)进行萃取。将有机层用水(2×100mL)进行洗涤,用无水硫酸钠干燥,并减压浓缩以获得粗物质。通过prep HPLC(使用TFA方法)纯化粗物质,以提供4-吗啉代哌啶-1-羧酸(E)-2-((2,6-二氨基吡啶-3-基)二氮烯基)苯基酯(0.094g,28.9%),为黄色固体。
LCMS:99.74%(m/z:426.24,[M+1]
1
按照实施例-35的反应条件(CDI、DMAP、DCM);已经合成了以下氨基甲酸酯前药,其中相应的仲胺是商业获得的。
实施例39:(E)-2-((2,6-二氨基吡啶-3-基)二氮烯基)苯基乙基碳酸酯的制备
在0℃温度下,向处于二氯甲烷(8mL)中的(E)-2-((2,6-二氨基吡啶-3-基)二氮烯基)苯酚盐酸盐(0.3g)的溶液中添加三乙胺(0.4mL)和氯甲酸乙酯(0.183g)。将所得反应混合物在室温下搅拌2h。反应完成后(通过TLC监测),用冷水(10mL)淬灭反应物质并用二氯甲烷(3×30mL)进行萃取。将合并的有机层用水(2×100mL)洗涤,经无水硫酸钠干燥,并减压浓缩,以得到粗品。通过快速色谱法(处于己烷中的70%乙酸乙酯)纯化粗品,以提供(E)-2-((2,6-二氨基吡啶-3-基)二氮烯基)苯基乙基碳酸酯(0.095g,28%),为橙色固体。
LCMS:97.86%(m/z:302.07,[M+1]
1
实施例40:(E)-N-(6-氨基-5-((2-((叔丁基二甲基硅基)氧基)苯基)二氮烯基)吡啶-2-基)乙酰胺的制备
步骤-01:(E)-3-((2-((叔丁基二甲基硅基)氧基)苯基)二氮烯基)吡啶-2,6-二胺的制备
在0℃温度下,向处于二氯甲烷(25mL)中的((E)-2-((2,6-二氨基吡啶-3-基)二氮烯基)苯酚(2.0g)的搅拌溶液中添加三乙胺(1.98mL)和叔丁基二甲基氯硅烷(2.2g)。将所得反应混合物在室温下另外搅拌4h。反应完成后(通过TLC监测),将混合物用水(20mL)稀释并用二氯甲烷(3×70mL)萃取。将合并的有机层经无水硫酸钠干燥并减压浓缩,以得到粗品。粗品通过组合快速(combi-flash)色谱法进行纯化(使用处于庚烷中的23%乙酸乙酯),以提供(E)-3-((2-((叔丁基二甲基硅基)氧基)苯基)二氮烯基)吡啶-2,6-二胺(2.6g,99%),为红色固体。
LCMS:99.69%(m/z:344.46),[M+1]
1
步骤-02:(E)-N-(6-氨基-5-((2-((叔丁基二甲基硅基)氧基)苯基)二氮烯基)吡啶-2-基)乙酰胺的制备
在0℃温度下,向处于二氯甲烷(6mL)中的(E)-3-((2-((叔丁基二甲基硅基)氧基)苯基)二氮烯基)吡啶-2,6-二胺(0.4g)的搅拌溶液中添加三乙胺(0.32mL)和乙酰氯(0.16mL)。将所得反应混合物在室温下搅拌3h。反应完成后(通过TLC监测),将混合物用水(20mL)稀释并用二氯甲烷(3×70mL)进行萃取。将合并的有机层用无水硫酸钠干燥并减压浓缩以获得粗品。通过组合快速色谱法(处于正庚烷中的18%乙酸乙酯)纯化粗品,以提供(E)-N-(6-氨基-5-((2-((叔丁基二甲基硅基)氧基)苯基)二氮烯基)吡啶-2-基)乙酰胺(0.350g,89%),为黄色固体。
分析细节:
LCMS:95.04%(m/z:385.19),[M+1]
步骤-03:(E)-N-(6-氨基-5-((2-((叔丁基二甲基硅基)氧基)苯基)二氮烯基)吡啶-2-基)乙酰胺的制备
在0℃下,向处于二氯甲烷(5mL)中的(E)-N-(6-氨基-5-((2-((叔丁基二甲基硅基)氧基)苯基)二氮烯基)吡啶-2-基)乙酰胺(0.35g)的搅拌溶液中加入三氟乙酸(4mL),然后将反应混合物在室温下搅拌3h。反应完成后(通过TLC监测),用二氯甲烷(2×20mL)对反应物质进行萃取,随后用水(2×50mL)洗涤。将合并的有机层用无水硫酸钠干燥并减压浓缩以获得粗品。通过乙醚洗涤来纯化粗品,以提供(E)-N-(6-氨基-5-((2-羟基苯基)二氮烯基)吡啶-2-基)乙酰胺(67mg,27.9%),为黄色固体。
LCMS:98.69%(m/z:271.11),[M+1]
1
实施例-41:(E)-(6-氨基-5-((2-羟基苯基)二氮烯基)吡啶-2-基)氨基甲酸甲酯的制备
步骤-01:(E)-3-((2-((叔丁基二甲基硅基)氧基)苯基)二氮烯基)吡啶-2,6-二胺的制备
在0℃温度下,向处于二氯甲烷(25mL)中的((E)-2-((2,6-二氨基吡啶-3-基)二氮烯基)苯酚(2g)的搅拌溶液中添加三乙胺(1.98mL)和叔丁基二甲基氯硅烷(2.2g)。将所得反应混合物在室温下另外搅拌4h。反应完成后(通过TLC监测),将混合物用水(20mL)稀释并用二氯甲烷(3×70mL)萃取。将收集的有机物用无水硫酸钠干燥并减压浓缩以获得粗品。通过组合快速色谱法(使用处于庚烷中的23%乙酸乙酯)纯化粗品,提供(E)-3-((2-((叔丁基二甲基硅基)氧基)苯基)二氮烯基)吡啶-2,6-二胺(2.6g,99%),为红色固体。
LCMS:99.69%(m/z:344.46),[M+1]
1
步骤-02:(E)-(6-氨基-5-((2-((叔丁基二甲基硅基)氧基)苯基)二氮烯基)吡啶-2-基)氨基甲酸甲酯的制备
在0℃温度下,向处于二氯甲烷(6mL)中的(E)-3-((2-((叔丁基二甲基硅基)氧基)苯基)二氮烯基)吡啶-2,6-二胺(0.3g)的搅拌溶液中添加三乙胺(0.2mL)和氯甲酸甲酯(0.165mL)。将所得反应混合物在室温下搅拌4h。反应完成后(通过TLC监测),将混合物用水(20mL)稀释并用二氯甲烷(3×70mL)进行萃取。将收集的有机物用无水硫酸钠干燥并减压浓缩以获得粗品。通过组合快速色谱法(处于正庚烷中的18%乙酸乙酯)纯化粗品,以提供(E)-(6-氨基-5-((2-((叔丁基二甲基硅基)氧基)苯基)二氮烯基)吡啶-2-基)氨基甲酸甲酯(0.16g,50.7%),为黄色固体。
分析细节:
LCMS:80.58%(m/z:402.47),[M+1]
步骤-03:(E)-(6-氨基-5-((2-羟基苯基)二氮烯基)吡啶-2-基)氨基甲酸甲酯的制备
在0℃温度下,向处于二氯甲烷(5mL)中的(E)-(6-氨基-5-((2-((叔丁基二甲基硅基)氧基)苯基)二氮烯基)吡啶-2-基)氨基甲酸甲酯(0.16g)的搅拌溶液中添加三氟乙酸(2mL),然后将反应混合物在室温下搅拌3h。反应完成后(通过TLC监测),将反应物质用二氯甲烷(2×20mL)进行萃取,随后用水(2×50mL)洗涤。将收集的有机物用无水硫酸钠干燥并减压浓缩以获得粗品。通过组合快速色谱法(使用处于正己烷中的25%乙酸乙酯)纯化粗品,以提供(E)-(6-氨基-5-((2-羟基苯基)二氮烯基)吡啶-2-基)氨基甲酸甲酯(40mg,36.3%),为黄色固体。
LCMS:98.84%(m/z:288.13),[M+1]
1
按照实施例-41的反应条件(氯甲酸酯、TEA、DCM);已经合成了以下氨基甲酸酯前药,其中相应的氯甲酸酯衍生物是商业获得的。
/>
实施例-46:(E)-(6-氨基-5-((2-羟基苯基)二氮烯基)吡啶-2-基)氨基甲酸辛酯的制备
步骤-01:(E)-(6-氨基-5-((2-((叔丁基二甲基硅基)氧基)苯基)二氮烯基)吡啶-2-基)氨基甲酸辛酯的制备
在0℃温度下,向处于二氯甲烷(12mL)中的(E)-3-((2-((叔丁基二甲基硅基)氧基)苯基)二氮烯基)吡啶-2,6-二胺(0.3g)的搅拌溶液中添加三光气(0.128g)和N,N’-二异丙基乙胺(0.25mL)。将该反应混合物在相同温度下另外搅拌15min。然后,将辛醇(0.12g)添加到上述混合物中,并在室温下继续搅拌另外16h。反应完成后(通过TLC监测:处于庚烷中的30%乙酸乙酯),用水(25mL)稀释混合物,随后用二氯甲烷(3×50mL)进行萃取。将收集的有机层用无水硫酸钠干燥并减压浓缩以获得粗品。将粗品通过快速色谱法纯化(使用处于正庚烷中的15%乙酸乙酯),以提供(E)-(6-氨基-5-((2-((叔丁基二甲基硅基)氧基)苯基)二氮烯基)吡啶-2-基)氨基甲酸辛酯(0.35g,85%),为棕色固体。
分析细节:
LCMS:97.04%(m/z:500.17),[M+1]
步骤-02:(E)-(6-氨基-5-((2-羟基苯基)二氮烯基)吡啶-2-基)氨基甲酸辛酯的制备
在0℃温度下,向处于二氯甲烷(10mL)中的(E)-(6-氨基-5-((2-((叔丁基二甲基硅基)氧基)苯基)二氮烯基)吡啶-2-基)氨基甲酸辛酯(0.3g)的搅拌溶液中加入三氟乙酸(1.1mL)并将所得溶液在室温下搅拌2h。通过TLC(处于庚烷中的70%乙酸乙酯)监测反应的完成。反应完成后,减压蒸发过量的溶剂,以获得粗品。通过用正戊烷洗涤、随后用二乙醚洗涤来纯化粗品,以得到(E)-(6-氨基-5-((2-羟基苯基)二氮烯基)吡啶-2-基)氨基甲酸辛酯(0.14g,31%),为红色固体。
LCMS:99.05%(m/z:386.09),[M+1]
1
按照实施例-46的反应条件(三光气、DIPEA、DCM);已经合成了以下氨基甲酸酯前药,其中相应的醇衍生物是商业获得的。
实施例-49:(E)-2-((2,6-二氨基吡啶-3-基)二氮烯基)苯基二氢磷酸酯的制备
步骤-01:(E)-二苄基((2-((2,6-二氨基吡啶-3-基)二氮烯基)苯基)磷酸酯的制备:
在0℃温度下,向处于四氢呋喃(20mL)中的叔丁基(E)-2-((2,6-二氨基吡啶-3-基)二氮烯基)苯酚盐酸盐(1g)的搅拌溶液中添加双(三甲基硅基)氨基钠(1M在THF中)并将所得混合物在相同温度下搅拌15min。之后,在相同温度下加入二磷酸四苄酯(4.6g)并将反应混合物在室温下继续搅拌30min。反应完成后(通过TLC监测:处于庚烷中的50%乙酸乙酯),用氯化铵淬灭混合物,随后用乙酸乙酯进行萃取。将收集的有机物用无水硫酸钠干燥并减压浓缩以获得粗品(2g,粗品)。粗品无需进一步纯化即可进行下一步骤。
LCMS:60.69%(m/z:491.20,[M+1]
步骤-02:(E)-2-((2,6-二氨基吡啶-3-基)二氮烯基)苯基二氢磷酸酯的制备
在0℃温度下,向处于二氯甲烷中的(E)-二苄基(2-((2,6-二氨基吡啶-3-基)二氮烯基)苯基)磷酸酯(1g)的搅拌溶液中添加三甲基溴硅烷(0.86mL),并且将所得反应混合物在室温下搅拌15min。反应完成后(通过TLC监测:处于庚烷中的50%乙酸乙酯),减压蒸发过量的溶剂以获得粗物质。通过prep-HPLC[(NH
LCMS:95.67%(m/z:310.04,[M+1]
1
实施例-50:(E)-(2-((2,6-二氨基吡啶-3-基)二氮烯基)苯氧基)甲基二氢磷酸酯的制备
步骤-01:(E)-二叔丁基((2-((2,6-二氨基吡啶-3-基)二氮烯基)苯氧基)甲基)磷酸酯的制备
向处于N,N’-二甲基甲酰胺(6mL)中的(E)-2-((2,6-二氨基吡啶-3-基)二氮烯基)苯酚氯化氢(0.3g)的搅拌溶液中添加碳酸钾(0.937g),然后添加二叔丁基(氯甲基)磷酸酯(1.16g)。将该溶液在60℃温度下搅拌另外6h。反应完成后(通过LCMS监测),用水(25mL)淬灭混合物并用乙酸乙酯(2×50mL)进行萃取。将合并的有机层用冷冻水洗涤,用无水硫酸钠进行干燥,并减压蒸发以获得(E)-二叔丁基((2-((2,6-二氨基吡啶-3-基)二氮烯基)苯氧基)甲基)磷酸酯(0.500g,粗品)。粗品无需进一步纯化即可用于下一步骤。
LCMS:37.14%(m/z:452.51),[M+1]
步骤-02:(E)-(2-((2,6-二氨基吡啶-3-基)二氮烯基)苯氧基)甲基二氢磷酸酯的制备
在0℃温度下,向处于二氯甲烷(10mL)中的(E)-二叔丁基((2-((2,6-二氨基吡啶-3-基)二氮烯基)苯氧基)甲基)磷酸酯(0.5g)的搅拌溶液中添加三氟乙酸(2mL)。将所得溶液在0℃温度下搅拌另外30min。反应完成后(通过LCMS监测),将混合物减压蒸发(低于25℃)以获得粗品。通过prep HPLC(使用碳酸氢铵作为缓冲剂)纯化粗品,以得到(E)-(2-((2,6-二氨基吡啶-3-基)二氮烯基)苯氧基)甲基二氢磷酸酯(0.012g,3.2%),为黄色固体。
LCMS:95.81%(m/z:340.05[M+1]
1
实施例-51:丁酸(E)-2,6-二氨基-5-(苯基二氮烯基)吡啶-3-基酯的制备步骤-01:2,6-二硝基吡啶-3-醇的制备
在0℃的温度下,向乙酸酐(14mL)和硝酸(6.2mL)的预混合溶液中滴加2-硝基吡啶-3-醇(5g)的溶液。将所得溶液在0℃下搅拌2h,然后在室温下搅拌另外2h。反应完成后(通过TLC监测),用冰水淬灭反应物质并用二氯甲烷(3×150mL)进行萃取。将合并的有机层用无水硫酸钠进行干燥,减压蒸发以得到粗品。将粗品用正戊烷研磨,然后减压干燥,以提供2,6-二硝基吡啶-3-醇(3.4g,51%),为黄色固体。
1
步骤-02:丁酸2,6-二氨基吡啶-3-基酯的制备
在0℃温度下,向处于二氯甲烷(100mL)中的2,6-二硝基吡啶-3-醇(1g)的搅拌溶液中添加三乙胺(1.7mL)和丁酰氯(1.09g)。然后,将所得混合物在50℃下搅拌2.5h。反应完成后(通过TLC监测),将反应物质冷却至室温,然后倒至冰水(300mL)上。用乙酸乙酯(3×150mL)对反应物质进行萃取,然后用水(2×100mL)洗涤。将合并的有机层用无水硫酸钠干燥,减压蒸发,以提供3-((4-甲氧基苄基)氧基)-2,6-二硝基吡啶(1g,粗品),为棕色油状物。粗产物无需纯化即可用于下一步骤。
1
步骤-03:丁酸2,6-二氨基吡啶-3-基酯
向处于乙酸(20mL)中的3-((4-甲氧基苄基)氧基)-2,6-二硝基吡啶(1g)的搅拌溶液中添加铁粉(1.5g),并将所得混合物在室温下搅拌1h。反应完成后(通过TLC监测),用乙酸乙酯(100mL)稀释反应物质。使反应物质通过硅藻土床以除去无机物,随后用乙酸乙酯洗涤数次。将合并的有机物用水(2×200mL)洗涤,用无水硫酸钠干燥,并减压浓缩,以获得3-((4-甲氧基苄基)氧基)吡啶-2,6-二胺,为棕色油状物(0.80g,粗品)。产物无需进一步纯化即可用于下一步骤。
LCMS:42.03%((m/z:195.22),[M+1]
1
步骤-04:丁酸(E)-2,6-二氨基-5-(苯基二氮烯基)吡啶-3-基酯的制备
向处于水(1.5mL)中的亚硝酸钠(0.28g)的搅拌溶液中添加苯胺(0.38g)和6N盐酸(0.56mL)。将所得混合物在室温下搅拌30min。此后,将3-((4-甲氧基苄基)氧基)吡啶-2,6-二胺(0.8g;溶解于水:5.0mL)添加至反应物质中,然后将其在室温下搅拌另外1h。反应完成后(通过TLC监测),将其用乙酸钠淬灭并搅拌15min。将反应混合物用乙酸乙酯(3×70mL)萃取,用水(2×200mL)洗涤,收集的有机物经无水硫酸钠干燥,减压蒸发以获得粗品。将粗品通过快速色谱法纯化(处于己烷中的50%乙酸乙酯),以提供丁酸(E)-2,6-二氨基-5-(苯基二氮烯基)吡啶-3-基酯(0.063g,5.16%),为红色固体。
LCMS:99.14%(m/z:300.07,[M+1]
1
程序:
/>
实施例-56:二乙基氨基甲酸(E)-2,6-二氨基-5-(苯基二氮烯基)吡啶-3-基酯的制备
步骤-01:二乙基氨基甲酸2,6-二硝基吡啶-3-基酯的制备
向处于二氯甲烷(100mL)中的2,6-二硝基吡啶-3-醇(0.5g)的搅拌溶液中添加N,N’-二异丙基乙胺(0.75mL),随后在0℃温度下添加三光气(0.4g)。将所得混合物在室温下搅拌10min。之后,将二乙胺(0.296g)添加到上述混合物中,并将其在室温下搅拌另外16h。反应完成后(通过TLC监测),向其中添加水(100mL)并用乙酸乙酯(3×150mL)萃取反应物质。将合并的有机层用无水硫酸钠干燥,过滤,并减压蒸发,以提供粗品。使用快速色谱法纯化粗品,以提供二乙基氨基甲酸2,6-二硝基吡啶-3-基酯(330mg,46%),为黄色固体。该化合物无需进一步纯化即可用于下一步骤。
步骤-02:二乙基氨基甲酸2,6-二氨基吡啶-3-基酯的制备
向处于乙酸(20mL)中的二乙基氨基甲酸2,6-二硝基吡啶-3-基酯(330mg)的搅拌溶液中添加铁粉(0.063g)并将所得混合物在室温下搅拌1h。反应完成后(通过TLC监测),将反应物质通过硅藻土过滤并减压浓缩以获得粗品。将粗品进一步干燥,以提供二乙基氨基甲酸2,6-二氨基吡啶-3-基酯(300,粗品),为棕色油状物。产物无需纯化即可用于下一步骤。
LCMS:76.50%((m/z:225.05),[M+1]
步骤-03:二乙基氨基甲酸(E)-2,6-二氨基-5-(苯基二氮烯基)吡啶-3-基酯的制备
在0℃温度下,向处于水(5mL)和6N盐酸(1mL)中的苯胺(0.381g)的搅拌溶液中滴加处于水(1.5mL)中的亚硝酸钠(0.27g)的溶液。将所得混合物在0℃温度下搅拌30min。之后,在0℃温度下添加二乙基氨基甲酸2,6-二氨基吡啶-3-基酯(0.3g),并将所得反应物质在室温下再搅拌1h。反应完成后(通过TLC监测),用乙酸钠淬灭反应物质并将反应物再搅拌15min。将反应混合物用乙酸乙酯(3×70mL)萃取,用水(2×50mL)洗涤。将收集的有机层用无水硫酸钠干燥,减压蒸发,以得到粗品。将粗品通过快速色谱法纯化(处于己烷中的50%乙酸乙酯),以提供丁酸(E)-2,6-二氨基-5-(苯基二氮烯基)吡啶-3-基酯(0.063g,5.2%),为红色固体。
LCMS:99.96%((m/z:329.23),[M+1]
1
/>
实施例60:(E)-2-氨基-N-(6-氨基-5-((2-羟基苯基)二氮烯基)吡啶-2-基)乙酰胺的制备
步骤-1:(E)-(2-((6-氨基-5-((2-羟基苯基)二氮烯基)吡啶-2-基)氨基)-2-氧代乙基)氨基甲酸叔丁酯的制备
在0℃温度下,向处于N,N’-二甲基甲酰胺(20mL)中的(E)-2-((2,6-二氨基吡啶-3-基)二氮烯基)苯酚(2g)的搅拌溶液中添加氢化钠(处于矿物油中60%w/w分散体,0.87g),然后将反应混合物在室温下搅拌15min。之后,将N-boc-甘氨酸N-羟基琥珀酰亚胺酯(9.5g)添加至上述溶液中。将所得反应混合物在室温下搅拌4h。反应完成后(通过TLC监测,处于正庚烷中的50%乙酸乙酯),用冰冷的水(50mL)淬灭混合物。使用乙酸乙酯(2×50mL)对反应物质进行萃取,以提供粗品。通过快速色谱法纯化粗品,以提供0.18g(E)-(2-((6-氨基-5-((2-羟基苯基)二氮烯基)吡啶-2-基)氨基)-2-氧代乙基)氨基甲酸叔丁酯,为黄色固体。
LCMS纯度:95.31%(m/z:387,[M+1],0.83min(运行4min),214nm)。
步骤-2:(E)-2-氨基-N-(6-氨基-5-((2-羟基苯基)二氮烯基)吡啶-2-基)乙酰胺的制备
在0℃温度和氮气气氛下,向处于二氯甲烷(5mL)中的(E)-(2-((6-氨基-5-((2-羟基苯基)二氮烯基)吡啶-2-基)氨基)-2-氧代乙基)氨基甲酸叔丁酯(0.25g)的搅拌溶液中添加处于1,4-二噁烷(1mL)中的4M HCl溶液。将所得反应混合物在室温下搅拌2h。反应完成后(通过TLC监测),将混合物减压蒸发以获得粗物质。使用三氟乙酸作为改性剂,通过制备型HPLC纯化粗化合物,以提供0.11g的(E)-2-氨基-N-(6-氨基-5-((2-羟基苯基)二氮烯基)吡啶-2-基)乙酰胺单三氟乙酸盐,为黄色固体。
LCMS纯度:99.22%(m/z:287,[M+1],4.66min(运行10min),214nm)。
1
实施例-61:环丙烷甲酸(E)-2-((2-氨基-6-(2-氨基乙酰氨基)吡啶-3-基)二氮烯基)苯基酯的制备
步骤-1:环丙烷甲酸(E)-2-((2-氨基-6-(2-((叔丁氧羰基)氨基)乙酰氨基)吡啶-3-基)二氮烯基)苯基酯的制备
向处于二氯甲烷(7.5mL)中的(E)-(2-((6-氨基-5-((2-羟基苯基)二氮烯基)吡啶-2-基)氨基)-2-氧代乙基)氨基甲酸叔丁酯(0.5g,如实施例-60步骤-1中制备)的搅拌溶液中添加三乙胺(0.45mL),并将所得混合物在0℃搅拌5min。将环丙基甲酰氯(0.15mL)加入到上述反应混合物中,并将反应物在室温下搅拌另外1h。通过TLC监测反应完成后,用冷水(10mL)淬灭反应混合物。将反应物质用二氯甲烷(3×15mL)进行萃取,随后用水(2×30mL)洗涤。收集有机物,用无水硫酸钠干燥,减压浓缩,以获得粗物质。通过组合快速色谱法纯化粗品,以提供0.32g的环丙烷甲酸(E)-2-((2-氨基-6-(2-((叔丁氧羰基)氨基)乙酰氨基)吡啶-3-基)二氮烯基)苯基酯,为黄色固体。
LCMS纯度:72.80%(m/z:455.2,[M+1]
步骤-2:环丙烷甲酸(E)-2-((2-氨基-6-(2-氨基乙酰氨基)吡啶-3-基)二氮烯基)苯基酯的制备
向处于二氯甲烷(6mL)中的环丙烷甲酸(E)-2-((2-氨基-6-(2-((叔丁氧羰基)氨基)乙酰氨基)吡啶-3-基)二氮烯基)苯基酯(0.30g)的搅拌溶液中加入三氟乙酸(0.5mL)并将所得反应混合物在室温下搅拌1h。通过TLC监测反应完成后,减压浓缩溶剂。将获得的粗物质用二乙醚研磨以获得0.15g的环丙烷甲酸(E)-2-((2-氨基-6-(2-氨基乙酰氨基)吡啶-3-基)二氮烯基)苯基酯,为黄色固体。
LCMS纯度:83.55%(m/z:355.0[M+1]
实施例-62:环丙烷甲酸(E)-2-((2-氨基-6-(2-氨基乙酰氨基)吡啶-3-基)二氮烯基)苯基酯的制备
步骤-1:丁酸(E)-2-((2-氨基-6-(2-((叔丁氧羰基)氨基)乙酰氨基)吡啶-3-基)二氮烯基)苯基酯的制备
向处于二氯甲烷(10mL)中的氨基甲酸(E)-(2-((6-氨基-5-((2-羟基苯基)二氮烯基)吡啶-2-基)氨基)-2-氧代乙基)酯(0.3g,如实施例-60步骤-1中制备)的溶液中添加二环己基碳二亚胺(0.698g)和4-(二甲基氨基)吡啶(0.068g),并在室温下搅拌反应物质15min,随后添加丁酸(0.118g)并将所得反应混合物在室温下搅拌另外2h。反应完成后(通过TLC监测),用冷水(10mL)淬灭反应物质并用二氯甲烷(3×30mL)萃取。将合并的有机层用水(2×10mL)洗涤,经无水硫酸钠干燥,并减压浓缩以获得粗物质,将其通过制备型HPLC纯化,以提供0.21g的(E)-2-((2-氨基-6-(2-((叔丁氧羰基)氨基)乙酰氨基)吡啶-3-基)二氮烯基)苯基,为黄色固体。
LCMS纯度:45.76%(m/z:457.2,[M+1]
步骤2:丁酸((E)-2-((2-氨基-6-(2-氨基乙酰氨基)吡啶-3-基)二氮烯基)苯基酯的制备
向处于二氯甲烷(4mL)中的丁酸(E)-2-((2-氨基-6-(2-((叔丁氧羰基)氨基)乙酰氨基)吡啶-3-基)二氮烯基)苯基酯(0.21g)的搅拌溶液中加入三氟乙酸(0.3mL)并将所得反应混合物在室温下搅拌1h。通过TLC监测反应完成后,减压浓缩溶剂。将获得的粗物质用二乙醚研磨,以提供0.080g的丁酸((E)-2-((2-氨基-6-(2-氨基乙酰氨基)吡啶-3-基)二氮烯基)苯基酯,为浅棕色固体。
LCMS纯度:94.48%(m/z:357.2[M+1]
实施例63:吗啉-4-甲酸(E)-2-((2-氨基-6-(2-氨基乙酰氨基)吡啶-3-基)二氮烯基)苯基酯的制备
步骤1:吗啉-4-碳酰氯的制备
在0℃温度和氮气气氛下,向处于二氯甲烷(40mL)中的吗啉(4g)的搅拌溶液中添加三乙胺(11.2mL)和三光气(20.44g)。将所得反应混合物在室温下搅拌3h。反应完成后(通过TLC监测),用冰冷的水稀释反应物质,然后用二氯甲烷(3×100mL)进行萃取。将有机层用水(3×100mL)洗涤,经无水硫酸钠干燥,并减压浓缩,以提供粗品。将粗品通过快速色谱法纯化,以获得4.52g的吗啉-4-碳酰氯,为浅黄色液体。
1
步骤-2:吗啉-4-甲酸(E)-2-((2,6-二氨基吡啶-3-基)二氮烯基)苯基酯的制备
向处于吡啶(15mL)和二氯甲烷(15mL)中的(E)-2-((2,6-二氨基吡啶-3-基)二氮烯基)苯酚(2g)的搅拌溶液中添加吗啉-4-碳酰氯(5.2g)。将所得反应混合物在室温下搅拌16h。反应完成后(通过TLC监测),用1M盐酸溶液淬灭反应物质,随后用二氯甲烷(2×50mL)进行萃取。将合并的有机层用水(50mL)洗涤,经无水硫酸钠干燥,并减压浓缩以获得粗物质。通过快速色谱法纯化粗品,以提供1.02g的吗啉-4-甲酸(E)-2-((2,6-二氨基吡啶-3-基)二氮烯基)苯基酯,为黄色固体。
LCMS纯度:96.92%(m/z:343.30,[M+1]
1
步骤-3:吗啉-4-甲酸(E)-2-((2-氨基-6-(2-((叔丁氧羰基)氨基)乙酰氨基)吡啶-3-基)二氮烯基)苯基酯的制备
在0℃温度下,向处于N,N-二甲基甲酰胺(7.0mL)中的吗啉-4-甲酸(E)-2-((2,6-二氨基吡啶-3-基)二氮烯基)苯基酯(0.65g)的搅拌溶液中分批添加氢化钠(处于矿物油中的60%w/w分散体,0.19g)。将所得反应混合物在0℃下搅拌15min。之后,将boc-甘氨酸N-羟基琥珀酰亚胺酯(2.07g)添加至上述溶液中并将反应混合物在室温下搅拌16h。反应完成后(通过TLC监测),用冰水淬灭反应物质,随后使用乙酸乙酯(3×50mL)进行萃取。将合并的有机层用水(50mL)洗涤,经无水硫酸钠干燥,并减压浓缩以获得粗物质。通过快速色谱法纯化粗品,以提供0.34g的吗啉-4-甲酸(E)-2-((2-氨基-6-(2-((叔丁氧羰基)氨基)乙酰氨基)吡啶-3-基)二氮烯基)苯基酯,为黄色固体。
LCMS纯度:93.16%(m/z:500.13[M+1]
步骤-4:吗啉-4-甲酸(E)-2-((2-氨基-6-(2-氨基乙酰氨基)吡啶-3-基)二氮烯基)苯基酯的制备
在0℃温度下,向处于二氯甲烷(2mL)中的吗啉-4-甲酸(E)-2-((2-氨基-6-(2-((叔丁氧羰基)氨基)乙酰氨基)吡啶-3-基)二氮烯基)苯基酯(0.3g)的搅拌溶液中加入处于二噁烷(1mL)中的4M HCl。将所得反应混合物在室温下搅拌2h。反应完成后(通过TLC监测),减压蒸发反应物质以获得粗物质。将粗品用二乙醚研磨,然后使用制备型HPLC纯化,提供0.12g的吗啉-4-甲酸(E)-2-((2-氨基-6-(2-氨基乙酰氨基)吡啶-3-基)二氮烯基)苯基酯,为黄色固体。
LCMS纯度:95.22%(m/z:400.42,[M+1]
1
实施例-64:乙酸(E)-2-((2-氨基-6-(2-氨基乙酰氨基)吡啶-3-基)二氮烯基)苯基酯的制备
步骤-1:乙酸(E)-2-((2-氨基-6-(2-((叔丁氧羰基)氨基)乙酰氨基)吡啶-3-基)二氮烯基)苯基酯的制备
在0℃下,向处于二氯甲烷(3mL)中的(E)-(2-((6-氨基-5-((2-羟基苯基)二氮烯基)吡啶-2-基)氨基)-2-氧代乙基)氨基甲酸叔丁酯(0.11g)的搅拌溶液中加入三乙胺(0.11mL),随后加入乙酰氯(0.026mL)。将所得反应混合物在室温下搅拌2h。通过TLC表明反应完成后,用冰水(10mL)稀释反应物质并用饱和碳酸氢钠溶液进行洗涤。将有机层经无水硫酸钠干燥并减压浓缩,以提供0.12g的乙酸(E)-2-((2-氨基-6-(2-((叔丁氧羰基)氨基)乙酰氨基)吡啶-3-基)二氮烯基)苯基酯,为橙色固体。
LCMS纯度:93.45%(m/z:427.2,[M-1]
步骤-2:乙酸(E)-2-((2-氨基-6-(2-氨基乙酰氨基)吡啶-3-基)二氮烯基)苯基酯的制备
在0℃温度和氮气气氛下,向处于二氯甲烷(3mL)中的乙酸(E)-2-((2-氨基-6-(2-((叔丁氧羰基)氨基)乙酰氨基)吡啶-3-基)二氮烯基)苯基酯(0.1g)的搅拌溶液中添加三氟乙酸(0.2mL)。将所得反应混合物在室温下搅拌1h。通过TLC表明反应完成后,将反应物质用冰水(10mL)稀释并用饱和碳酸氢钠溶液洗涤。将有机层经无水硫酸钠干燥并减压浓缩,以提供0.075g的乙酸(E)-2-((2-氨基-6-(2-氨基乙酰氨基)吡啶-3-基)二氮烯基)苯基酯,为黄色固体。
LCMS纯度:94.83%(m/z:329.0[M+1]
实施例-65:(E)-2-氨基-N-(6-氨基-5-((2-((5-甲基-2-氧代-1,3-二氧杂环戊烯-4-基)甲氧基)苯基)二氮烯基)吡啶-2-基)乙酰胺的制备
步骤-1:(E)-(2-((6-氨基-5-((2-((5-甲基-2-氧代-1,3-二氧杂环戊烯-4-基)甲氧基)苯基)二氮烯基)吡啶-2-基)氨基)-2-氧代乙基)氨基甲酸叔丁酯的制备
在0℃温度和氮气气氛下,向处于四氢呋喃(3mL)中的(E)-(2-((6-氨基-5-((2-羟基苯基)二氮烯基)吡啶-2-基)氨基)-2-氧代乙基)氨基甲酸酯(0.1g,如实施例-60步骤-1中制备)的搅拌溶液中依次加入三苯基膦(0.088g)和偶氮二甲酸二异丙酯(0.068g),并将所得反应混合物在室温下搅拌10min,然后添加4-(羟甲基)-5-甲基-1,3-二氧杂环戊烯-2-酮(0.050g)。通过TLC表明反应完成后,用冰水(10mL)稀释反应物质并用乙酸乙酯(2×10mL)进行萃取。将有机层经无水硫酸钠干燥并减压浓缩,以得到粗物质,将其通过柱色谱法使用处于石油醚中的10%乙酸乙酯进行纯化,以提供0.03g的(E)-(2-((6-氨基-5-((2-((5-甲基-2-氧代-1,3-二氧杂环戊烯-4-基)甲氧基)苯基)二氮烯基)吡啶-2-基)氨基)-2-氧代乙基)氨基甲酸叔丁酯,为棕色半固体。
LCMS纯度:31.28%(m/z:497.2,[M-1]
步骤-2:(E)-2-氨基-N-(6-氨基-5-((2-((5-甲基-2-氧代-1,3-二氧杂环戊烯-4-基)甲氧基)苯基)二氮烯基)吡啶-2-基)乙酰胺的制备
在0℃温度下,向处于二氯甲烷(1.5mL)中的(E)-(2-((6-氨基-5-((2-((5-甲基-2-氧代-1,3-二氧杂环烯-4-基)甲氧基)苯基)二氮烯基)吡啶-2-基)氨基)-2-氧代乙基)氨基甲酸叔丁酯(0.03g)的搅拌溶液中添加三氟乙酸(0.1mL)。将所得反应混合物在室温下搅拌1h。通过TLC表明反应完成后,用冰水(10mL)稀释反应物质并用饱和碳酸氢钠溶液洗涤。将有机层经无水硫酸钠干燥并减压浓缩,以提供0.015g的(E)-2-氨基-N-(6-氨基-5-((2-((5-甲基-2-氧代-1,3-二氧杂环戊烯-4-基)甲氧基)苯基)二氮烯基)吡啶-2-基)乙酰胺,为橙色固体。
LCMS纯度:29.11%(m/z:399.0,[M+1]
实施例-66:(E)-2-((2-氨基-6-(2-氨基乙酰氨基)吡啶-3-基)二氮烯基)苯基异丁基碳酸酯的制备
步骤-1:(E)-(2-((6-氨基-5-((2-((异丁氧羰基)氧基)苯基)二氮烯基)吡啶-2-基)氨基)-2-氧代乙基)氨基甲酸叔丁酯的制备
在0℃温度和氮气气氛下,向处于二氯甲烷(3mL)中的(E)-(2-((6-氨基-5-((2-羟基苯基)二氮烯基)吡啶-2-基)氨基)-2-氧代乙基)氨基甲酸酯(0.10g;如实施例-60步骤-1中制备)的搅拌溶液中添加三乙胺(0.1mL),随后添加氯甲酸异丁酯(0.045mL)。将所得反应混合物在室温下搅拌2h。通过TLC表明反应完成后,用冰冷的水(10mL)稀释反应物质并用饱和碳酸氢钠溶液洗涤。将有机层用无水硫酸钠干燥并减压浓缩,以提供0.10g的(E)-(2-((6-氨基-5-((2-((异丁氧羰基)氧基)苯基)二氮烯基)吡啶-2-基)氨基)-2-氧代乙基)氨基甲酸叔丁酯,为浅棕色固体。
LCMS纯度:97.15%(m/z:487.0,[M+1]
步骤-2:(E)-2-((2-氨基-6-(2-氨基乙酰氨基)吡啶-3-基)二氮烯基)苯基异丁基碳酸酯的制备
在0℃温度和氮气气氛下,向处于二氯甲烷(3mL)中的(E)-(2-((6-氨基-5-((2-((异丁氧羰基)氧基)苯基)二氮烯基)吡啶-2-基)氨基)-2-氧代乙基)氨基甲酸叔丁酯(0.1g)的搅拌溶液中添加三氟乙酸(0.3mL)。将所得反应混合物在室温下搅拌1h。通过TLC表明反应完成后,用冰冷的水(10mL)稀释反应物质并用饱和碳酸氢钠溶液洗涤。将有机层用无水硫酸钠干燥并减压浓缩,以提供0.07g的(E)-2-((2-氨基-6-(2-氨基乙酰氨基)吡啶-3-基)二氮烯基)苯基异丁基碳酸酯,为黄色固体。
LCMS纯度:94.27%(m/z:487.1[M+1]
实施例67:(E)-2,6-二氨基-5-(苯基二氮烯基)吡啶-3-基甲基碳酸酯的制备
步骤-1:2-吗啉代乙酸(E)-2-((2-氨基-6-(2-((叔丁氧羰基)氨基)乙酰氨基)吡啶-3-基)二氮烯基)苯基酯
在0℃温度下,向处于二氯甲烷(4mL)中的(E)-(2-((6-氨基-5-((2-羟基苯基)二氮烯基)吡啶-2-基)氨基)-2-氧代乙基)氨基甲酸叔丁酯(0.10g,如实施例-60步骤-1中制备)的搅拌溶液中依次加入二环己基碳二亚胺(0.13g)和4-(二甲基氨基)吡啶(0.039g)。将所得反应混合物在相同温度下搅拌30min,然后添加2-吗啉代乙酸(0.056g)并在室温下搅拌另外2h。通过TLC表明反应完成后,用冰冷的水(50mL)稀释反应物质并用乙酸乙酯(3×200mL)进行萃取。将合并的有机层经无水硫酸钠干燥并减压浓缩,得到粗物质,将其进行柱色谱法,使用处于石油醚中的30%乙酸乙酯作为流动相,其提供0.11g的2-吗啉代乙酸(E)-2-((2-氨基-6-(2-((叔丁氧羰基)氨基)乙酰氨基)吡啶-3-基)二氮烯基)苯基酯,为黄色固体。
LCMS纯度:69%(m/z:514.1[M+1]
步骤-2:2-吗啉代乙酸(E)-2-((2-氨基-6-(2-氨基乙酰氨基)吡啶-3-基)二氮烯基)苯基酯的制备
在0℃温度和氮气气氛下,向处于二氯甲烷(3mL)中的2-吗啉代乙酸(E)-2-((2-氨基-6-(2-((叔丁氧羰基)氨基)乙酰氨基)吡啶-3-基)二氮烯基)苯基酯(0.1g)的搅拌溶液中添加三氟乙酸(0.3mL)。将所得反应混合物在室温下搅拌1h。通过TLC表明反应完成后,用冰冷的水(10mL)稀释反应物质并用饱和碳酸氢钠溶液洗涤。将有机层用无水硫酸钠干燥并减压浓缩,以提供0.06g的2-吗啉代乙酸(E)-2-((2-氨基-6-(2-氨基乙酰氨基)吡啶-3-基)二氮烯基)苯基酯,为深橙色固体。
LCMS纯度:35.50%(m/z:412.2[M-1]
实施例-68:(E)-2,6-二氨基-5-(苯基二氮烯基)吡啶-3-基甲基碳酸酯的制备
步骤1:(E)-3-溴-5-(苯基二氮烯基)吡啶-2,6-二胺的合成
在0℃温度下,向处于水(5mL)中的苯胺(2.9g)的搅拌溶液中添加6N盐酸(3.01g),随后添加亚硝酸钠(2.2g),并将所得悬浮液在0℃下搅拌另外30min,然后滴加处于6N HCl(25mL)中的3-溴吡啶-2,6-二胺(5g)的溶液。将所得反应混合物在室温下搅拌另外2h。通过TLC监测反应完成后,用乙酸钠(30mL)淬灭反应物质并搅拌15min。用乙酸乙酯(100mL×3)对反应物质进行萃取,然后用水(300mL×2)洗涤。将合并的有机层经无水硫酸钠干燥,并减压浓缩以获得粗品。通过快速色谱法纯化粗品,以提供2.8g的(E)-3-溴-5-(苯基二氮烯基)吡啶-2,6-二胺,为红色固体。
LCMS纯度:86.73%(m/z:293.92,[M+2]
1
步骤2:(E)-3-(苯基二氮烯基)-5-(4,4,5,5-四甲基-1,3,2-二氧杂环戊硼烷-2-基)吡啶-2,6-二胺的合成
在室温下,向处于二噁烷(30mL)中的(E)-3-溴-5-(苯基二氮烯基)吡啶-2,6-二胺(2.8g)的搅拌溶液中添加双(频哪醇合)二硼(14.4g)、乙酸钾(1.41g)和三环己基膦四氟硼酸盐(0.353g)。接下来的15分钟使用连续的氩气流对反应混合物进行脱气。此后,加入Pd
LCMS纯度:75%(m/z:340.33,[M+1]
1
步骤3:(E)-2,6-二氨基-5-(苯基二氮烯基)吡啶-3-醇的合成
向处于四氢呋喃(40mL)中的(E)-3-(苯基二氮烯基)-5-(4,4,5,5-四甲基-1,3,2-二氧杂环戊硼烷-2-基)吡啶-2,6-二胺(2.2g)的搅拌溶液中添加过氧化氢(0.424g)并将所得溶液在室温下搅拌2h。通过TLC监测反应完成后,用乙酸乙酯(40mL)稀释反应物质。将有机层用水(100mL×2)洗涤,经无水硫酸钠干燥并减压浓缩,以得到粗品。使用三氟乙酸作为改性剂,通过制备型HPLC纯化粗品,以提供0.44g的(E)-2,6-二氨基-5-(苯基二氮烯基)吡啶-3-醇,为红色固体。
LCMS纯度:97%(m/z:230.03,[M+1]
1
步骤-4:(E)-2,6-二氨基-5-(苯基二氮烯基)吡啶-3-基甲基碳酸酯的合成
在0℃温度下,向处于二氯甲烷(3mL)中的(E)-2,6-二氨基-5-(苯基二氮烯基)吡啶-3-醇(0.08g)的搅拌溶液中加入三乙胺(0.045g)和氯甲酸甲酯(0.039g),并将所得反应混合物在室温下搅拌10min。通过TLC监测反应完成后,用水(3mL)淬灭反应物质,用二氯甲烷(3×30mL)进行萃取,用盐水(2×100mL)洗涤,经无水硫酸钠干燥,并减压浓缩以获得粗品。使用三氟乙酸作为改性剂,通过制备型HPLC纯化粗品,以得到0.018g的(E)-2,6-二氨基-5-(苯基二氮烯基)吡啶-3-基甲基碳酸酯,为棕色固体。
LCMS纯度:98.09%(m/z:288.10[M+1]
1
实施例69.基因表达分析
研究设计:为了评估测试化合物对疼痛相关标志物的效果,我们利用了人尿路上皮“RT4(人类膀胱癌细胞)”细胞,用测试化合物与环磷酰胺(CYP)组合处理,并进行了疼痛相关基因的基因表达分析。对于基因表达研究,用环磷酰胺和测试化合物处理0.5×10
4h孵育结束时,从所有孔中除去培养基并用无菌PBS洗涤细胞一次。按照制造商的说明使用Qiagen RNA迷你试剂盒(Cat#74104)进行RNA分离。随后利用Superscript IIIFirst strand cDNA合成系统(Thermo fisher,Cat No 18080051)每个样品使用3μg的总RNA进行cDNA合成。使用SYBR green master mix(PCR Biosystems,Cat No:-PB20.14)和P2RX3基因特异性引物进行qRT-PCR。PCR完成后,使用CFX管理软件分析数据。使用以下公式计算表达的倍数变化:2^-ddCT=2^-((CT靶标–CT内部对照)
结果:如图1所示,当与对照细胞(未处理)相比时,对RT4细胞进行CYP处理4h增加了P2RX3的基因表达。有趣的是,当与CYP对照相比时,暴露于10μM浓度的测试化合物IX的CYP处理的细胞表现出P2RX3基因表达的降低。
实施例70.疼痛评估-CFA模型
研究设计:为了评估测试化合物对炎性疼痛的效果,我们使用弗氏完全佐剂(Complete Freund’s Adjuvant,CFA)作为诱导剂来产生炎症,并基于缩爪阈值(PWT)测量疼痛反应。将CFA乳剂(75μg/150μL/大鼠)注射到大鼠后爪足底内的表面以诱导机械性痛觉超敏。CFA注射后,使用Von Frey仪器测量基线同侧PWT(iPWT)。在开始实验之前和Von Frey测试之前,使大鼠适应测试室~30min。为了确定最低机械阈值,将2g细丝施加到后爪,如果长达3秒没有缩回反应,将提示使用具有增加的力的下一个细丝。相反,3秒内的后爪缩回反应将提示使用具有减小的力的下一个细丝。iPWT≤4g的动物被认为是痛觉过敏的并被用于研究。组1:溶媒对照,组2:CFA对照(75μg/150μL,经口服(PO)),组3:CFA+化合物XI(20mg/kg,PO),组4:CFA+化合物XI(5mg/kg,IV),组5:CFA+萘普生(20mg/kg,PO),组6:CFA+加巴喷丁(100mg/kg,IP)。以萘普生和加巴喷丁为参考标准。在给药后0.5小时和4小时时记录PWT。使用以下公式计算机械性痛觉超敏的最大可能效果(MPE)%:
其中15=鼠的机械性痛觉超敏的截止阈值(最大的力)。
%MPE如图2所示
实施例71.疼痛评估-SNL模型
研究设计:为了评估测试化合物对神经性疼痛的效果,我们使用脊髓神经结扎(Spinal Nerve Ligation,SNL)技术并基于缩爪阈值(PWT)测量疼痛反应。
在较低的腰椎和骶骨水平上进行纵向切口,然后去除肌肉和结缔组织。在SNL组中去除覆盖L4和L5脊神经的L6横突并紧密结扎L5和L6脊神经,而在Sham对照组中仅进行纵向切口。手术后进行抗生素处理。为了测量PWT,将传感器的尖端放置在大鼠后爪的中部并线性施加压力直至观察到明显的缩回。每只动物重复3次,每次读数间隔5min,平均压力值被认为是最终的PWT记录。SNL手术后第6天,将大鼠置于动态足底触觉计仪器内30min以适应环境,并记录基线反应,选择对~20g疼痛力有反应的动物并随机分为不同组,如组1:Sham对照(不进行SNL),组2:溶媒对照(SNL),组3:SNL+化合物XI(20mg/kg,PO),组4:SNL+化合物XI(5mg/kg,IV),组5:SNL+加巴喷丁(100mg/kg,PO)。单次服用化合物XI后,在手术后第7天,使大鼠适应15min,并使用动态足底触觉计在给药后1小时和4小时记录机械痛觉超敏-缩爪阈值(PWT)。最大力为50g,波形时间为20秒。完成平均PWT记录后,计算与溶媒对照相比,平均PWT的变化%(图3)。
实施例72-化合物针对急性的环磷酰胺(CYP)诱导的膀胱炎模型的功效
实验系统-在雌性SD大鼠(6-8周龄)上进行研究。
研究方案-通过以150mg/kg体重的剂量单次腹膜内给予环磷酰胺,在实验动物中诱导急性间质性膀胱炎。表中提到了研究组、剂量和给予途径(RoA)。向正常对照组给予盐水。在环磷酰胺给予之前30min给予测试化合物。在环磷酰胺给予前1h给予参考化合物布洛芬。对照组仅给予溶媒。在注射CYP或盐水之前(基底,0h)和CYP给予后4h评估伤害性反应。将增加力的Von Frey细丝施加到下腹部,靠近膀胱。处死动物,通过腹部下中线切口取出膀胱。
/>
图4显示了CYP给药后4小时伤害性疼痛评分的AUC。
实施例73-化合物XI对慢性的环磷酰胺(CYP)诱导的膀胱炎模型的功效
实验系统-在雌性SD大鼠(6-7周龄)上进行研究。
研究方案-通过在第0天、第3天和第6天以50mg/kg体重的剂量腹膜内给予环磷酰胺来诱导慢性间质性膀胱炎。向正常对照组给予盐水。在第7天,通过使用Von Frey细丝对动物进行伤害性反应评估。在测试之前,将每只动物的下腹部区域剃毛以进行机械刺激。连续地施加分级力为1g至60g的Von Frey细丝,并根据疼痛行为等级量表记录疼痛行为。基于体重作为主要标准和疼痛评分作为次要标准,将动物随机分为不同的处理组。选择表现出最佳疼痛反应的动物进行随机分组,发现各组的平均疼痛评分在±10%以内。在下表中提及了测试项目、剂量、给药频率和给予途径。从第8天到第10天为期3天,给予测试化合物或参考化合物。如研究设计表中所示,通过口服途径每天给予一次或两次测试化合物。每天口服给予一次布洛芬,持续3天。对照组仅给予溶媒。在第10天最后一次给药后,在给药后1h记录伤害性行为。
IP-腹膜内注射,QD-每天一次,BID-每天两次
图5显示了第10天(处理三天后)的AUC疼痛评分。
实施例74-化合物对急性的环磷酰胺(CYP)诱导的膀胱炎模型的功效
实验系统-在雌性SD大鼠(6-7周龄)上进行研究。
研究方案-通过以150mg/kg体重的剂量单次腹膜内给予环磷酰胺,在实验动物中诱导急性间质性膀胱炎。在表中提及了研究组、剂量和给予途径(RoA)。向正常对照组给予盐水。在环磷酰胺给予前30min给予测试化合物。在环磷酰胺给予前1h给予参考化合物布洛芬。对照组仅给予溶媒。在注射CYP或盐水之前(基底,0h)和CYP给予后4h评估伤害性反应。将增加力的Von Frey细丝施加到下腹部,靠近膀胱。处死动物,通过腹部下中线切口取出膀胱。
图6显示了CYP注射4h后的AUC疼痛评分。
实施例-化合物对急性的环磷酰胺(CYP)诱导的膀胱炎模型的功效
实验系统-在雌性SD大鼠(6-7周龄)上进行研究。
研究方案-通过以150mg/kg体重的剂量单次腹膜内给予环磷酰胺,在实验动物中诱导急性间质性膀胱炎。在表中提及了研究组、剂量和给予途径(RoA)。向正常对照组给予盐水。在环磷酰胺给予前30min给予测试化合物。在环磷酰胺给予前1h给予参考化合物布洛芬。对照组仅给予溶媒。在注射CYP或盐水之前(基底,0h)和CYP给予后4h评估伤害性反应。将增加力的Von Frey细丝施加到下腹部,靠近膀胱。处死动物,通过腹部下中线切口取出膀胱。
图7显示了CYP注射4h后的AUC疼痛评分。
实施例75:大鼠中的药代动力学数据
该研究的目的是评价在雌性Sprague Dawley大鼠中每次口服给予31.2mg/kg的化合物No.12、32.8mg/kg的化合物No.43、34.8mg/kg的化合物No.446、25mg/kg的化合物No.449、21.2mg/kg的化合物XIII和27mg/kg的化合物No.54后,化合物XI与化合物No.12/化合物No.43/化合物No.446/化合物No.449/化合物XIII/化合物No.54的药代动力学暴露。这项研究是在禁食过夜的雌性Sprague Dawley大鼠中进行的。给药制剂是在给药当天新鲜配制的。使用0.1%的tween-80+99.9%的0.5%CMC作为制备给药制剂的溶媒。
在给药前、给药后0.25h、0.5h、1h、2h、4h、8h、12h和24h通过眼眶后丛穿刺收集血样。在每个时间点,抽取血液并转移至预先标记的含有抗凝剂的微型离心管中。通过翻转管轻轻混合管以促进抗凝剂与血液混合。所有血液/血浆样本均使用适合目的的LC-MS/MS方法进行分析。
使用标准非房室分析(
表:在向Sprague Dawley大鼠以单次口服途径给予化合物No.12/化合物No.43/化合物No.446/化合物No.449/化合物XIII/化合物No.54之后,化合物No.12/化合物No.43/化合物No.446/化合物No.449/化合物XIII/化合物No.54和化合物XI的算术平均药代动力学参数。
/>
NC-未计算实施例76-化合物XI对膀胱压力的效果
实验系统-在雌性SD大鼠(7-9周龄)上进行研究。
研究方案-通过以150mg/kg体重的剂量单次腹膜内给予环磷酰胺在实验动物中诱导急性间质性膀胱炎。表中提及了研究组、剂量和给予途径(RoA)。向正常对照组给予生理盐水。在给予环磷酰胺之前30分钟给予测试化合物。对照组仅给予溶媒。将大鼠麻醉,通过温度探头和加热灯持续监测体温并将其保持在36.5℃±0.2℃。膀胱被插管并连接到一侧的压力传感器和另一侧的输液泵(哈佛仪器)。压力传感器通过放大器连接到数据采集系统和计算机。使用Lab chart Version 8.8.2软件用于记录。启动输液泵,以100μL/min输注盐水。因此,膀胱以恒定速率充满,记录平均膀胱压力并示于图14中。