苯并呋喃2,3-c吡咯-1,3(2H)-二酮类化合物及其合成方法
文献发布时间:2024-04-18 20:00:50
技术领域
本发明属于有机合成技术领域,具体涉及苯并呋喃[2,3-c]吡咯-1,3(2H)-二酮类化合物及其合成方法。
背景技术
马来酰亚胺具备良好的亲电性,其所具备的独特的马来酰亚胺骨架结构广泛存在于天然产物与生物活性分子中,且在药物、功能材料合成领域有着广泛的应用。马来酰亚胺是a、b-不饱和酰亚胺基团,酰亚胺部分为聚合物带来了良好的热稳定性,可作为荧光材料的重要骨架。除此之外,马来酰亚胺类化合物还显示出多种生物活性,例如抗菌、抗真菌和抗癌活性等。
苯并呋喃[2,3-c]吡咯母核类化合物在医药和农业、工业、林业方面有着显著作用。已有文献报道,3-(3-氧代-1,3-二氢-2H-苯并呋喃[2,3-c]吡咯-2-基)哌啶-2,6-二酮,可以作为有效的小脑蛋白(CRBN)配体,进一步可以合成相应的PROTACs分子。
2021年,国际公布号为WO 2021/143816 A1的专利公开了一种苯并呋喃[2,3-c]吡咯母核类化合物的制备方法如下:
2021年,Min Xiang,Chen-Yi Li等人报道了采用羟基马来酰亚胺与苯醌作为原料,通过咪唑作用发生迈克尔加成反应生成苯并呋喃并[2,3-c]吡咯类化合物。该反应过程可用下述的反应式表示:
发明内容
第一方面,本发明旨在提供一种苯并呋喃[2,3-c]吡咯-1,3(2H)-二酮类化合物。
第二方面,本发明旨在提供一种苯并呋喃[2,3-c]吡咯-1,3(2H)-二酮类化合物的合成方法。
为实现上述发明目的,本发明通过以下技术方案实现:
一种苯并呋喃[2,3-c]吡咯-1,3(2H)-二酮类化合物,其结构式如通式(4)所示:
其中,R
作为优选,R
作为优选,R
作为优选,R
作为优选,R
作为优选,R
如上所述的苯并呋喃[2,3-c]吡咯-1,3(2H)-二酮类化合物的合成方法,包括以下步骤:(S.1)将化合物a和化合物b与碱混合并加入干燥的甲苯或干燥的苯,加热搅拌,反应结束后取反应液,分离纯化后得到中间产物;
(S.2)将步骤(S.1)中得到的中间产物与催化剂、氧化剂混合并加入无水乙酸,氮气保护下进行加热搅拌,反应结束后取反应液,分离纯化后得到通式(4)所示的产物c。
作为优选,所述步骤(S.1)中化合物a、化合物b、中间产物的结构式依次如通式(1)、通式(2)、通式(3)所示:
作为优选,所述步骤(S.1)中的碱为碳酸铯、叔丁醇钾、叔丁醇钠中的任意一种。
作为优选,所述步骤(S.2)中的催化剂为醋酸钯、氯化钯、硝酸钯中的任意一种。
作为优选,所述步骤(S.2)中的氧化剂为过硫酸钾、醋酸铜、硝酸银中的任意一种。
作为优选,所述步骤(S.1)中的反应温度为45~60℃,反应时间为1~6h。
作为优选,所述步骤(S.2)中的反应温度为90~110℃,反应时间为10~18h。
作为优选,所述步骤(S.1)中添加的化合物a、化合物b以及碱的摩尔比为(1.05~1.2):1:(1.05~1.2)。
作为优选,所述步骤(S.2)中添加的中间产物、催化剂以及氧化剂的摩尔比为1:(0.05~0.2):(3~5)。
作为优选,苯并呋喃[2,3-c]吡咯-1,3(2H)-二酮类化合物的合成路线如下所示:
作为优选,所述步骤(S.1)中的反应过程通过TLC监测反应进程,当通式(2)所示的化合物b反应完全时,TLC监测反应结束。
作为优选,所述步骤(S.2)中的反应过程通过TLC监测反应进程,当通式(3)所示的中间产物反应完全时,TLC监测反应结束。
作为优选,所述步骤(S.1)中反应液分离纯化过程包括以下步骤:
利用旋蒸仪将反应液中的甲苯或苯旋干,再往其中加水,用二氯甲烷萃取2~3次,合并有机相并用无水硫酸镁干燥、过滤、滤液旋干得粗产品。再将粗产品重结晶(EA:PE)纯化得到中间产物。
作为优选,所述步骤(S.2)中反应液分离纯化过程包括以下步骤:
向反应液中加入水,用乙酸乙酯萃取2~3次,合并有机相并用无水硫酸镁干燥、过滤、滤液减压浓缩。残留物用硅胶柱层析纯化,收集含有产物c的洗脱剂,蒸除溶剂得到苯并呋喃[2,3-c]吡咯-1,3(2H)-二酮类化合物(即通式(4)所示的产物c)。硅胶柱层析纯化的洗脱剂采用体积比为10:1的石油醚、乙酸乙酯混合溶剂。
因此,本发明具有以下有益效果:
(1)本发明通过以通式(1)、通式(2)所示的化合物a、化合物b为原料,通过偶联、环化,高效制得苯并呋喃[2,3-c]吡咯-1,3(2H)-二酮类化合物;
(2)本发明的合成方法具有反应高效、操作简单易行、原料便宜易得、收率高的优异特点,对各种取代基都有很好的兼容性;
(3)本发明制得的苯并呋喃[2,3-c]吡咯-1,3(2H)-二酮类化合物具有抗癌抗肿瘤的活性,在制备抗癌药物中具有广泛的应用前景。
附图说明
图1为实施例1中产物c的
图2为实施例1中产物c的
图3为实施例6中产物c的
图4为实施例6中产物c的
图5为实施例9中产物c的
图6为实施例9中产物c的
图7为实施例10中产物c的
图8为实施例10中产物c的
具体实施方式
下面结合说明书附图以及具体实施例对本发明做进一步描述。本领域普通技术人员在基于这些说明的情况下将能够实现本发明。此外,下述说明中涉及到的本发明的实施例通常仅是本发明一部分的实施例,而不是全部的实施例。因此,基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动的前提下所获得的所有其他实施例,都应当属于本发明保护的范围。
实施例1
一种苯并呋喃[2,3-c]吡咯-1,3(2H)-二酮类化合物的合成方法,包括以下步骤:
(S.1)在25mL单口干燥烧瓶中,加入0.5mmol化合物a、0.55mmol化合物b、0.55mmol碳酸铯、8mL干燥的甲苯(即无水甲苯),在60℃油浴下搅拌反应,TLC监测反应进程。反应结束后,利用旋蒸仪将反应液中的甲苯旋干,再往其中加入水,用二氯甲烷萃取2~3次,饱和食盐水洗涤。合并有机相并用无水硫酸镁干燥、过滤、滤液旋干得粗产品。再将粗产品重结晶(EA:PE)纯化得到中间产物;
(S.2)将0.3mmol步骤(S.1)中得到的中间产物、3eq过硫酸钾、10%醋酸钯、5mL干燥的乙酸(即无水乙酸)置于25mL两口圆底干燥烧瓶中,N
产物c的
1
13
实施例2
本实施例与实施例1的区别在于:
本实施例中提供一种苯并呋喃[2,3-c]吡咯-1,3(2H)-二酮类化合物的合成方法,其中,作为底物1的化合物a、作为终产物的产物c的结构式不相同。其他都与实施例1中相同。本实施例中作为底物1的化合物a、作为底物2的化合物b以及作为终产物的产物c的结构式见下表1所示。产率为77%。
产物c的
1
13
实施例3
本实施例与实施例1的区别在于:
本实施例中提供一种苯并呋喃[2,3-c]吡咯-1,3(2H)-二酮类化合物的合成方法,其中,作为底物1的化合物a、作为终产物的产物c的结构式不相同。其他都与实施例1中相同。本实施例中作为底物1的化合物a、作为底物2的化合物b以及作为终产物的产物c的结构式见下表1所示。产率为85%。
产物c的
1
13
实施例4
本实施例与实施例1的区别在于:
本实施例中提供一种苯并呋喃[2,3-c]吡咯-1,3(2H)-二酮类化合物的合成方法,其中,作为底物1的化合物a、作为终产物的产物c的结构式不相同。其他都与实施例1中相同。本实施例中作为底物1的化合物a、作为底物2的化合物b以及作为终产物的产物c的结构式见下表1所示。产率为95%。
产物c的
1
13
实施例5
本实施例与实施例1的区别在于:
本实施例中提供一种苯并呋喃[2,3-c]吡咯-1,3(2H)-二酮类化合物的合成方法,其中,作为底物1的化合物a、作为终产物的产物c的结构式不相同。其他都与实施例1中相同。本实施例中作为底物1的化合物a、作为底物2的化合物b以及作为终产物的产物c的结构式见下表1所示。产率为92%。
产物c的
1
13
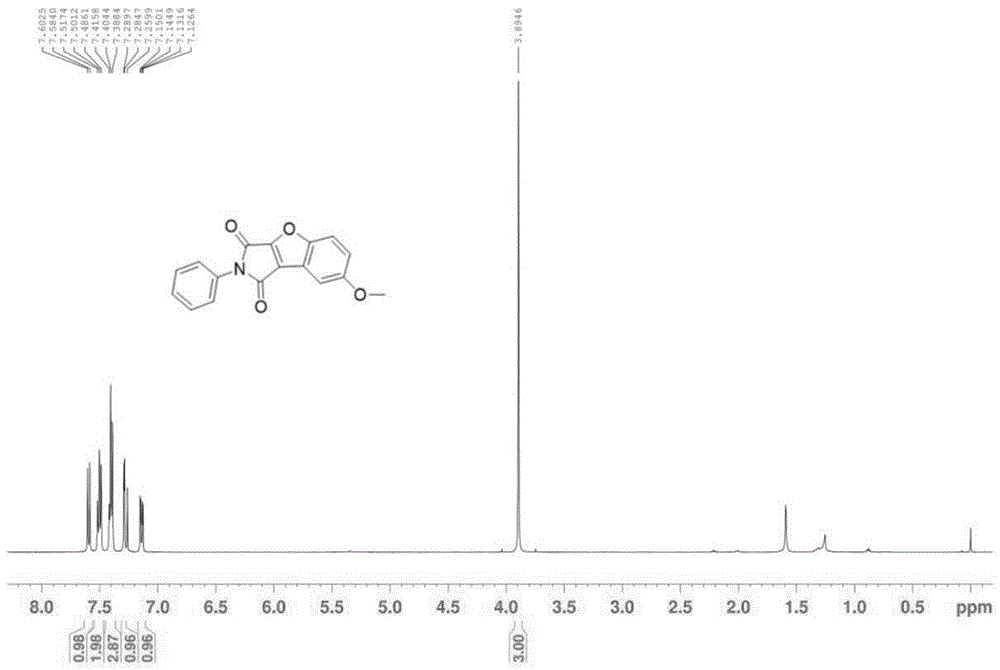
实施例6
本实施例与实施例1的区别在于:
本实施例中提供一种苯并呋喃[2,3-c]吡咯-1,3(2H)-二酮类化合物的合成方法,其中,作为底物1的化合物a、作为终产物的产物c的结构式不相同。其他都与实施例1中相同。本实施例中作为底物1的化合物a、作为底物2的化合物b以及作为终产物的产物c的结构式见下表1所示。产率为70%。图3所示为本实施例中产物c的
产物c的
1
13
实施例7
本实施例与实施例1的区别在于:
本实施例中提供一种苯并呋喃[2,3-c]吡咯-1,3(2H)-二酮类化合物的合成方法,其中,作为底物1的化合物a、作为终产物的产物c的结构式不相同。其他都与实施例1中相同。本实施例中作为底物1的化合物a、作为底物2的化合物b以及作为终产物的产物c的结构式见下表1所示。产率为87%。
产物c的
1
13
实施例8
本实施例与实施例1的区别在于:
本实施例中提供一种苯并呋喃[2,3-c]吡咯-1,3(2H)-二酮类化合物的合成方法,其中,作为底物1的化合物a、作为终产物的产物c的结构式不相同。其他都与实施例1中相同。本实施例中作为底物1的化合物a、作为底物2的化合物b以及作为终产物的产物c的结构式见下表1所示。产率为96%。
产物c的
1
13
实施例9
本实施例与实施例1的区别在于:
本实施例中提供一种苯并呋喃[2,3-c]吡咯-1,3(2H)-二酮类化合物的合成方法,其中,作为底物2的化合物b、作为终产物的产物c的结构式不相同。其他都与实施例1中相同。本实施例中作为底物1的化合物a、作为底物2的化合物b以及作为终产物的产物c的结构式见下表1所示。产率为88%。图5所示为本实施例中产物c的
产物c的
1
13
实施例10
本实施例与实施例1的区别在于:
本实施例中提供一种苯并呋喃[2,3-c]吡咯-1,3(2H)-二酮类化合物的合成方法,其中,作为底物2的化合物b、作为终产物的产物c的结构式不相同。其他都与实施例1中相同。本实施例中作为底物1的化合物a、作为底物2的化合物b以及作为终产物的产物c的结构式见下表1所示。产率为86%。图7所示为本实施例中产物c的
产物c的
1
13
实施例11
本实施例与实施例1的区别在于:
本实施例中提供一种苯并呋喃[2,3-c]吡咯-1,3(2H)-二酮类化合物的合成方法,其中,作为底物2的化合物b、作为终产物的产物c的结构式不相同。其他都与实施例1中相同。本实施例中作为底物1的化合物a、作为底物2的化合物b以及作为终产物的产物c的结构式见下表1所示。产率为88%。
产物c的
1
13
实施例12
本实施例与实施例1的区别在于:
本实施例中提供一种苯并呋喃[2,3-c]吡咯-1,3(2H)-二酮类化合物的合成方法,其中,作为底物2的化合物b、作为终产物的产物c的结构式不相同。其他都与实施例1中相同。本实施例中作为底物1的化合物a、作为底物2的化合物b以及作为终产物的产物c的结构式见下表1所示。产率为63%。
产物c的
1
13
实施例13
本实施例与实施例1的区别在于:
本实施例中提供一种苯并呋喃[2,3-c]吡咯-1,3(2H)-二酮类化合物的合成方法,其中,作为底物2的化合物b、作为终产物的产物c的结构式不相同。其他都与实施例1中相同。本实施例中作为底物1的化合物a、作为底物2的化合物b以及作为终产物的产物c的结构式见下表1所示。产率为96%。
产物c的
1
13
实施例14
本实施例与实施例1的区别在于:
本实施例中提供一种苯并呋喃[2,3-c]吡咯-1,3(2H)-二酮类化合物的合成方法,其中,作为底物2的化合物b、作为终产物的产物c的结构式不相同。其他都与实施例1中相同。本实施例中作为底物1的化合物a、作为底物2的化合物b以及作为终产物的产物c的结构式见下表1所示。产率为83%。
产物c的
1
13
实施例1~14中作为反应底物1的化合物a、作为反应底物2的化合物b以及作为终产物的产物c的结构式以及产物c的产率如下表1所示。
表1
/>
实施例15
按实施例6中的合成方法制备的苯并呋喃[2,3-c]吡咯-1,3(2H)-二酮类化合物可进一步用于合成作为新型大麻素受体调节物的3-(羟甲基)-5-甲氧基-N-苯基苯并呋喃-2-甲酰胺化合物。本实施例中提供一种可行简便的3-(羟甲基)-5-甲氧基-N-苯基苯并呋喃-2-甲酰胺化合物合成反应路线如下:
上述合成反应路线中的反应条件如下:
N
实施例16
按实施例9中的合成方法制备的苯并呋喃[2,3-c]吡咯-1,3(2H)-二酮类化合物还可以进一步用于合成一种小脑蛋白E3泛素连接酶蛋白质结合配体化合物。本实施例中提供一种可行简便的小脑蛋白E3泛素连接酶蛋白质结合配体化合物合成反应路线如下:
/>
上述合成反应路线中的反应条件如下:
室温下,将五氟化锑(0.5g,2.3mmol)加入到2-苯基-1H-苯并呋喃并[2,3-c]吡咯-1,3(2H)-二酮(0.05g,0.2mmol)的CF
以上所述仅是对本发明的优选实施例及原理进行了详细说明,对本领域的普通技术人员而言,依据本发明提供的思想,在具体实施方式上会有改变之处,而这些改变也应视为本发明的保护范围。