一种新型联苯类衍生物及其制备方法与医药用途
文献发布时间:2024-04-18 20:01:23
本发明属于药物化学领域,具体涉及一类具有PD-1/PD-L1抑制活性的联苯类衍生物,制备方法,以及含有这些化合物的药物组合物及其在治疗肿瘤中的用途。
近几年来肿瘤免疫疗法已成为肿瘤治疗领域的焦点,与直接针对肿瘤细胞的传统治疗手段不同,肿瘤免疫疗法是利用人体自身免疫系统对肿瘤细胞进行杀伤。免疫检查点通路的活化会抑制T细胞的激活,防止人体免疫系统的过度激活,维持正常机体的免疫耐受,避免自身免疫疾病的发生。肿瘤通过使自身和一些淋巴细胞过度激活免疫检查点通路导致肿瘤免疫逃逸的发生。在这些免疫检查点当中,PD-1/PD-L1的过度激活对于肿瘤的发展起着至关重要的作用。阻断PD-1/PD-L1相互作用可以重新激活免疫系统,杀死肿瘤细胞,在临床黑色素瘤、结肠癌、非小细胞肺癌等肿瘤治疗中展示出较好的疗效(Clinical and Translational Oncology,2019,21:702–712;The Oncologist,2019,24(Suppl 1):S31-S41;Human Vaccines&Immunotherapeutics,2014,10(11):3111-6;Journal of Medicinal Chemistry,2020,63(22):13825–13850)。
自2014年来,已有多种PD-1或PD-L1单抗药物经批准上市,并且这些单抗药物在多种肿瘤的临床治疗中取得了突破性进展。许多肿瘤患者的生存期显著延长,部分患者得到完全缓解。虽然单抗药物的临床效果显著,但由于单抗类药物的半衰期较长,且与靶点的结合时间久,因而会导致严重的免疫相关不良反应。单抗类药物的生产工艺复杂,价格昂贵且不便于储存运输,普通患者只能望“药”兴叹。与单克隆抗体相比,小分子药物的价格低廉、可口服给药、可跨越生物屏障、便于运输和储存、具有良好的膜通透性和非免疫原性等优势。国际申请WO2015034820A公开了一种PD-1/PD-L1小分子抑制剂,但是对其B环衍生的基团研究有限。而在缺乏现有技术启示的情况下,本发明人令人意外地开发出了具有PD-1/PD-L1抑制活性的新型联苯类衍生物,包括含有1,2,3-三氮唑杂环的衍生物。
发明内容
本发明提供了具有PD-1/PD-L1抑制活性的联苯类衍生物、其制备方法以及作为PD-1/PD-L1抑制剂的制药应用等。
在本发明中,PD-1/PD-L1抑制作用指的是对PD-1与PD-L1之间的相互作用的降低或者阻断。例如,可以通过现有技术或本发明实施例22所列的方法来定量地测定化合物对PD-1/PD-L1抑制作用的大小。
具体而言,本发明提供了一种如通式(I)所示的联苯类衍生物或其药学上可接受的盐:
其中:
X、Y各自代表O、NH、S或CH
A代表取代的苯基或芳杂环基,所述芳杂环基是含有1~3个O、N或S原子的五元或六元芳香环,所述的取代基是H、F、Cl、Br、CN、NH
L代表-CH
R
其中R
R
所述取代的C
R
R
R
本发明的通式(I)所示的联苯类衍生物优选是1,2,3-三氮唑杂环衍生物,
其中,X和Y优选O;m优选1,n优选2或3,p优选1;
A优选1,4-取代的1,2,3-三氮唑环;
L优选-CH
R
R
R
R
R
本发明的化合物中,X和Y代表O;m=1,p=1;A代表1,4-取代的1,2,3-三氮唑环;L代表-CH
其中,n=2或3;
R
R
进一步,R
更进一步,R
R
本发明的通式(I)所示的联苯类衍生物优选是II-1、II-2、II-3、II-4、II-5、II-7、II-8、II-9、II-10、II-11、II-12、II-13、II-14、II-15、II-16、II-18、II-19、II-20或II-21。
上述化合物或其药学上可接受的盐,其中药学上可接受的盐为通式(I)化合物与下列酸形成的酸加成盐:氯化氢、溴化氢、硫酸、碳酸、草酸、柠檬酸、琥珀酸、酒石酸、磷酸、乳酸、丙酮酸、乙酸、马来酸、甲磺酸、苯磺酸、对甲苯磺酸或阿魏酸。
本发明化合物(I)可用下列方法制备:
当X和Y代表O;m=1,n=2或3;p=1;A代表1,4-取代的1,2,3-三氮唑环;R
中间体IX的制备:
其中,R
化合物III首先与硫酸成盐后,再与亚硝酸钠反应得到重氮盐,最后水解得到化合物IV。反应温度选自0至110℃,优选25~100℃。
由化合物IV与1,3-二溴丙烷或1,4-二溴丁烷反应制备化合物V,所用碱选自氢氧化钠、氢氧化钾、碳酸钾、碳酸钠、碳酸铯、碳酸氢钠或碳酸氢钾,优选碳酸钾;所用溶剂选自丙酮、乙腈、二氧六环、N,N-二甲基甲酰胺(DMF)、二甲基亚砜(DMSO)或任意两者组成的混合溶剂,优选丙酮。
由化合物V与叠氮基三甲基硅烷制备化合物VI,所用溶剂选自丙酮、乙腈、二氧六环、N,N-二甲基甲酰胺、二甲基亚砜或任意两者组成的混合溶剂,优选N,N-二甲基甲酰胺;反应温度选自50至120℃,优选60℃~100℃。
由化合物VI与3,3-二乙氧基-1-丙炔经Click反应制备化合物VII,所用的催化剂为硫酸铜、氯化铜、溴化铜、氯化亚铜、溴化亚铜、碘化亚铜或铜粉,优选碘化亚铜。所用的溶剂为四氢呋喃、1,4-二氧六环、乙腈、乙醇、甲醇、乙二醇二甲醚、二氯甲烷、N,N-二甲基甲酰胺、N,N-二甲基乙酰胺(DMAc)或上述溶剂与水组成的混合溶剂,优选乙腈和水。反应温度选自0至80℃,优选20~50℃。
由化合物VII在酸作用下制备化合物VIII,所用的酸选自氯化氢、溴化氢、硫酸、磷酸、三氟乙酸、甲磺酸、苯磺酸、对甲苯磺酸,优选三氟乙酸;所用溶剂选自乙酸乙酯、四氢呋喃、二氯甲烷、丙酮、乙腈、二氧六环、N,N-二甲基甲酰胺、二甲基亚砜或任意两者组成的混合溶剂,优选二氯甲烷。反应温度选自0至80℃,优选25~40℃。
由化合物VIII与联硼酸频那醇酯制备化合物IX,所用溶剂选自甲苯、N,N-二甲基甲酰胺、N,N-二甲基乙酰胺、乙二醇二甲醚、1,4-二氧六环、四氢呋喃、乙醇、乙腈、丙酮、水或任意两者组成的混合溶剂,优选1,4-二氧六环;所用碱选自乙醇钠、乙酸钠、乙酸钾、磷酸钾、碳酸氢钾、碳酸钾或三乙胺,优选乙酸钾;所用催化剂选自四(三苯基膦)钯(Pd(PPh
化合物II的制备:
其中,R
化合物X与SOCl
由化合物XI经还原制备化合物XII,所用溶剂选自二氯甲烷、四氢呋喃、甲醇、乙醇、N,N-二甲基乙酰胺或二氧六环,优选甲醇;所选还原剂选自四氢锂铝、硼氢化钠或硼氢化钾,优选四氢锂铝。反应温度选自-5℃至50℃,优选0~25℃。
由化合物XII经溴代反应制备化合物XIII,所用溴代试剂选自N-溴代琥珀酰亚胺(NBS)、三溴化磷、四溴化碳或氢溴酸,优选三溴化磷。所用溶剂选自二氯甲烷、氯仿、四氯化碳、四氢呋喃、乙腈或上述溶剂组成的混合溶剂,优选二氯甲烷。反应温度选自-5℃至50℃,优选0~25℃。
由化合物XIII与5-氯-2,4-二羟基苯甲醛在碱性条件下经Williamson成醚反应制备化合物XIV,所用溶剂选自丙酮、N,N-二甲基甲酰胺、二甲基亚砜、乙腈、四氢呋喃或上述溶剂组成的混合溶剂,优选乙腈;所用碱选自氢化钠、叔丁醇钾、乙醇钠、甲醇钠、碳酸钾、碳酸钠、碳酸铯、氢氧化钠、氢氧化钾、氢氧化锂、碳酸氢钠或碳酸氢钾,优选碳酸氢钠。反应温度选自0℃至100℃,优选25~80℃。
由化合物XIV与化合物IX经suzuki反应制备化合物XV,所用溶剂选自甲苯、N,N-二甲基甲酰胺、N,N-二甲基乙酰胺、乙二醇二甲醚、1,4-二氧六环、四氢呋喃、甲醇、乙醇、乙腈、丙酮、水或任意两种溶剂组成的混合溶剂,优选1,4-二氧六环和水的混合溶剂;所用碱选自乙醇钠、乙酸钠、乙酸钾、磷酸钾、碳酸氢钾、碳酸钾或三乙胺,优选碳酸钾;所用催化剂选自Pd(PPh
由化合物XV与碘甲烷或5-(氯甲基)烟腈在碱性条件下经Williamson反应制备化合物XVI,所用溶剂选自丙酮、N,N-二甲基甲酰胺、乙腈、四氢呋喃或任意两种溶剂组成的混合溶剂,优选乙腈;所用碱选自氢化钠、甲醇钠、乙醇钠、碳酸钾、碳酸钠、碳酸铯、氢氧化钠、氢氧化钾、碳酸氢钠、碳酸氢钾或氢氧化锂,优选碳酸钾。反应温度选自0℃至100℃,优选25~80℃。
由化合物XVI与胺类化合物发生还原胺化反应制备目标化合物II,所用溶剂选自甲苯、N,N-二 甲基甲酰胺、二氯甲烷、二氯乙烷、三氯甲烷、四氯化碳、甲醇、1,4-二氧六环、四氢呋喃、乙醇、乙腈、丙酮或上述两种溶剂组成的混合溶剂,优选二氯甲烷和甲醇组成的混合溶剂;所用还原剂选自三乙酰氧基硼氢化钠、氰基硼氢化钠、硼氢化钠、硼氢化钾或保险粉,优选三乙酰氧基硼氢化钠。反应温度选自0℃至80℃,优选0~50℃。
本发明还提供了一种药物组合物,其含有上述通式(I)化合物(包括手性异构体)或其药学上可接受的盐及药学上可接受的载体。所述的化合物可以添加药学上可接受的载体制成常见的药用制剂,如片剂、胶囊、糖浆、悬浮剂、注射剂,可以加入香料、甜味剂、液体或固体填料或稀释剂等常用药用辅料。
本发明的药物组合物可通过所属领域技术人员所熟知的给药方式来进行给药,例如口服、直肠、舌下、肺部、透皮、离子透入、阴道及鼻内给药。本发明的药物组合物优选胃肠道外给药,如皮下、肌内或静脉内注射。给药剂量根据制剂形式和期望的作用时间以及治疗对象的情况而有所变化,实际治疗所需的治疗有效量可以由医师根据实际情况(如,病人的病情、体重等)而确定。对于一般的成人,本发明的药物组合物的剂量,以通式(I)化合物计,可以是每kg成人体重1ng–10g,优选为每kg体重10μg-100mg。
本发明所述的通式(I)化合物或其水合物、溶剂合物、结晶或药学上可接受的盐在制备PD-1/PD-L1蛋白-蛋白相互作用抑制剂药物中的应用也在本发明的保护范围内。相应地,本发明还提供了一种治疗方法,其包括向需要抑制PD-1/PD-L1相互作用的受治疗者给予治疗有效量的本发明的通式(I)化合物或其水合物、溶剂合物、结晶或药学上可接受的盐。
本发明还提供了本发明的通式(I)化合物或其水合物、溶剂合物、结晶或药学上可接受的盐在制备抗肿瘤药物中的用途。相应地,本发明还提供了一种治疗方法,其包括向患有肿瘤的受治疗者给予治疗有效量的本发明的通式(I)化合物或其水合物、溶剂合物、结晶或药学上可接受的盐。
优选地,本发明的PD-1/PD-L1抑制剂可用于制备治疗非小细胞肺癌、结肠癌或黑色素瘤等癌症的药物。
与现有技术相比,本发明具有如下显著有益效果:
(1)本发明的新型联苯类衍生物可以显著抑制PD-1/PD-L1的相互作用,且活性显著优于已知的BMS-202。尤其重要的是,本发明的联苯类化合物可以显著地阻断PD-L1对CD3
(2)本发明的联苯类衍生物的合成路线设计巧妙、简便易行,原料便宜易得,合成工艺安全、环保,易于规模化生产。
(3)适用性广,其作为活性成分的药物可用于治疗与免疫检查点PD-1/PD-L1相关的多种癌症或肿瘤。
(4)本发明的联苯类衍生物的用药安全性高。
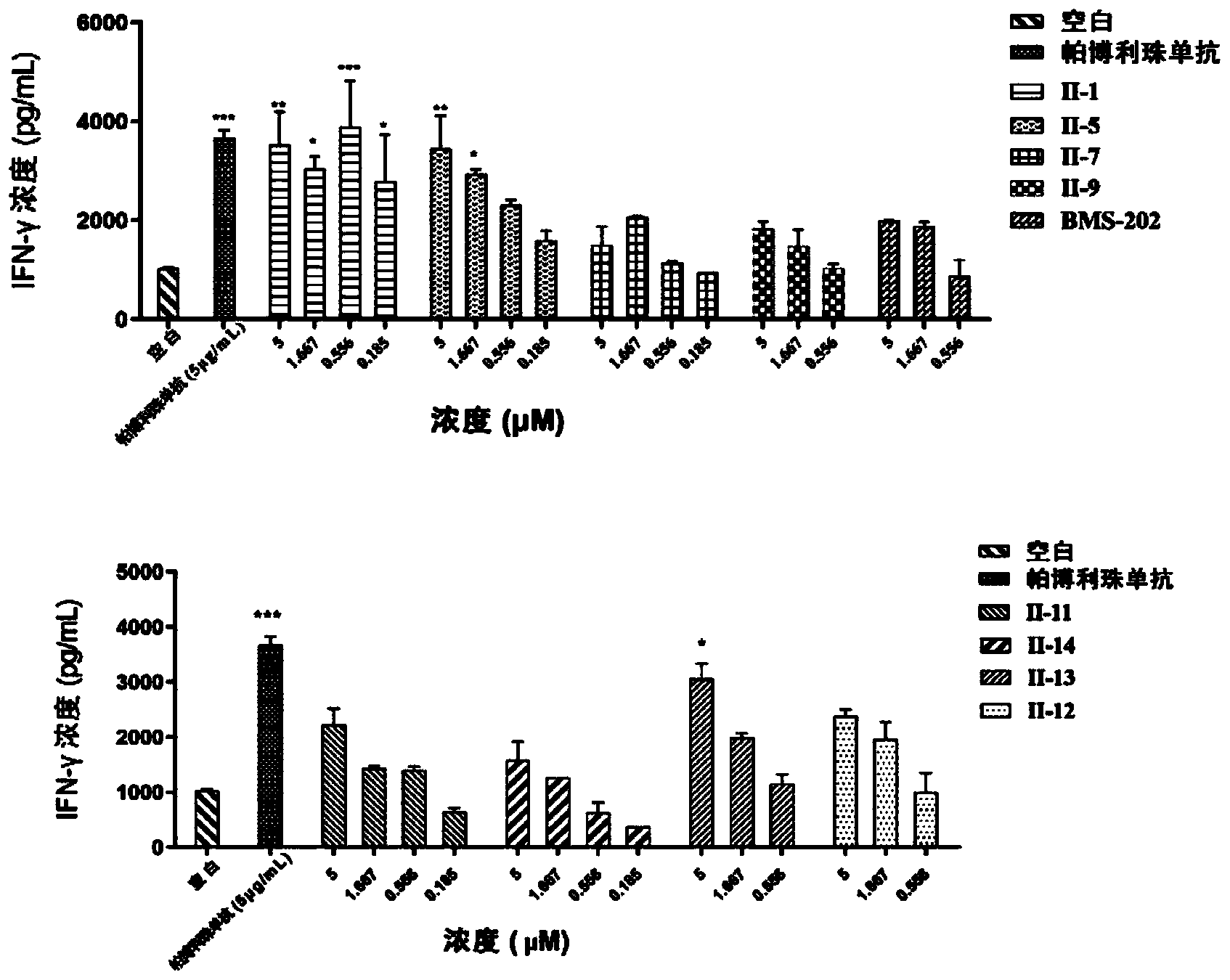
图1是共培养实验中化合物对INF-γ表达的影响;
图2是本发明的化合物的急性毒性实验结果;
图3是化合物II-1和II-18对人源化MC38-hPD-L1细胞移植瘤模型C57BL/6小鼠体内抗肿瘤效果 (肿瘤体积的影响);
图4是化合物II-1和II-18对人源化MC38-hPD-L1细胞移植瘤模型C57BL/6小鼠体内抗肿瘤效果(肿瘤质量的影响);
图5是小鼠体内实验中,化合物II-1(25mg/kg,ip,bid)和II-18(25mg/kg,po,bid)对小鼠体内CD3+CD4+T细胞分群的促进作用;
图6是小鼠体内实验中,化合物II-1(25mg/kg,ip,bid)和II-18(25mg/kg,po,bid)对小鼠体内CD3+CD8+T细胞分群的促进作用。
下面结合具体实施例对本发明作出详细说明。
实施例1
(4-((3'-(3-(4-(((1-羧基-2-羟乙基)氨基)甲基)-1H-1,2,3-三氮唑-1-基)丙氧基)-2,2'-二甲基-[1,1'-联苯]-3-基)甲氧基)-5-氯-2-((5-氰基吡啶-3-基)甲氧基)苄基)丝氨酸(II-1:R
3-溴-2-甲基苯酚(IV-1)的合成
将3-溴-2-甲基苯胺(5.00g,26.9mmol)加入三颈瓶中,搅拌下缓慢滴入硫酸溶液(65mL,1mmol/L),有大量不溶性白色固体析出。滴毕后,将反应液内温降至0~5℃,缓慢滴加30%亚硝酸钠溶液(2.23g,32.3mmol),有黄色不溶物生成,滴毕后降温至0~5℃搅拌30分钟。加入甲苯(30mL),升温至100℃反应1小时。反应液澄清透明后停止反应,冷至室温,乙酸乙酯(50mL×3)萃取,合并有机相,分别用水(30mL×2)和饱和氯化钠溶液(20mL)洗涤,无水硫酸钠干燥,抽滤,减压除去溶剂,经柱层析纯化(石油醚:乙酸乙酯=100:1),得白色针状结晶4.83g,收率为96.1%。m.p.95.0-98.0℃.
1
1-(1-溴-3-(3-溴丙氧基)-2-甲基苯(V-1)的合成
将3-溴-2-甲基苯酚(5.00g,26.7mmol)、1,3二溴丙烷(8.13mL,80.1mmol)、碳酸钾(9.23g,66.8mmol)溶于丙酮(50mL)中,升温至回流,反应4小时后,TLC(石油醚:乙酸乙酯=15:1)监测原料反应完全。停止加热,冷至室温。将反应液倒入冰水(100mL)中,用乙酸乙酯(50mL×3)萃取,合并有机相,分别用水(30mL×2)和饱和氯化钠溶液(20mL×2)洗涤,无水硫酸钠干燥,抽滤,减压除去溶剂得到粗品,经柱层析纯化(石油醚100%),得无色透明液体6.98g,收率为84.8%。
1
1-(3-叠氮丙氧基)-3-溴-2-甲基苯(VI-1)的合成
将化合物V-1(6.00g,19.5mmol)、叠氮三甲基硅烷(3.08mL,23.4mmol),CsF(4.44g,29.3mmol)和DMF(30mL)依次加入茄形瓶中,升温至50℃。反应4小时后,TLC监测(石油醚:乙酸乙酯=30:1)原料反应完全,停止加热,冷至室温。将反应液缓慢倒入冰水(60mL)中,乙酸乙酯(40mL×3)萃取,合并有机相,分别用水(40mL×2)和饱和氯化钠溶液(30mL×2)洗涤,无水硫酸钠干燥,抽滤,减压除去溶剂得粗品,经柱层析纯化得无色透明液体4.91g,收率为93.4%。
1
1-(3-(3-溴-2-甲基苯氧基)丙基)-4-(二乙氧基甲基)-1H-1,2,3-三氮唑(VII-1)的合成
将化合物VI-1(4.50g,16.7mmol)、3,3-二乙氧基-1-丙炔(2.88mL,20.1mmol)、碘化亚铜(0.16g,0.84mmol)和乙腈(30mL)依次加入茄形瓶中,升温至30℃反应。TLC(石油醚:乙酸乙酯=8:1)监测(约12小时)原料反应完全,停止加热,冷至室温。抽滤除去不溶物,将滤液缓慢倒入水(50mL)中,用乙酸乙酯(30mL×3)萃取,合并有机相,分别用水(30mL×2)和饱和氯化钠溶液(30mL×2)洗涤,无水硫酸钠干燥,抽滤,减压除去溶剂得粗品,经柱层析纯化得无色透明液体4.89g,收率73.8%,直接用于下一步反应。
1-(3-(3-溴-2-甲基苯氧基)丙基)-1H-1,2,3-三氮唑-4-甲醛(VIII-1)的合成
将化合物VII-1(4.50g,11.3mmol)和二氯甲烷(30mL)依次加到茄形瓶中,缓慢滴加三氟乙酸(3.54mL,47.6mmol)。室温搅拌1小时后,TLC(石油醚:乙酸乙酯=4:1)监测原料反应完全后,用饱和碳酸氢钠调节pH至7。用二氯甲烷(30mL×2)萃取,合并有机相,分别用水(30mL×2)和饱和氯化钠溶液(30mL×2)洗涤,无水硫酸钠干燥,抽滤,减压除去溶剂得粗品,经柱层析(石油醚:乙酸乙酯=6:1)纯化得白色固体粉末3.25g,收率89.0%。m.p.94.0-96.0℃.
1
1-(3-(2-甲基-3-(4,4,5,5-四甲基-1,3,2-二氧杂硼烷-2-基)苯氧基)丙基)-1H-1,2,3-三氮唑-4-甲醛(IX-1)的合成
将化合物VIII-1(3.00g,9.25mmol)、联硼酸频那醇酯(2.83g,11.1mmol)、乙酸钾(2.28g,23.2mmol)溶于1,4-二氧六环(15mL)中,N
1
3-溴-2-甲基苯甲酸甲酯(XI-1)的合成
将3-溴-2-甲基苯甲酸(X-1)(10.0g,46.7mmol)溶于甲醇(50mL)中,降温至0℃。缓慢滴加氯化亚砜(4.07mL,56.1mmol),滴毕,升温至65℃回流反应3小时。TLC(石油醚:乙酸乙酯=8:1)监测原料反应完全,停止加热,冷至室温。减压蒸除溶剂得白色固体粉末10.65g,收率99.9%。m.p.31.0-33.0℃。(文献值:30~31℃(1)Zhang,Peng;Journal of Medicinal Chemistry 2010,V53(16),P6112-6121CAPLUS)。
1
3-溴-2-甲基苯甲醇(XII-1)的合成
将化合物XI-1(10.50g,46.1mmol)溶于无水四氢呋喃(50mL),氮气保护下,降至0℃,缓慢分批加入LiAlH
1
1-溴-3-(溴甲基)-2-甲基苯(XIII-1)的合成
将化合物XII-1(2.58g,12.8mmol)溶于二氯甲烷(20mL)中,降温至0℃。缓慢加入三溴化磷(1.82mL,19.2mmol),室温反应过夜。TLC监测(石油醚:乙酸乙酯=15:1-8:1)反应完全,有小极性点生成。使用饱和碳酸氢钠调节pH至碱性,用二氯甲烷(20mL×3)萃取,饱和氯化钠溶液(30mL×3)洗涤,水(30mL×3)洗涤,无水硫酸钠干燥,减压蒸除溶剂得无色透明油状液体3.30g,收率97.4%。m.p.30.0-31.5℃。
1
4-((3-溴-2-甲基苄基)氧基)-5-氯-2-羟基苯甲醛(XIV-1)的合成
将化合物XIII-1(5.00g,18.9mmol)、5-氯-2,4-二羟基苯甲醛(3.27g,18.9mmol)、碳酸氢钠(3.97g,47.3mmol)溶于乙腈(50mL)中,升温至80℃,反应12小时后,TLC(石油醚:乙酸乙酯=8:1~4:1)监测有新点生成,停止反应。将反应液倒入水中(150mL),抽滤得白色粉末固体,真空干燥,粗品经柱层析得白色固体粉末1.87g,收率27.5%。m.p.178.0-181.0℃.
1
1-(3-((3'-((2-氯-4-甲酰基-5-羟基苯氧基)甲基)-2,2'-二甲基-[1,1'-联苯]-3-基)氧基)丙基)-1H-1,2,3-三氮唑-4-甲醛(XV-1)的合成
将化合物XIV-1(5.20g,14.7mmol)、化合物IX-1(4.97g,13.4mmol)和1,4-二氧六环(60mL)依次加入茄形瓶,碳酸钾(5.18g,37.5mmol)溶于水(6mL)中加入反应液,氮气保护下加入Pd(PPh
1
5-((4-氯-2-甲酰基-5-((3'-(3-(4-甲酰基-1H-1,2,3-三氮唑-1-基)丙氧基)-2,2'-二甲基-[1,1'-联苯]-3-基)甲氧基)苯氧基)甲基)烟腈(XVI-1)的合成
将化合物XV(2.88g,5.54mmol)、5-(氯甲基)烟腈(1.15g,6.10mmol)、碳酸铯(5.40g,16.7mmol)、碘化钾(0.05g,0.28mmol)和N,N-二甲基甲酰胺(15mL)依次加入茄形瓶中,升温至80℃反应6小时。TLC监测原料反应完全后,停止加热,冷至室温。将反应液缓慢倒入冰水(50mL)中,抽滤有固体析出,然后滤液用乙酸乙酯(20mL×3)萃取,合并有机相,分别用水(20mL×2)和饱和氯化钠(20mL×2)洗涤,无水硫酸钠干燥,抽滤,减压除去溶剂得棕黑色固体,经柱层析纯化(石油醚:乙酸乙酯=15:1—8:1),得黄色固体粉末2.29g,收率为65.0%。m.p.57.0-59.0℃。
1
(4-((3'-(3-(4-(((1-羧基-2-羟乙基)氨基)甲基)-1H-1,2,3-三氮唑-1-基)丙氧基)-2,2'-二甲基-[1,1'-联苯]-3-基)甲氧基)-5-氯-2-((5-氰基吡啶-3-基)甲氧基)苄基)丝氨酸(II-1)的合成
将化合物XVI-1(0.28g,0.44mmol)和丝氨酸甲酯盐酸盐(274mg,1.76mmol)溶于二氯甲烷(10mL)中,室温搅拌30min,缓慢加入NaBH(OAc)
1H NMR(300MHz,DMSO-d
HRMS(ESI):m/z[M+H]
实施例2
5-((4-氯-2-(((2-羟乙基)氨基)甲基)-5-((3'-(3-(4-(((2-羟乙基)氨基)甲基)-1H-1,2,3-三氮唑-1-基)丙氧基)-2,2'-二甲基-[1,1'-联苯]-3-基)甲氧基)苯氧基)甲基)烟腈(II-2:R
以化合物XVI-1(0.20g,0.31mmol)和乙醇胺(57μL,0.95mmol)溶于二氯甲烷(4mL)和甲醇(2mL)混合溶液中,室温搅拌30min,缓慢加入NaBH(OAc)
1
实施例3
N-(2-((4-((3'-(3-(4-(((2-乙酰氨基乙基)氨基)甲基)-1H-1,2,3-三氮唑-1-基)丙氧基)-2,2'-二甲基-[1,1'-联苯]-3-基)甲氧基)-5-氯-2-((5-氰基吡啶-3-基)甲氧基)苄基)氨基)乙基)乙酰胺(II-3:R
将化合物XVI-1(0.20g,0.31mmol)和N-乙酰基乙二胺(0.10g,0.95mmol)溶于二氯甲烷(5mL)依次加入反应瓶中,室温搅拌30min,缓慢加入NaBH(OAc)
1
实施例4
1-(4-((3'-(3-(4-((2-氨基甲酰基吡咯烷-1-基)甲基)-1H-1,2,3-三氮唑-1-基)丙氧基)-2,2'-二甲基-[1,1'-联苯]-3-基)甲氧基)-5-氯-2-((5-氰基吡啶-3-基)甲氧基)苄基)吡咯烷-2-羧酰胺(II-4:R
以化合物XVI-1(0.20g,0.31mmol)和L-脯氨酰胺(0.11g,0.95mmol)溶于二氯甲烷(5mL)依次加入反应瓶中,室温搅拌30min。缓慢加入NaBH(OAc)
1
实施例5
(4-((3'-(3-(4-(((羧甲基)氨基)甲基)-1H-1,2,3-三氮唑-1-基)丙氧基)-2,2'-二甲基-[1,1'-联苯]-3-基)甲氧基)-5-氯-2-((5-氰基吡啶-3-基)甲氧基)苄基)甘氨酸(II-5:R
以化合物XVI-1(0.20g,0.31mmol)和甘氨酸甲酯盐酸盐(0.11g,0.95mmol)为原料,操作同化合物II-1,得黄色固体粉末35mg,收率14.8%。在甲醇中,用氯化氢的甲醇溶液饱和成盐,得黄色固体粉末39.56mg,收率98.9%。
1
实施例6
(4-((3'-(2-(4-(((1-羧基-2-羟乙基)氨基)甲基)-1H-1,2,3-三氮唑-1-基)乙氧基)-2,2'-二甲基-[1,1'-联苯]-3-基)甲氧基)-5-氯-2-((5-氰基吡啶-3-基)甲氧基)苄基)丝氨酸(II-6:R
1-溴-3-(2-溴乙氧基)-2-甲基苯(V-2)的合成
以化合物IV-1(4.20g,18.3mmol)和1,2-二溴乙烷(4.75mL,54.9mmol)为原料,操作同化合物V-1,无色透明液体3.94g,收率为73.7%。
MS(ESI):m/z[M+H]
1
1-(2-叠氮基乙氧基)-3-溴-2-甲基苯(VI-2)
以化合物V-2(5.00g,17.1mmol)和TMSN
1
1-(2-(3-溴-2-甲基苯氧基)乙基)-4-(二乙氧基甲基)-1H-1,2,3-三氮唑(VII-2)
以化合物VI-2(4.00g,15.6mmol)和3,3-二乙氧基-1-丙炔(2.00g,15.6mmol)为原料,操作同化合物VII-1,得白色固体4.32g。未经处理直接投下一步。
1-(2-(3-溴-2-甲基苯氧基)乙基)-1H-1,2,3-三氮唑-4-甲醛(VIII-2)
以化合物VII-2(4.32g,11.2mmol)为原料,操作同化合物VIII-1,得白色固体2.53g,两步收率为52.3%。m.p 102.0-104.0℃。
1
1-(2-(2-甲基-3-(4,4,5,5-四甲基-1,3,2-二氧杂硼烷-2-基)苯氧基)乙基)-1H-1,2,3-三氮唑-4-甲醛(IX-2)
将化合物VIII-2(2.50g,8.06mmol)为原料,操作同化合物IX-1,得白色固体粉末2.45g,收率为85.3%。m.p.106.5-109.0℃.
1
1-(2-((3'-((2-氯-4-甲酰基-5-羟基苯氧基)甲基)-2,2'-二甲基-[1,1'-联苯]-3-基)氧基)乙基)-1H-1,2,3-三氮唑-4-甲醛(XV-2)
以化合物XIV(1.00g,2.81mmol)和化合物IX-2(1.21g,3.37mmol)为原料,操作同化合物XV-1,经柱层析纯化(石油醚:乙酸乙酯=2:1),得黄色固体粉末0.85g,收率为59.7%。m.p.138.0-140.0℃.
1
5-((4-氯-2-甲酰基-5-((3'-(2-(4-甲酰基-1H-1,2,3-三氮唑-1-基)乙氧基)-2,2'-二甲基-[1,1'-联苯]-3-基)甲氧基)苯氧基)甲基)烟腈(XVI-2)
将化合物XV-2(0.70g,1.38mmol)、5-(氯甲基)烟腈(0.31g,1.66mmol)、碳酸铯(1.34g,4.14mmol)、碘化钾(11mg,0.07mmol)和N,N-二甲基甲酰胺(15mL)依次加入茄形瓶中,升温至80℃反应6小时。TLC监测原料反应完全,停止加热,冷至室温。将反应液缓慢倒入冰水中,用乙酸乙酯(20mL×3)萃取,合并有机相,分别用水(20mL×2)和饱和氯化钠溶液(20mL×2)洗涤,无水硫酸钠干燥,抽滤,减压除去溶剂得棕黑色固体,经柱层析纯化(石油醚:乙酸乙酯=15:1~8:1)得淡黄色固体粉末0.48g,收率为55.8%。m.p.69.0-71.0℃.
1
(4-((3'-(2-(4-(((1-羧基-2-羟乙基)氨基)甲基)-1H-1,2,3-三氮唑-1-基)乙氧基)-2,2'-二甲基-[1,1'-联苯]-3-基)甲氧基)-5-氯-2-((5-氰基吡啶-3-基)甲氧基)苄基)丝氨酸(II-6)的合成
将化合物XVI-2(0.30g,0.48mmol)和丝氨酸甲酯盐酸盐(0.27g,1.93mmol)溶于二氯甲烷(5mL)依次加入反应瓶中,室温搅拌30min,缓慢加入NaBH(OAc)
1
实施例7
(4-((3'-(3-(4-(((1-羧基-2-羟乙基)氨基)甲基)-1H-1,2,3-三氮唑-1-基)丙氧基)-2,2'-二甲基-[1,1'-联苯]-3-基)甲氧基)-5-氯-2-甲氧基苄基)丝氨酸(II-7:R
1-(3-((3'-((2-氯-4-甲酰基-5-甲氧基苯氧基)甲基)-2,2'-二甲基-[1,1'-联苯]-3-基)氧基)丙基)-1H-1,2,3-三氮唑-4-甲醛(XVI-3)的合成
将化合物XV-1(0.72g,1.38mmol)、CH
1
(4-((3'-(3-(4-(((1-羧基-2-羟乙基)氨基)甲基)-1H-1,2,3-三氮唑-1-基)丙氧基)-2,2'-二甲基-[1,1'-联苯]-3-基)甲氧基)-5-氯-2-甲氧基苄基)丝氨酸(II-7)的合成
以化合物XVI-3(250mg,0.47mmol)和丝氨酸甲酯盐酸盐(182mg,1.17mmol)为原料,操作同化合物II-1,得棕色固体粉末42mg,收率12.6%。
1
实施例8
1-(4-((3'-(3-(4-((2-氨基甲酰基吡咯烷-1-基)甲基)-1H-1,2,3-三氮唑-1-基)丙氧基)-2,2'-二甲基-[1,1'-联苯]-3-基)甲氧基)-5-氯-2-甲氧基苄基)吡咯烷-2-甲酰胺(II-8:R
以化合物XVI-3(250mg,0.47mmol)和L-脯氨酰胺(134mg,1.17mmol)为原料,操作同化合物II-4,得棕色固体粉末49mg,收率14.3%。
1
HRMS(ESI):m/z[M+Na]
实施例9
2-(((1-(3-((3'-((2-氯-4-(((2-羟乙基)氨基)甲基)-5-甲氧基苯氧基)甲基)-2,2'-二甲基-[1,1'-联苯基]-3-基)氧基)丙基)-1H-1,2,3-三氮唑-4-基)甲基)乙烷-1-醇(II-9:R
以化合物XVI-3(200mg,0.38mmol)和乙醇胺(69.4mg,1.12mmol)为原料,操作同化合物II-2,得黄色固体粉末35mg,收率14.8%。
1
HRMS(ESI):m/z[M+Na]
实施例10
N-(2-((4-((3'-(3-(4-(((2-乙酰氨基乙基)氨基)甲基)-1H-1,2,3-三氮唑-1-基)丙氧基)-2,2'-二甲基-[1,1'-联苯]-3-基)甲氧基)-5-氯-2-甲氧基苄基)氨基)乙基)乙酰胺(II-10:R
以化合物XVI-3(200mg,0.37mmol)和N-乙酰基乙二胺(151mg,1.48mmol)为原料,操作同化合物II-3,得黄色固体粉末72mg,收率27.2%。m.p.48.0~50.0℃.
1
HRMS(ESI):m/z[M+H]
实施例11
(R)-1-((1-(3-((3'-((2-氯-4-(((R)-3-羟基吡咯烷-1-基)甲基)-5-甲氧基苯氧基)甲基)-2,2'-二甲基-[1,1'-联苯]-3-基)氧基)丙基)-1H-1,2,3-三氮唑-4-基)甲基)吡咯烷-3-醇(II-11:R
以化合物XVI-3(300mg,0.56mmol)和R-3-吡咯烷醇盐酸盐(0.35mg,2.81mmol)为原料,操作同化合物II-3,得白色固体粉末120mg,收率32.9%。
1
HRMS(ESI):m/z[M+H]
实施例12
3-((4-((3'-(3-(4-(((2-羧乙基)氨基)甲基)-1H-1,2,3-三氮唑-1-基)丙氧基)-2,2'-二甲基-[1,1'-联苯]-3-基)甲氧基)-5-氯-2-甲氧基苄基)氨基)丙酸(II-12:R
以化合物XVI-3(210mg,0.39mmol)和β-丙氨酸乙酯盐酸盐(0.52mg,1.57mmol)为原料,操作同化合物II-7,得黄色固体粉末157mg,收率57.6%。
1
HRMS(ESI):m/z[M+H]
实施例13
1-(4-((3'-(3-(4-((2-羧基哌啶-1-基)甲基)-1H-1,2,3-三氮唑-1-基)丙氧基)-2,2'-二甲基-[1,1'-联苯]-3-基)甲氧基)-5-氯-2-甲氧基苄基)哌啶-2-羧酸(II-13:R
以化合物XVI-3(300mg,0.56mmol)和哌啶-2-甲酸甲酯盐酸盐(0.43mg,2.81mmol)为原料,操作同化合物II-7,得黄色固体粉末106mg,收率24.8%。
1
HRMS(ESI):m/z[M+H]
实施例14
(R)-1-((1-(3-((3'-((2-氯-4-(((R)-3-羟基哌啶-1-基)甲基)-5-甲氧基苯氧基)甲基)-2,2'-二甲基-[1,1'-联苯]-3-基)氧基)丙基)-1H-1,2,3-三氮唑-4-基)甲基)哌啶-3-醇(II-14:R
以化合物XVI-3(200mg,0.37mmol)和R-哌啶-2-甲酸甲酯盐酸盐(0.23mg,1.50mmol)为原料,操作同化合物II-3,得黄色固体粉末179mg,收率67.86%。
1
实施例15
(4-((3'-(3-(4-(((1-羧乙基)氨基)甲基)-1H-1,2,3-三唑-1-基)丙氧基)-2,2'-二甲基-[1,1'-联苯]-3-基)甲氧基)-5-氯-2-甲氧基苄基)丙氨酸(II-15:R
以化合物XVI-3(210mg,0.39mmol)和β-丙氨酸乙酯盐酸盐(241mg,1.57mmol)为原料,操作同化合物II-3,经柱层析(二氯甲烷:甲醇=80:1~30:1)纯化得棕黄色固体粉末157mg,收率为57.6%。
1
HRMS(ESI):m/z[M+H]
实施例16
(4-((3'-(3-(4-(((1-羧基-2-羟乙基)氨基)甲基)-1H-1,2,3-三氮唑-1-基)丙氧基)-2,2'-二氯-[1,1'-联苯]-3-基)甲氧基)-5-氯-2-((5-氰基吡啶-3-基)甲氧基)苄基)丝氨酸(II-16:R
1-溴-3-(3-溴丙氧基)-2-氯苯(V-3)的合成
以3-溴-2-氯苯酚(10.00g,48.2mmol)和1,3-二溴丙烷(29.40g,144.6mmol)为原料,操作同化合物V-1,经柱层析纯化得无色透明油状液体14.57,收率92.0%。
1
1-(3-叠氮丙氧基)-3-溴-2-氯苯(VI-3)的合成
以化合物V-3(10.00g,30.7mmol)和TMSN
1
1-(3-(3-溴-2-氯苯氧基)丙基)-4-(二乙氧基甲基)-1H-1,2,3-三氮唑(VII-3)的合成
以化合物VI-3(4.82g,16.6mmol)和丙醛二乙基乙缩醛(2.34g,18.26mmoL)为原料,操作同化合物VII-1,经柱层析纯化得无色透明粘稠液体6.68g,直接用于下一步反应。
1-(3-(3-溴-2-氯苯氧基)丙基)-1H-1,2,3-三氮唑-4-甲醛(VIII-3)的合成
以化合物VII-3(6.68g,16.0mmol)和三氟乙酸(7mL)为原料,操作同化合物VIII-1,得白色固体粉末4.17g,收率75.9%。m.p.112-114℃.
1
1-(3-(2-氯-3-(4,4,5,5-四甲基-1,3,2-二氧杂环戊烷-2-基)苯氧基)丙基)-1H-1,2,3-三氮唑-4-甲醛(IX-3)的合成
以化合物VIII-3(6.42g,18.6mmol)和B
1
3-溴-2-氯苯甲酸甲酯(XI-2)的合成
以3-溴-2-氯苯甲酸(5.22g,22.17mmol)和SOCl
1
(3-溴-2-氯苯基)甲醇(XII-2)的合成
以化合物XI-2(12.84g,51.5mmol)和LiAlH
1
1-溴-3-(溴甲基)-2-氯苯(XIII-2)的合成
以化合物XII-2(6.40g,28.9mmol)和PBr
1
4-((3-溴-2-氯苄基)-氧基)-5-氯-2-羟基苯甲醛(XIV-2)的合成
以化合物XIII-2(5.00g,17.6mmol)和5-氯-2,4-二羟基苯甲醛(3.03g,17.6mmol)为原料,操作同化合物XIV-1的制备,得白色体粉末2.35g,收率35.5%。m.p.158.0-160.0℃.
1
5-((4-氯-5-((2,2'-二氯-3'-(3-(4-甲酰基-1H-1,2,3-三氮唑-1-基)丙氧基)-[1,1'-联苯基]-3-基)甲氧基)-2-甲酰基苯氧基)甲基)烟腈(XVI-4)的合成
5-((5-((3-溴-2-氯苄基)氧基)-4-氯-2-甲酰基苯氧基)甲基)烟腈(1)的合成
将化合物XIV-2(1.00g,2.66mmol)、5-(氯甲基)烟腈(0.60g,3.19mmol)、K
1
5-((4-氯-5-((2,2'-二氯-3'-(3-(4-甲酰基-1H-1,2,3-三氮唑-1-基)丙氧基)-[1,1'-联苯基]-3-基)甲氧基)-2-甲酰基苯氧基)甲基)烟腈(XVI-4)的合成
以化合物IX-2(1.29g,2.62mmol)和化合物1(1.23g,3.14mmol)为原料,操作同化合物XVI-1,得黄白色固体粉末0.87g,收率49.0%。m.p.58.0-60.0℃.
1
(4-((3'-(3-(4-(((1-羧基-2-羟乙基)氨基)甲基)-1H-1,2,3-三氮唑-1-基)丙氧基)-2,2'-二氯-[1,1'-联苯]-3-基)甲氧基)-5-氯-2-((5-氰基吡啶-3-基)甲氧基)苄基)丝氨酸(II-15)的合成
以化合物XVI-4(200mg,0.30mmol)和丝氨酸甲酯盐酸盐(184mg,1.18mmol)为原料,操作同化合物II-1,得褐色固体粉末83mg,收率32.9%。
1
实施例17
(4-((3'-(3-(4-(((羧甲基)氨基)甲基)-1H-1,2,3-三氮唑-1-基)丙氧基)-2,2'-二氯-[1,1'-联苯基]-3-基)甲氧基)-5-氯-2-((5-氰基吡啶-3-基)甲氧基)苄基)甘氨酸(II-17:R
以化合物XVI-4(200mg,0.30mmol)和甘氨酸甲酯盐酸盐(148mg,1.18mmol)为原料,操作同化合物II-1,得褐色固体粉末76mg,收率32.3%。
1
实施例18
(5-氯-2-((5-氰基吡啶-3-基)甲氧基)-4-((3'-(3-(4-((((R)-1-乙氧基-3-羟基-1-氧代丙烷-2-基)氨基)甲基)-1H-1,2,3-三氮唑-1-基)丙氧基)-2,2'-二甲基-[1,1'-联苯]-3-基)甲氧基)苄基)-D-丝氨酸乙酯(II-18:R
以化合物XVI-1(0.20g,0.31mmol)、三乙胺(0.26mL,1.89mmol)和D-丝氨酸乙酯盐酸盐(0.32g,1.89mmol)为原料,操作同II-2,得黄色固体粉末,0.14g产物,收率46.1%。
1
HRMS(ESI):m/z[M+H]
实施例19
(5-氯-2-((5-氰基吡啶-3-基)甲氧基)-4-((3'-(3-(4-((((R)-1-甲氧基-3-羟基-1-氧代丙烷-2-基)氨基)甲基)-1H-1,2,3-三氮唑-1-基)丙氧基)-2,2'-二甲基-[1,1'-联苯]-3-基)甲氧基)苄基)-D-丝氨酸甲酯(II-19:R
以化合物XVI-1(0.28g,0.44mmol)、三乙胺(0.24mL,1.76mmol)和D-丝氨酸甲酯盐酸盐(274mg,1.76mmol)为原料,操作同II-2,得黄色固体粉末0.12g,收率32.4%。
1
HRMS(ESI):m/z[M+H]
实施例20
(5-氯-2-((5-氰基吡啶-3-基)甲氧基)-4-((3'-(3-(4-(((2-乙氧基-2-氧代乙基)氨基)甲基)-1H-1,2,3-三氮唑-1-基)丙氧基)-2,2'-二甲基-[1,1'-联苯]-3-基)甲氧基)苄基)甘氨酸乙酯(II-20:R
以化合物XVI-1(0.20g,0.31mmol)、甘氨酸乙酯盐酸盐(0.26g,1.89mmol)和三乙胺(0.26mL,1.89mmol)为原料,操作同II-2,得黄色粘稠物0.11g,收率40.1%。
1
HRMS(ESI):m/z[M+H]
实施例21
((1-(3-((3'-((2-氯-4-((((R)1-乙氧基-3-羟基-1-氧代丙-2-基)氨基)甲基)-5-甲氧基苯氧基)甲基)-2,2'-二甲基-[1,1'-联苯]-3-基)氧基)丙基)-1H-1,2,3-三氮唑-4-基)甲基)-D-丝氨酸乙酯(II-21:R
以化合物XVI-3(0.20g,0.37mmol)、D-丝氨酸乙酯盐酸盐(0.38g,2.22mmol)和三乙胺(0.31mL,2.22mmol)为原料,操作同II-2,得淡黄色粘稠物0.16g,收率57.1%。
1
HRMS(ESI):m/z[M+H]
实施例22
本发明的部分化合物的药理学实验及结果如下:
1.本发明的化合物对hPD-1/hPD-L1相互作用的抑制活性评价
实验目的:使用hPD-1/hPD-L1 binding assay kit检测试剂盒(CISBIO公司),检测本发明化合物对PD-1/PD-L1的抑制活性。
实验原理:HTRF(均相时间分辨荧光,Homogeneous Time-Resolved Fluorescence)是一种用来检测纯液相体系中待测物的技术。其主要利用两种荧光基团的能量转移,这两种荧光基团分为能量供体铕(Eu+)和能量受体。当供体被外来激发(例如闪光灯或激光),若与受体在足够近的距离之内,可以将能量共振转移到受体上,受体被激发出特定的波长。利用HTRF技术,该测定法能够以高通量形式对化合物和抗体阻滞剂进行简单、快速的表征。通过使用标记有Eu
实验材料:试剂盒购买自CISBIO公司的hPD-1/hPD-L1 binding assay kits;96孔板:购买自CISBIO公司。
测试仪器:Perkin Elmer,型号:EnVision。
受试化合物:本发明制备所得化合物II-1至II-21。使用DMSO溶解,diluent buffer稀释;DMSO浓度不超过0.5%。
实验过程:使用hPD-1/hPD-L1 binding assay kits。设置阴性组、阳性组和给药组,每组2个复孔。阳性对照组,向96孔板中加入2μL diluent;按照说明书稀释后的4μL PD-L1和4uL PD-1;阴性对照组,向96孔板中加入6μL diluent和4μL PD-L1;给药组将2μL本发明化合物(或者阳性化合物BMS-202)、4μL PD-L1和4μL PD-1依次加入到96孔板中。使用封板膜封板,1000rpm离心1分钟,室温孵育15分钟。而后将Buffer稀释后的Anti-Tag-Eu3
A表示0.10-10nM;B表示10.01-100nM;C表示100.01nM~1μM。
表1化合物对hPD-1/hPD-L1相互作用的抑制活性
实验结果表明,本发明的化合物具有显著的PD-1/PD-L1抑制活性。预示本发明的联苯类化合物可用于制备PD-1/PD-L1抑制剂类抗肿瘤药物。
2.本发明的化合物与hPD-L1蛋白亲和力测试
实验原理:当入射光以临界角入射到两种不同折射率的介质界面(比如玻璃表面的金或银镀层)时,可引起金属自由电子的共振,由于共振致使电子吸收了光能量,从而使反射光在一定角度内大大减弱。其中,使反射光在一定角度内完全消失的入射角称为SPR角。SPR角随表面折射率的变化而变化,而折射率的变化又和结合在金属表面的生物分子质量成正比。分子间可逆的结合/解离造成金膜附近折光率的实时变化,这一现象被Biacore实时记录。
实验试剂:
Fc-hPD-L1蛋白购自Acro Biosystems公司,PBS-P+buffer 10x缓冲液、10mM glycine pH 1.5与Series S Sensor Chip Protein A芯片购自GE Healthcare Bio-sciences公司。
实验方法:
将受试化合物用DMSO溶解配成终浓度为10mM的储备液,再用1.05x PBS-P+稀释至1x,接着使用含有5%DMSO的1x PBS-P+buffer 4倍稀释;将Fc-PD-L1蛋白用含有5%DMSO的1x PBS-P+溶液稀释至10μg/mL,以流速10μl/min,时间120s捕获在Protein A芯片上,每次实验参用双通道,其中一个捕获配体,另一个作为参比通道;分析物现用现配,每个分析物至少设5个浓度,浓度的选择因分析物而异,分析物以30μl/min,90s流过两个通道;溶剂校正用于排除DMSO的影响,50%DMSO的1x PBS-P+溶液用于冲洗流路,30μl/min,30s的10mM glycine pH 1.5用于芯片表面再生,实验温度为25℃;使用Biacore T200 Evaluation software,1:1 binding model计算KD值。数据显示在表2。A表示0.10-100nM;B表示100.01-1000nM。
表2.化合物和hPD-L1亲和力数值
实验结果显示,本发明的化合物都可以和PD-L1蛋白结合,具有非常高的亲和力,预示本发明的联苯类化合物对PD-L1蛋白具有强的抑制活性。
3.本发明的化合物阻断PD-L1抑制T细胞分泌INF-γ表达作用的实验
实验原理:Hep3B-OS8-hPDL1细胞(上海睿智化学研究有限公司)表面稳定表达hPD-L1蛋白;CD3+T细胞(上海睿智化学研究有限公司)表面表达PD-1;当两株细胞被共培养的时候,Hep3B-OS8-hPDL1细胞表面的hPD-L1会与蛋白CD3+T细胞表面PD-1蛋白相互作用,从而抑制CD3+T细胞的激活、增殖、免疫因子INF-γ的表达。当化合物阻断PD-1/PD-L1相互作用的时候,将解除对CD3+T细胞的抑制,从而促进INF-γ的表达。
实验过程:用EDTA抗凝管盛放全血,密度梯度离心法分离PBMC。用EasySepTM Human T Cell Isolation Kit从PBMC中进一步分离得到CD3+T细胞,并用RPMI-1640完全培养液重悬细胞调整浓度为5×10
结果如图1所示。注:显著性差异使用one-way analysis of variance(ANOVA)计算得出;*P<0.05;**P<0.01;***P<0.001vs Blank。实验结果明显可以看出,本发明的化合物均可以通过阻断PD-1/PD-L1相互作用,从而解除对CD3+T细胞的抑制作用,显著促进INF-γ的表达。本发明的化合物剂量依赖性的促进INF-γ的表达,并且显著高于BMS-202的作用,仅仅略低于pembrolizumab(5μg/mL)的效果,因而具有增强T细胞抗肿瘤的功效;因此,本发明的联苯类化合物作为免疫检查点PD-1/PD-L1抑制剂可用于制备肿瘤免疫治疗的药物。
4.本发明的化合物的急性毒性实验
实验目的:为了获得化合物的安全给药剂量,我们对化合物II-1和II-18(250mg/kg和125mg/kg)和BMS-202(125mg/kg)分别腹腔注射给药,观察小鼠的状态。
实验过程:使用C57BL/6小鼠,检疫隔离室饲养3天。随机分为五组,每组5只;腹腔注射给药(0.2mL/20g),观察动物状态,测量体重。
实验结果:如图2,腹腔注射后,化合物II-1和II-18(250mg/kg和125mg/kg)组的小鼠均没有出现死亡,体重也没有下降。腹腔注射BMS-202(125mg/kg)后,小鼠出现明显抽搐、颤抖等体征,小鼠体重显著下降。因此本发明的化合物相比于BMS-202显示出更高的安全性。
5.本发明的化合物的小鼠体内抗肿瘤效果
实验目的:为了准确评估化合物的体内抗肿瘤效果,我们使用了人源化C57BL/6小鼠的MC38-hPDL1细胞移植瘤模型(上海南方模式生物科技股份有限公司)进行化合物的抗肿瘤效果评价
实验过程:结肠癌细胞MC38-hPDL1培养在37℃、5%CO
取对数生长期细胞,重悬于PBS中,进行细胞计数,调整细胞浓度到1.0×10
建模成功后,将小鼠置于检疫隔离室饲养3天,于SPF实验环境中饲养。用游标卡尺测量小鼠移植瘤大小,将符合实验要求的小鼠挑选出来,随机分成4组,每笼5只,每组平均肿瘤体积约为54mm
实验结果:由图3可知,相比于模型空白组,化合物II-1(腹腔注射给药组)和II-18(灌胃给药组)均能显著抑制结肠癌的生长(相对肿瘤抑制率分别为45.8%和49.6%),而化合物II-1(灌胃给药组)对肿瘤生长的抑制作用很弱。通过比较化合物II-1和II-18的化学结构可以发现,II-1分子中的两个羧基成酯后得到前药型的II-18,其口服抗肿瘤效果得到明显的提高。由图4可知,肿瘤重量也体现出化合物II-1(腹腔注射组)和II-18(灌胃组)优异的抗肿瘤效果(*p<0.05)。图5和图6分别显示所申请化合物II-1(腹腔注射组)和II-18(灌胃组)可以促进CD3