一种阿兹夫定中间体的合成工艺
文献发布时间:2024-04-18 20:00:50
技术领域
本发明属于阿兹夫定中间体制备技术领域,更具体的,涉及一种阿兹夫定中间体的合成工艺。
背景技术
阿兹夫定(Azvudine),又称阿滋福啶,由常俊标教授发明,河南真实生物科技有限公司、郑州大学、河南师范大学、河南省科学院高新技术研究中心共同研发。它是一种艾滋病毒逆转录酶(RT)抑制剂,通过抑制艾滋病毒逆转录酶的活性,阻断病毒复制和感染细胞的能力,从而达到治疗艾滋病的目的,并且具有强效的抗逆转录酶活性,在低浓度下就能有效抑制病毒的复制,属世界先进、国内首创的新一代治疗艾滋病药物。
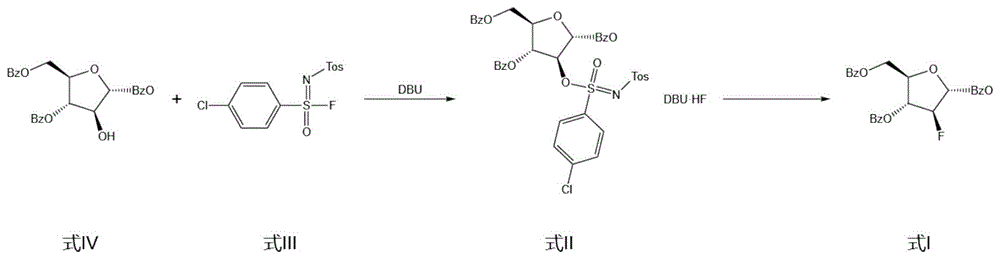
其中,2-脱氧-2-氟-1,3,5-三苯甲酰基-α-D-阿拉伯呋喃糖是合成阿兹夫定化合物的关键中间体,其化学结构式如下:
在专利文献CN101531695中,公开了一种以咪唑磺酰基活化化合物2位羟基后,再通过HF或四丁基氟化铵等氟代试剂进行亲核氟代反应,从而得到目标产物的制备工艺,该方案中氟代反应的副产物多,导致总体收率偏低,同时由于产物不易纯化,使得目标产物的纯度也较低,不适宜工业化生产。并且反应过程中所使用的氟代试剂HF或四丁基氟化铵,其中HF在常压下为剧毒气体,且具有强腐蚀性,四丁基氟化铵则价格昂贵,不仅提高了生产成本,参与氟代反应的选择性也较差,两种氟代试剂的后处理产物的难度也较高,该方法工艺路线如下:
在专利文献CN101555267中,公开了一种以DAST(二乙胺基三氟化硫)为氟代试剂进行一步制得目标化合物的方法。但DAST热稳定性差且易爆炸,在生产、储存、运输过程中如遇碰撞或高温极易发生爆炸,在遇水分解后则会放出大量二氧化硫和HF气体,不仅含有剧毒且容易污染环境,同时DAST的腐蚀性强对设备要求高生产难度大,反应过程中也容易产生难以处理的副产物,该方法工艺路线如下:
上述两个以公开专利文献中的脱氢氟化反应中氟代试剂价格昂贵、稳定性差、后处理难度高且最终制得的产物收率与纯度都较差,因此,目前亟需寻找一种生产成本低、稳定性好、生产难度低、产物收率与产物纯度高的制备方法。
发明内容
本发明针对以上现有技术中存在的缺陷,提供一种阿兹夫定中间体的合成工艺,解决的问题是如何实现降低生产成本与生产难度、提高产物收率与纯度的制备方法。
本发明以式III化合物为4-氯-N-[(4-甲基苯基)磺酰]苯磺胺酰氟为氟代试剂,该氟代试剂稳定性好且反应性较强,在DBU的催化下使式IV化合物发生脱氧氟化,整个反应流程不长,同时反应过程皆可在室温下进行,并且反应速率较大、操作较为简单,由于氟代试剂的选择性较强,因此副产物生成量少,容易处理后再次利用,产物收率与产物纯度也较高,总合成路线为:
该方法包括以下步骤:
S1:在有机溶剂中,将起始原料式IV化合物与氟化试剂即式Ⅲ化合物在催化剂DBU的作用下进行脱氧氟化反应第一阶段,制备得到式Ⅱ化合物;
S2:将式Ⅱ化合物进行脱氧氟化反应第二阶段,制备得到式I化合物即最终产物阿兹夫定中间体。
进一步的,所述步骤S1中,所述式III化合物为4-氯-N-[(4-甲基苯基)磺酰]苯磺胺酰氟。
作为优选,相对于现存的氟化试剂,4-氯-N-[(4-甲基苯基)磺酰]苯磺胺酰氟的反应性强,更加温和、易得、反应时间短、处理简单,还具有较高的消除选择性。
进一步的,所述S1步骤中,所述有机溶剂可选用甲苯、二氯甲烷或四氢呋喃等其中一种。
作为优选,选用甲苯作为反应溶剂时反应速率较大。
进一步的,所述S1步骤中,所述式IV化合物与所述DBU的质量分数比为1:1.2~1.6。
作为优选,式IV化合物与DBU的质量分数比为1:1.2时,消耗的催化剂DBU最少,当质量分数比为1:1.6时,DBU消耗量较大产物收率最大产物纯度降低。
进一步的,所述S1步骤中,所述式IV化合物与所述式III化合物的质量分数比为1:1.2~1.4。
作为优选,式IV化合物与式III化合物的质量分数比为1:1.2时,反应速度最快,式IV化合物与式III化合物的质量分数比为1:1.4时,反应容易产生副产物。
进一步的,所述S1步骤中,所述脱氧氟化反应第一阶段在室温下进行。
作为优选,室温下反应更温和,避免高温催化产生副产物。
进一步的,所述S1步骤中,所述脱氧氟化反应第一阶段反应时间为1~3min。
进一步的,所述S2步骤中,所述脱氧氟化反应第二阶段在室温下进行。
进一步的,所述S2步骤中,所述脱氧氟化反应第二阶段反应时间为5~10min。
进一步的,所述S2步骤中,副产物4-氯-硝基苯磺酰胺的铵盐沉淀通过过滤回收后可重新用于式Ⅲ化合物的合成。
综上所述,本发明与现有技术相比,具有以下优点:
本发明中通过使用4-氯-N-[(4-甲基苯基)磺酰]苯磺胺酰氟作为氟化试剂,在DBU的催化下促使起始原料式IV化合物进行脱氧氟化反应,相较于如HF、DAST等常规氟化试剂,4-氯-N-[(4-甲基苯基)磺酰]苯磺胺酰氟的反应性强,能够极大缩短反应时间,同时更加温和、易得、反应时间短、处理简单,还具有较高的消除选择性,不易生成副产物,保障了较高的反应收率,并且较为安全,操作难度较低。
附图说明
图1为本发明合成路线。
具体实施方式
下面通过具体实施例,对本发明的技术方案作进一步具体的说明,但是本发明并不限于这些实施例。
实施例1
三口瓶中放入43g式IV化合物,加入300mL甲苯,室温搅拌15mim,加入41.74g4-氯-N-[(4-甲基苯基)磺酰]苯磺胺酰氟,滴加18.27gDBU,滴加完毕室温搅拌反应3min,反应结束后,室温静置反应10min,底物完全耗尽后反应停止。减压脱溶,使用300mL饱和食盐水洗涤有机相,使用二氯甲烷重结晶,过滤后烘干,得到39.45g最终产物式I化合物即阿兹夫定中间体,产物收率为91.3%,产物纯度为99.4%。
实施例2
三口瓶中放入43g式IV化合物,加入300mL甲苯,室温搅拌15mim,加入41.74g4-氯-N-[(4-甲基苯基)磺酰]苯磺胺酰氟,滴加24.36gDBU,滴加完毕室温搅拌反应3min,反应结束后,室温静置反应10min,底物完全耗尽后反应停止。减压脱溶,使用300mL饱和食盐水洗涤有机相,使用二氯甲烷重结晶,过滤后烘干,得到40.2g最终产物式I化合物即阿兹夫定中间体,产物收率为93%,产物纯度为98.6%。
实施例3
三口瓶中放入43g式IV化合物,加入300mL甲苯,室温搅拌15mim,加入48.69g4-氯-N-[(4-甲基苯基)磺酰]苯磺胺酰氟,滴加18.27gDBU,滴加完毕室温搅拌反应3min,反应结束后,室温静置反应10min,底物完全耗尽后反应停止。减压脱溶,使用300mL饱和食盐水洗涤有机相,使用二氯甲烷重结晶,过滤后烘干,得到38.98g最终产物式I化合物即阿兹夫定中间体,产物收率为90.2%,产物纯度为99.3%。
实施例4
三口瓶中放入43g式IV化合物,加入300mL甲苯,室温搅拌15mim,加入41.74g4-氯-N-[(4-甲基苯基)磺酰]苯磺胺酰氟,滴加18.27gDBU,滴加完毕室温搅拌反应1min,反应结束后,室温静置反应5min,底物完全耗尽后反应停止。减压脱溶,使用300mL饱和食盐水洗涤有机相,使用二氯甲烷重结晶,过滤后烘干,得到37.77g最终产物式I化合物即阿兹夫定中间体,产物收率为87.4%,产物纯度为99%。
实施例5
三口瓶中放入43g式IV化合物,加入300mL二氯甲烷,室温搅拌15mim,加入41.74g4-氯-N-[(4-甲基苯基)磺酰]苯磺胺酰氟,滴加24.36gDBU,滴加完毕室温搅拌反应3min,反应结束后,室温静置反应10min,底物完全耗尽后反应停止。减压脱溶,使用300mL饱和食盐水洗涤有机相,使用甲苯重结晶,过滤后烘干,得到38.04g最终产物式I化合物即阿兹夫定中间体,产物收率为89.9%,产物纯度为98.8%。
实施例6
三口瓶中放入43g式IV化合物,加入300mL四氢呋喃,室温搅拌15mim,加入41.74g4-氯-N-[(4-甲基苯基)磺酰]苯磺胺酰氟,滴加24.36gDBU,滴加完毕室温搅拌反应3min,反应结束后,室温静置反应10min,底物完全耗尽后反应停止。减压脱溶,使用200mL乙酸乙酯萃取,合并有机相,使用300mL饱和食盐水洗涤有机相,脱溶液,使用甲苯重结晶,过滤后烘干,得到38.12g最终产物式I化合物即阿兹夫定中间体,产物收率为90.1%,产物纯度为99%。
对比例
本对比例为已公开专利文献CN115947768A的实施例:
(a)化合物II的合成:
向反应容器中加入化合物III(50g,108mmol)、固体氢氧化钾(3.03g,54mmol)和100ml甲苯,将混合物搅拌至完全溶解。然后再加入碘甲烷(5.11g,36mmol),在100℃下搅拌4h。反应完成后,冷却至室温(约25℃),过滤,将有机相分离,通过蒸馏干燥得到15.49g化合物I1,收率90.3%,纯度99.2%。
(b)化合物I的合成:
向反应瓶中加入步骤(a)中制备的化合物II(24.3g,51mmol)、丙酰氟(7.8g,102mmol)和50ml正庚烷,冷却至20℃,搅拌全溶。然后使用注射器加入三氟化硼四氢呋喃配位化合物(0.7g,5.1mmol),在20℃下搅拌16h。反应结束后,向反应混合物中加入水,分取有机层,无水硫酸钠干燥,然后精馏得到20.8g化合物I,收率87.8%,纯度99.1%。
对比本发明中的制备方法,此实施例中使用三氟化硼作为氟化试剂,不仅腐蚀性强操作难度大,在生产过程中还需要高温进行催化,大规模生产成本高且不易控制副产物生成,容易降低产物收率,且在后处理过程中副产物较难与溶液分离。
本发明的实施方式并不限于上述实施例所述,在不偏离本发明的精神和范围的情况下,本领域普通技术人员可以在形式和细节上对本发明做出各种改变和改进,而这些均被认为落入了本发明的保护范围。