一种含吩噁嗪并噁唑类结构的化合物及其应用
文献发布时间:2024-04-18 20:00:50
技术领域
本发明涉及一种含吩噁嗪并噁唑类结构的化合物及其应用,属于有机电致发光技术领域。
背景技术
有机电致发光器件(OLED)是近年来国际上平板显示领域的一个研究热点。其具有结构简单,成品率高、成本低,主动发光、响应速度快,分辨率高等优点;并且驱动电压低、全固态、非真空器件,具有抗振荡、耐低温(-40℃)性能,被认为是未来最有可能替代液晶显示器,引起了人们极大关注。在OLED的制备和优化中,发光材料的选择至关重要,其性质是决定器件性能的重要因素之一。
有机电致发光器件(OLED)具有包括发光层和在发光层两侧的一对电极所构成。当在两个电极之间施加适当电场时,电子由负极注入,空穴由正极注入,在发光层中电子和空穴重新结合形成激发态,当激发态返回到基态时产生的能量发射光。
发光材料在OLED中是最重要的材料。选择发光材料必须满足以下的要求:(1)高量子效率的荧光发射,荧光光谱主要集中在400~700nm的可见光区域;(2)具有良好的导电性,或者空穴迁移能力,即能让载流子有效地在发光材料里迁移;(3)良好的热稳定性,即需要较高的分解温度和玻璃化温度,防止在加工和器件运行过程中由于受热或者器件内部产生的焦耳热而变质或者结晶。因此,开发稳定高效的有机电致发光材料显得尤为重要。
发明内容
本发明针对现有技术存在的不足,提供一种含吩噁嗪并噁唑类结构的化合物及其应用,所述含吩噁嗪并噁唑类结构的化合物能提高有机电致发光器件的效率和寿命。
本发明解决上述技术问题的技术方案如下:
一种含吩噁嗪并噁唑类结构的化合物,所述含吩噁嗪并噁唑类结构的化合物的结构式如下[结构式I]所示:
[结构式I];
R表示为被氘取代或未取代的苯基、联苯基、C10~C14的稠环芳基、芘基、呋喃基、噻吩基、苯基咔唑基、甲基芴基、苯基芴基、螺二芴基、噁唑基、嘧啶基、三嗪基中的任意一种。
进一步的,所述含吩噁嗪并噁唑类结构的化合物选自如下化学式中的任意一个:
其中,X选自氢、氘、腈基或甲基,Y选自O或S;
其中,D
进一步的,所述含吩噁嗪并噁唑类结构的化合物选自如下结构式中的任意一个:
本发明还公开了所述含吩噁嗪并噁唑类结构的化合物的应用,所述含吩噁嗪并噁唑类结构的化合物应用于有机电致发光器件;在有机电致发光器件中应用时,[结构式I]所表示的化合物可以使用其中一种化合物,也可以同时使用[结构式I]中的两种或两种以上化合物。
进一步的,所述的有机电致发光器件包括阴极层、阳极层和有机层,所述含吩噁嗪并噁唑类结构的化合物应用于有机层,所述有机层包括空穴注入层、空穴传输层、发光层、空穴阻挡层、电子注入层、电子传输层中的至少一层。
进一步的,所述有机层包括发光层,所述含吩噁嗪并噁唑类结构的化合物应用于发光层,所述发光层为蓝色发光层,所述含吩噁嗪并噁唑类结构的化合物作为蓝色主体,可得到高效率、高分辨率、高亮度及长寿命的有机电致发光器件。
进一步的,所述有机层包括电子传输层,所述含吩噁嗪并噁唑类结构的化合物应用于电子传输层。
进一步的,所述有机层包括发光层和电子传输层,所述发光层和电子传输层均含有所述化合物,所述发光层和电子传输层中含吩噁嗪并噁唑类结构的化合物相同或者不同。
进一步的,所述含吩噁嗪并噁唑类结构的化合物应用于有机电致发光器件,所述含吩噁嗪并噁唑类结构的化合物单独使用,或者与其他化合物混合使用。
本发明的有益效果是:
本发明所述的含吩噁嗪并噁唑类结构的化合物,其结构特性使化合物具有优良的空穴注入和电子传输能力。将吩噁嗪基团和噁唑相结合,能提高化合物的电子输注和传输能力。所合成的含氮化合物LUMO能级电子云和HOMO能级轨道分离,拓宽载流子复合区域,提高发光性;含氮化合物能在保持较高的T1(三线态能级)值的前提下,具有平衡的空穴和电子传输能力,进而改善器件发光效率以及使用寿命。
本发明所述含吩噁嗪并噁唑类结构的化合物应用在有机电致发光器件中,可以有效提升有机电致发光器件的发光效率和使用寿命。
附图说明
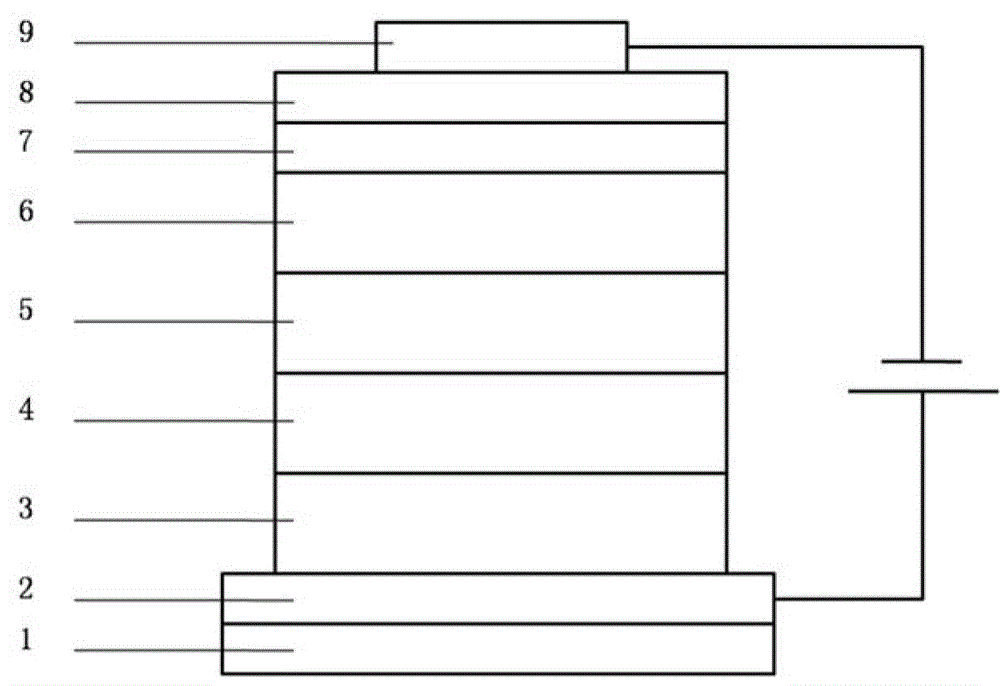
图1为实施例中所述有机电致发光器件结构层图;
图中,1、基板;2、阳极;3、空穴注入层;4、空穴传输层;5、发光层;6、空穴阻挡层;7、电子传输层;8、电子注入层;9、阴极。
具体实施方式
为使本发明的上述目的、特征和优点能够更加明显易懂,下面对本发明的具体实施方式做详细的说明。在下面的描述中阐述了很多具体细节以便于充分理解本发明。但是本发明能够以很多不同于在此描述的其它方式来实施,本领域技术人员可以在不违背本发明内涵的情况下做类似改进,因此本发明不受下面公开的具体实施例的限制。
除非另有定义,本文所使用的所有的技术和科学术语与属于本发明的技术领域的技术人员通常理解的含义相同。本文中在本发明的说明书中所使用的术语只是为了描述具体的实施方式的目的,不是旨在于限制本发明。
实施例中,中间体化合物H,10-苯基-7H-噁唑[5,4-b]吡嗪[2,3-h]吩噁嗪,其具体合成方法如下所示:
;
(1)中间体C的制备:
在氮气保护条件下,向装有机械搅拌、温度计、球形冷凝管的三口瓶中依次加入化合物A(50.0g,318.3mmol),化合物B(41.3g,381.9mmol),1,1'-双(二苯基膦)二茂铁(8.8g,15.9mmol)和二甲苯500.0mL。加料毕,搅拌升温至130~140℃回流保温12h。反应毕,待温度降至70~80℃后静置分层,将上层有机相水洗至中性。水洗毕,经干燥、过柱、脱溶剂、甲苯/乙醇重结晶得到固体化合物44.1g,收率为65%。根据LC-MS分析,该固体化合物鉴定为中间体C。LC-MS:M/Z 213.06。
(2)中间体D的制备:
在氮气保护条件下,向装有机械搅拌、温度计、球形冷凝管的三口瓶中加入浓硫酸150mL,并将体系降温至-10~0℃,搅拌状态下缓慢加入化合物C(40.0g,187.6mmol)。加料毕,控温-10~0℃缓慢滴加浓硝酸(27.3g,281.4mmol,65%含量)。滴加毕,控温-10~0℃保温2h。反应毕,将反应液控温30℃以下,缓慢倒入500mL冰水中水解搅拌20min。水解毕,过滤、水煮至中性后,经甲苯/乙醇重结晶得到固体化合物34.4g,收率为71%。根据LC-MS分析,该固体化合物鉴定为中间体D。LC-MS:M/Z 258.04。
(3)中间体F的制备:
在氮气保护条件下,向装有机械搅拌、温度计、球形冷凝管的三口瓶中依次加入化合物E(50.0g,312.2mmol),二氯乙烷500.0mL。加料毕,搅拌降温至-5~5℃后,开始缓慢滴加三溴化硼(93.8g,374.6mmol)。滴加毕,控温-5~5℃保温4h。反应毕,控温30℃以下将反应液缓慢倒入500mL冰水中水解搅拌20min。水解毕,升温至40~50℃保温至全溶后,静置分层,将下层有机相水洗至中性。水洗毕,经干燥、过柱、脱溶剂、甲苯/乙醇重结晶得到固体化合物34.7g,收率为76%。根据LC-MS分析,该固体化合物鉴定为中间体F。LC-MS:M/Z 146.05。
(4)中间体G的制备:
在氮气保护条件下,向装有机械搅拌、温度计、球形冷凝管的三口瓶中依次加入化合物D(30.0g,116.2mmol),化合物F(18.7g,127.8mmol),碳酸钾(48.2g,348.6mmol)和N,N-二甲基甲酰胺300.0mL。加料毕,搅拌升温至130~135℃保温12h。反应毕,待温度降至70℃以下,过硅藻土,滤饼用N,N-二甲基甲酰胺充分淋洗后,将滤液脱溶剂。脱溶剂毕,经甲苯/乙醇重结晶得到固体化合物33.0g,收率为74%。根据LC-MS分析,该固体化合物鉴定为中间体G。LC-MS:M/Z 384.09。
(5)中间体H的制备:
在氮气保护条件下,向装有机械搅拌、温度计、球形冷凝管的三口瓶中依次加入化合物G(30.0g,78.1mmol),亚磷酸三乙酯(64.8g,390.3mmol)和邻二氯苯300.0mL。加料毕,搅拌升温至130~135℃保温12h。反应毕,待温度降至70℃以下,向反应体系中加入100mL纯化水水解。水解毕,静置分层,将下层有机相水洗至中性。水洗毕,经干燥、过柱、脱溶剂、甲苯/乙醇重结晶得到固体化合物18.4g,收率为67%。根据LC-MS分析,该固体化合物鉴定为中间体H。LC-MS:M/Z 352.10。
合成实施例
1. 化合物3-2的制备
;
在氮气保护条件下,向装有机械搅拌、温度计、球形冷凝管的三口瓶中依次加入化合物H(10.0g,28.4mmol),间溴苯腈(6.2g,34.1mmol),叔丁醇钠(4.1g,42.6mmol),双(二亚苄基丙酮)钯(0.1632g,0.28mmol),2-二环己基膦基-2′,6′-二甲氧基联苯基(0.2330g,0.57mmol)和甲苯300mL。加料毕,搅拌升温至100~110℃保温12h。反应毕,待温度降至70℃以下,向反应体系中加入100mL纯化水水解。水解毕,静置分层,将上层有机相水洗至中性。水洗毕,经干燥、过柱、脱溶剂、甲苯/乙酸乙酯重结晶得到固体化合物10.6g,收率为82%。根据LC-MS分析,该固体化合物鉴定为化合物3-2。LC-MS:M/Z 453.45,化合物3-2经NMR测试结果为:
2. 化合物3-5的制备
;
在氮气保护条件下,向装有机械搅拌、温度计、球形冷凝管的三口瓶中依次加入化合物H(10.0g,28.4mmol),氘代溴苯(5.5g,34.1mmol),叔丁醇钠(4.1g,42.6mmol),双(二亚苄基丙酮)钯(0.1632g,0.28mmol),2-二环己基膦基-2′,6′-二甲氧基联苯基(0.2330g,0.57mmol)和甲苯300mL。加料毕,搅拌升温至100~110℃保温12h。反应毕,待温度降至70℃以下,向反应体系中加入100mL纯化水水解。水解毕,静置分层,将上层有机相水洗至中性。水洗毕,经干燥、过柱、脱溶剂、甲苯/乙酸乙酯重结晶得到固体化合物10.5g,收率为85%。根据LC-MS分析,该固体化合物鉴定为化合物3-5。LC-MS:M/Z 433.16,化合物3-5经NMR测试结果为:
3. 化合物3-16的制备
;
在氮气保护条件下,向装有机械搅拌、温度计、球形冷凝管的三口瓶中依次加入化合物H(10.0g,28.4mmol),4-溴二苯并呋喃(8.4g,34.1mmol),叔丁醇钠(4.1g,42.6mmol),双(二亚苄基丙酮)钯(0.1632g,0.28mmol),2-二环己基膦基-2′,6′-二甲氧基联苯基(0.2330g,0.57mmol)和甲苯300mL。加料毕,搅拌升温至100~110℃保温12h。反应毕,待温度降至70℃以下,向反应体系中加入100mL纯化水水解。水解毕,静置分层,将上层有机相水洗至中性。水洗毕,经干燥、过柱、脱溶剂、甲苯/乙酸乙酯重结晶得到固体化合物11.8g,收率为80%。根据LC-MS分析,该固体化合物鉴定为化合物3-16。LC-MS:M/Z518.14,化合物3-16经NMR测试结果为:
4. 化合物3-33的制备
;
在氮气保护条件下,向装有机械搅拌、温度计、球形冷凝管的三口瓶中依次加入化合物H(10.0g,28.4mmol),4-溴-9,9-二苯基-9H-芴(13.5g,34.1mmol),叔丁醇钠(4.1g,42.6mmol),双(二亚苄基丙酮)钯(0.1632g,0.28mmol),2-二环己基膦基-2′,6′-二甲氧基联苯基(0.2330g,0.57mmol)和甲苯300mL。加料毕,搅拌升温至100~110℃保温12h。反应毕,待温度降至70℃以下,向反应体系中加入100mL纯化水水解。水解毕,静置分层,将上层有机相水洗至中性。水洗毕,经干燥、过柱、脱溶剂、甲苯/乙酸乙酯重结晶得到固体化合物13.7g,收率为72%。根据LC-MS分析,该固体化合物鉴定为化合物3-33。LC-MS:M/Z 668.74,化合物3-33经NMR测试结果为:
5. 化合物3-38制备
;
在氮气保护条件下,向装有机械搅拌、温度计、球形冷凝管的三口瓶中依次加入化合物H(10.0g,28.4mmol),2-([1,1'-二联苯]-4-基)-4-氯-6-苯基-1,3,5-三嗪(11.7g,34.1mmol),叔丁醇钠(4.1g,42.6mmol),双(二亚苄基丙酮)钯(0.1632g,0.28mmol),2-二环己基膦基-2′,6′-二甲氧基联苯基(0.2330g,0.57mmol)和甲苯300mL。加料毕,搅拌升温至100~110℃保温12h。反应毕,待温度降至70℃以下,向反应体系中加入100mL纯化水水解。水解毕,静置分层,将上层有机相水洗至中性。水洗毕,经干燥、过柱、脱溶剂、甲苯/乙酸乙酯重结晶得到固体化合物14.0g,收率为75%。根据LC-MS分析,该固体化合物鉴定为化合物3-38。LC-MS:M/Z659.69,化合物3-38经NMR测试结果为:
6. 化合物3-41制备
;
在氮气保护条件下,向装有机械搅拌、温度计、球形冷凝管的三口瓶中依次加入化合物H(10.0g,28.4mmol),1-溴芘(9.6g,34.1mmol),叔丁醇钠(4.1g,42.6mmol),双(二亚苄基丙酮)钯(0.1632g,0.28mmol),2-二环己基膦基-2′,6′-二甲氧基联苯基(0.2330g,0.57mmol)和甲苯300mL。加料毕,搅拌升温至100~110℃保温12h。反应毕,待温度降至70℃以下,向反应体系中加入100mL纯化水水解。水解毕,静置分层,将上层有机相水洗至中性。水洗毕,经干燥、过柱、脱溶剂、甲苯/乙酸乙酯重结晶得到固体化合物12.2g,收率为78%。根据LC-MS分析,该固体化合物鉴定为化合物3-41。LC-MS:M/Z 552.58,化合物3-41经NMR测试结果为:
7. 化合物3-1~3-46制备
化合物3-1~3-46的制备方法同制备化合物3-2、3-5、3-16、3-33、3-38、3-41,所用原料为中间体化合物H和其它溴或氯代物原料,具体如下表1:
表1 制备化合物3-1~3-46的原料表
/>
/>
/>
/>
/>
/>
/>
/>
器件实施例:
通过热蒸镀的方式制备了15个有机电致发光器件,各个具体的器件结构如下:
比较实验例1
将涂层厚度为1500Å的ITO玻璃基板1放在蒸馏水中清洗2次,超声波洗涤30分钟,再用蒸馏水反复清洗2次,超声波洗涤10分钟,蒸馏水清洗结束后,采用异丙醇、丙酮、甲醇溶剂按顺序进行超声波洗涤后干燥,将干燥后的基板1转移到等离子体清洗机里,将上述基板1洗涤5分钟后送到蒸镀机里。
已经清洗好的ITO作为基板1,基板1具有阳极2表面,阳极2表面上依次蒸镀空穴注入层3(2-TNATA蒸镀厚度为500Å)、空穴传输层4(a-NPD蒸镀厚度为300Å)、发光层5(AND 9,10-二(2-萘基)蒽)和5%的EGl,蒸镀厚度为300Å)、空穴阻挡层6及电子传输层7(TPBi蒸镀厚度为400Å,此处空穴阻挡层6和电子传输层7二者一体)、电子注入层8(LiF蒸镀厚度为5Å)和A1蒸镀厚度为2000Å形成阴极9;上述过程有机物蒸镀速度保持1Å/sec,LiF蒸镀速度为0.2Å/sec,A1蒸镀速度为3~7 Å/sec。
其中器件实施例中涉及的化学结构式如下:
。
比较实验例2-比较实验例5:
采用比较实验例1相同的方法制备有机电致发光器件,不同之处在于,分别用EG2、EG3、EG4、EG5替换比较实验例1中的EG1。
器件实施例1-器件实施例15
采用比较实验例1相同的方法制备有机电致发光器件,不同之处在于,分别用本发明所述含吩噁嗪并噁唑类结构的化合物替换比较实施例1中的EG1。
器件实施例和比较实验例制备得到的有机电致发光器件的性能检测结果如下表2。
表2 有机电致发光器件的性能检测结果
从上述表结果中,采用本发明提供的有机电致器件用化合物制备有机电致器件,有机电致器件的发光效率和寿命特性显著提高。
以上所述实施例的各技术特征可以进行任意的组合,为使描述简洁,未对上述实施例中的各个技术特征所有可能的组合都进行描述,然而,只要这些技术特征的组合不存在矛盾,都应当认为是本说明书记载的范围。
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。