4-((R)-2-{6-((S)-3-甲氧基-吡咯烷-1-基)-2-苯基-嘧啶-4-羰基-氨基}-3-膦酰基-丙酰基)-哌嗪-1-羧酸丁酯的合成方法
文献发布时间:2024-04-18 20:01:55
技术领域
本发明涉及4-((R)-2-{[6-((S)-3-甲氧基-吡咯烷-1-基)-2-苯基-嘧啶-4-羰基]-氨基}-3-膦酰基-丙酰基)-哌嗪-1-羧酸丁酯(下文中亦称作“化合物”,亦称作塞拉格瑞(selatogrel))或其盐酸盐的合成方法;关于4-((R)-2-{[6-((S)-3-甲氧基-吡咯烷-1-基)-2-苯基-嘧啶-4-羰基]-氨基}-3-膦酰基-丙酰基)-哌嗪-1-羧酸丁酯盐酸盐(下文中亦称作“化合物·HCl”)的晶型,及关于用作药剂或用于制备药剂的化合物·HCl的晶型。
背景技术
化合物的制备及医疗用途述于WO 2009/069100;WO 2018/167139;Baldoni D等人,Clin Drug Investig(2014),34(11),807-818;Caroff E等人,J.Med.Chem.(2015),58,9133-9153;Storey R F等人,European Heart Journal,ehz807,doi:10.1093/eurheartj/ehz807;及Sinnaeve P R等人,J Am Coll Cardiol(2020),75(20),2588-97(doi.org/10.1016/j.jacc.2020.03.059)中。
例如,化合物·HCl可根据反应图1中所示的程序制备:化合物3可通过在存在偶合剂(诸如T3P或EDC、HOBt)下使哌嗪-1-羧酸丁酯或其盐酸盐与(R)-2-叔丁氧羰基氨基-3-(二乙氧基-膦酰基)-丙酸(化合物2)的酰胺偶合获得。化合物3中的氨基保护基可在适宜酸性条件(诸如含TFA的DCM或含HCl的二噁烷中)下移除,以得到4-[(R)-2-氨基-3-(二乙氧基-膦酰基)-丙酰基]-哌嗪-1-羧酸丁酯(化合物4)。化合物4可在存在如EDC、HOBt的偶合试剂下偶合至(S)-6-(3-甲氧基吡咯烷-1-基)-2-苯基嘧啶-4-羧酸钠盐(化合物6),以得到化合物7。化合物6可(例如)通过(S)-6-(3-甲氧基吡咯烷-1-基)-2-苯基嘧啶-4-甲腈(化合物5)与如氢氧化钠水溶液的碱于如2-丙醇之类的溶剂中的皂化来获得。
反应图1:合成化合物·HCl
化合物可(例如)自4-((R)-3-(二乙氧基-膦酰基)-2-{[6-((S)-3-甲氧基-吡咯烷-1-基)-2-苯基-嘧啶-4-羰基]-氨基}-丙酰基)-哌嗪-1-羧酸丁酯(化合物7)通过用TMSBr于乙腈中处理及利用逆相管柱层析法纯化来制备(Caroff E等人,J.Med.Chem.(2015),58,9133-9153)。针对大规模合成,此方法具有使用大量昂贵TMSBr来脱去保护基及基于管柱层析法的纯化步骤的缺点。这样的缺点可通过以下来克服:如WO 2018/055016中所述,通过4-((R)-3-(二乙氧基-膦酰基)-2-{[6-((S)-3-甲氧基-吡咯烷-1-基)-2-苯基-嘧啶-4-羰基]-氨基}-丙酰基)-哌嗪-1-羧酸丁酯利用浓盐酸水溶液于DCM/THF混合物中脱去保护基及将所获得的产物结晶来制备化合物·HCl。即使此方法更优选适用于大规模合成,但仍发现化合物在这种反应条件下在显著程度上水解成(R)-2-(6-((S)-3-甲氧基吡咯烷-1-基)-2-苯基嘧啶-4-甲酰氨基)-3-膦酰基丙酸(下文中亦称作“水解产物”)。例如,含于DCM(4vol)中的4-((R)-3-(二乙氧基-膦酰基)-2-{[6-((S)-3-甲氧基-吡咯烷-1-基)-2-苯基-嘧啶-4-羰基]-氨基}-丙酰基)-哌嗪-1-羧酸丁酯的溶液及32%w/w HCl水溶液(3.6vol)于RT下搅拌4小时后得到2.8%-a/a水解产物及93.5%-a/a化合物·HCl的混合物,如通过HPLC所分析得到。于20小时后,混合物中的水解产物的量甚至增加至15.2%-a/a。因此,于浓盐酸水溶液中脱去保护基具有所需产物快速分解的缺点,需要更强纯化及导致更低产率。
发明内容
出入意料地,发现若利用盐酸于仅含有催化量的水的特定有机溶剂中进行反应,则可显著减少副产物(R)-2-(6-((S)-3-甲氧基吡咯烷-1-基)-2-苯基嘧啶-4-甲酰氨基)-3-膦酰基丙酸的量及可以高产率及良好反应速率获得化合物·HCl。然而该反应极慢及/或产生大量副产物(例如,利用于如庚烷、乙腈、2-甲基-四氢呋喃或乙醇的溶剂中的HCl),但是利用含于甲苯、丙酮、羧酸酯及尤其羧酸(例如,乙酸)中的HCl其产生出入意料良好结果。
附图说明
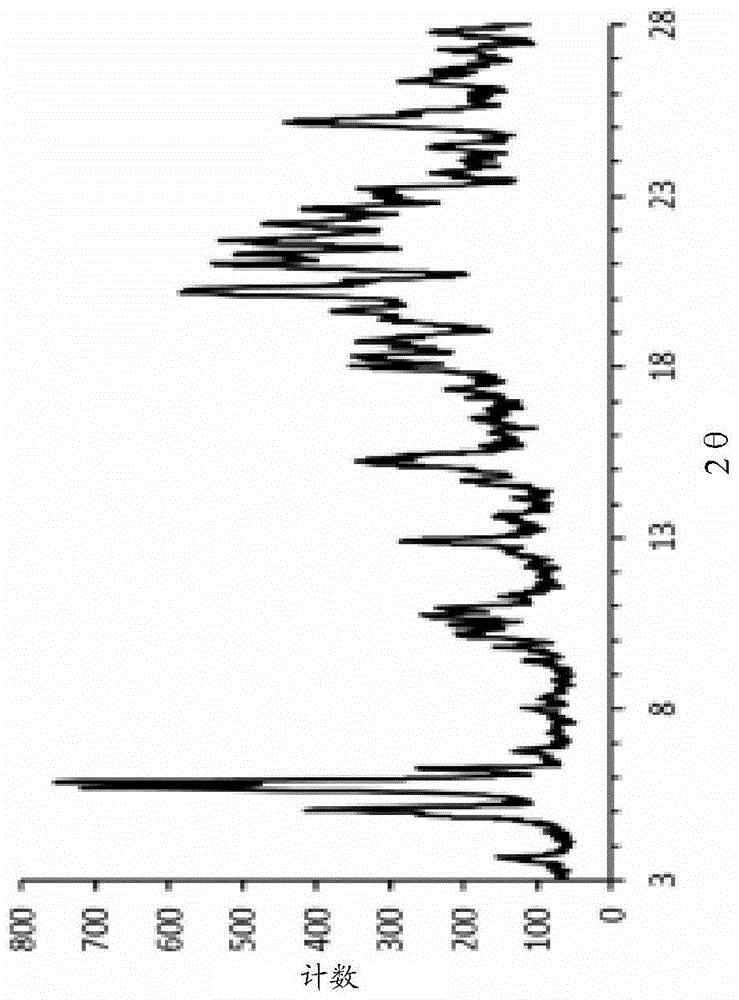
图1显示呈晶型(I)的化合物·HCl的X-射线粉末衍射图,其中该X-射线粉末衍射图利用实验部分中所述的XRPD方法测量及针对Cu Kα辐射显示。该X-射线衍射图显示如与图中的最强峰相比,在指定绕射角2θ处具有下列百分比的相对强度(相对峰强度于括号内提供)的峰(报告范围3至28°2θ与大于10%相对强度的所选峰):3.7°(12%)、5.1°(50%)、5.7°(93%)、5.9°(100%)、10.2°(17%)、10.4°(16%)、10.7°(25%)、11.0°(20%)、12.9°(28%)、14.7°(13%)、15.2°(32%)、15.4°(26%)、18.0°(26%)、18.3°(23%)、18.8°(26%)、19.4°(21%)、19.6°(31%)、20.2°(58%)、21.0°(54%)、21.3°(49%)、22.2°(45%)、22.6°(33%)、25.2°(40%)、及26.4°(18%)。
于图1的X-射线衍射图中,将绕射角2θ(2θ)在水平轴在线绘图及在竖直轴在线计数。
为避免任何怀疑,以上所列的峰描述图1中所示的X-射线粉末绕射的实验结果。应了解,与以上峰列表相反,仅要求选择特征峰以充分且明确地表征呈本发明的各自晶型的化合物·HCl。
具体实施方式
于以下中,将描述本发明及呈现本发明的各种实施例。
1)于第一实施例中,本发明涉及一种制造4-((R)-2-{[6-((S)-3-甲氧基-吡咯烷-1-基)-2-苯基-嘧啶-4-羰基]-氨基}-3-膦酰基-丙酰基)-哌嗪-1-羧酸丁酯(化合物)或其盐酸盐(化合物·HCl)的方法
该方法包括式(I)化合物
其中R
与盐酸于包含有机溶剂和水的混合物中的反应,
其中该有机溶剂为丙酮、甲苯、R
且其中该水的量小于相对于式(I)化合物的量的12当量。
除非另有明确阐述定义提供更宽或更窄定义,否则意指本文中所提供的定义在整篇描述及权利要求中统一应用。应充分理解,术语的定义或优选定义独立于(及与以下组合)如本文中所定义的任何或所有其他术语的任何定义或优选定义,定义及可替代各自术语。
应了解,于反应中需要的盐酸可源自盐酸的任何适宜来源,其不增加反应混合物中的水的量至相对于式(I)化合物的量的12当量或更多水。例如,可将盐酸以氯化氢气体或以含于溶剂中的溶液添加至反应混合物中,其中该溶剂为有机溶剂(实例:二噁烷、乙醇及异丙醇,尤其二噁烷),或水(尤其含于水中的溶液,及尤其含于水中的浓溶液);或可通过亲电氯化物源(即,于与亲核试剂的反应中释放氯离子的化合物)与质子亲核试剂(即,包含连接至氢原子的杂原子的官能基的化合物,其中该杂原子具有一或多个自由电子对)的反应原位产生。亲电氯化物源的实例为羧酸氯化物(尤其(C
短语“其中该有机溶剂为丙酮、甲苯、R
如于“第一化合物的量为相对于第二化合物的量的“X”当量”的上下文中所用,术语“当量”意指给定混合物含有相对于第二化合物的量(以与分子数目相关的任何单位)的“X”倍的第一化合物的量(以相同单位提供)。例如,术语“当量”于“其中水的量小于相对于式(I)化合物的量的12(或,或者介于值“x”与值“y”之间)当量”的上下文中意指反应混合物含有相对于式(I)化合物的量(以与分子数目相关的任何单位)的给定范围当量的水的量(以相同单位提供)。例如,如果反应混合物中的水的量被定义为小于相对于式(I)化合物的量的12当量,则这意指反应混合物中的水与式(I)化合物之间的摩尔比率低于12比1;及如果反应混合物中的水的量被定义为相对于式(I)化合物的量的0.5当量与3.0当量之间,则此意指反应混合物中的水与式(I)化合物之间的摩尔比率为1比2、3比1或其之间的任何值。
优选地,水的量为相对于式(I)化合物的量的0.2当量与9.5当量之间(更优选地介于0.5当量与3.0当量之间及最佳地介于0.5当量与2.0当量之间)。应了解,水的量是指存在于反应混合物中的水的总量,即,所添加的水的量连同存在于试剂、溶剂、反应容器及水的其他来源中的水的量。
除非关于温度使用,否则放在数值“X”之前的术语“约”于本申请中是指自X减去X的10%扩展至X加上X的10%的间隔,尤其是指自X减去X的5%扩展至X加上X的5%的间隔及尤其是指自X减去X的2%扩展至X加上X的2%的间隔。在温度的特定情况下,放在温度“Y”之前的术语“约”于本申请中是指自温度Y减去10℃扩展至Y加上10℃的间隔,尤其是指自Y减去5℃扩展至Y加上5℃的间隔,及尤其是指自Y减去3℃扩展至Y加上3℃的间隔。室温意指约25℃的温度。
无论何时将词语“之间”或“至”用于描述数值范围,应了解,指定范围的端点明确包含于该范围内。例如:若描述温度范围为介于40℃与80℃之间(或40℃至80℃),此意指端点40℃及80℃包含于该范围内;或若将变量定义为介于1与4之间(或1至4),则此意指该变量为整数1、2、3或4。
表述%w/w是指与所考虑的组合物的总重量相比的重量%。同样,表述v/v是指所考虑的两种组分的体积比率。
单独或以组合使用的术语“烷基”是指含有1至4个碳原子的直链或分支链饱和烃链。术语“(C
单独或以组合使用的术语“(C
单独或以组合使用的术语“((C
单独或以组合使用的术语“(C
单独或以组合使用的术语“(C
单独或以组合使用的术语“(C
单独或以组合使用的术语“((C
2)另一实施例涉及如实施例1)的方法,其中该方法为一种用于制造4-((R)-2-{[6-((S)-3-甲氧基-吡咯烷-1-基)-2-苯基-嘧啶-4-羰基]-氨基}-3-膦酰基-丙酰基)-哌嗪-1-羧酸丁酯盐酸盐(化合物·HCl)的方法。
3)另一实施例涉及如实施例1)或2)中任一项的方法,其中R
4)另一实施例涉及如实施例1)至3)中任一项的方法,其中R
5)另一实施例涉及如实施例1)或2)中任一项的方法,其中R
6)另一实施例涉及如实施例1)或2)中任一项的方法,其中R
7)另一实施例涉及如实施例1)至6)中任一项的方法,其中将该盐酸以氯化氢气体添加至反应混合物中或通过亲电氯化物源与质子亲核试剂的反应原位产生。
8)另一实施例涉及如实施例1)至6)中任一项的方法,其中该盐酸通过亲电氯化物源与质子亲核试剂的反应原位产生。
9)另一实施例涉及如实施例7)或8)中任一项的方法,其中该亲电氯化物源选自羧酸氯化物(尤其(C
10)另一实施例涉及如实施例7)或8)中任一项的方法,其中该亲电氯化物源选自(C
11)另一实施例涉及如实施例7)或8)中任一项的方法,其中该亲电氯化物源为乙酰氯(CH
12)另一实施例涉及如实施例7)至11)中任一项的方法,其中该质子亲核试剂选自水、烷醇(尤其(C
13)另一实施例涉及如实施例7)至11)中任一项的方法,其中该质子亲核试剂选自水及(C
14)另一实施例涉及如实施例7)至11)中任一项的方法,其中该质子亲核试剂选自(C
15)另一实施例涉及如实施例7)或8)中任一项的方法,其中该盐酸通过选自(C
16)另一实施例涉及如实施例7)或8)中任一项的方法,其中该盐酸通过乙酰氯(CH
17)另一实施例涉及如实施例7)至16)中任一项的方法,其中该亲电氯化物源(尤其(C
18)另一实施例涉及如实施例7)至16)中任一项的方法,其中该亲电氯化物源(尤其(C
19)另一实施例涉及如实施例7)至16)中任一项的方法,其中该亲电氯化物源(尤其(C
20)另一实施例涉及如实施例7)至19)中任一项的方法,其中该质子亲核试剂(尤其水及(C
21)另一实施例涉及如实施例7)至19)中任一项的方法,其中该质子亲核试剂(尤其水及(C
22)另一实施例涉及如实施例7)至19)中任一项的方法,其中该质子亲核试剂(尤其水及(C
23)另一实施例涉及如实施例1)至6)中任一项的方法,其中该盐酸通过((C
24)另一实施例涉及如实施例7)的方法,其中该盐酸以氯化氢气体添加至反应混合物中。
25)另一实施例涉及如实施例1)至16)、23)或24)中任一项的方法,其中以氯化氢气体添加至反应混合物中;或通过亲电氯化物源与质子亲核试剂的反应原位产生;或通过((C
26)另一实施例涉及如实施例1)至16)或24)中任一项的方法,其中以氯化氢气体添加至反应混合物中;或通过亲电氯化物源与质子亲核试剂的反应原位产生的盐酸的量为相对于式(I)化合物的量的0.9当量与5.0当量之间(尤其1.0与3.0当量之间)。
27)另一实施例涉及如实施例1)至16)或24)中任一项的方法,其中以氯化氢气体添加至反应混合物中;或通过亲电氯化物源与质子亲核试剂的反应原位产生的盐酸的量为相对于式(I)化合物的量的1.0当量与2.0当量之间(尤其约1.5当量)。
28)另一实施例涉及如实施例1)至27)中任一项的方法,其中该有机溶剂为甲苯、R
29)另一实施例涉及如实施例1)至27)中任一项的方法,其中该有机溶剂为R
30)另一实施例涉及如实施例1)至29)中任一项的方法,其中R
31)另一实施例涉及如实施例1)至30)中任一项的方法,其中R
32)另一实施例涉及如实施例1)至27)中任一项的方法,其中该有机溶剂为乙酸(CH
33)另一实施例涉及如实施例1)至27)中任一项的方法,其中该有机溶剂为乙酸(CH
34)另一实施例涉及如实施例1)至33)中任一项的方法,其中该有机溶剂的体积为1.5与20之间升/千克式(I)化合物。有机溶剂的体积的下限为1.5、2.0、2.5及2.8升/千克式(I)化合物,上限为20、10、5.0及3.3升/千克式(I)化合物。应了解,可将各下限与各上限组合。因此,本文应公开所有组合。
35)另一实施例涉及如实施例1)至33)中任一项的方法,其中该有机溶剂的体积为2.0与10之间(尤其2.5与5.0之间)升/千克式(I)化合物。
36)另一实施例涉及如实施例1)至33)中任一项的方法,其中该有机溶剂的体积为2.5与3.3之间(尤其约2.9)升/千克式(I)化合物。
37)另一实施例涉及如实施例1)至33)中任一项的方法,其中该式(I)化合物于有机溶剂中的浓度介于20与30%w/w之间(尤其约25%w/w)。
38)另一实施例涉及如实施例1)至37)中任一项的方法,其中水的量为相对于式(I)化合物的量的0.2当量与9.5当量之间。水的量的下限为0.2、0.3、0.5及0.8当量,上限为9.5、5.0、3.0及2.0当量。应了解,可将各下限与各上限组合。因此,本文应公开所有组合。
39)另一实施例涉及如实施例1)至37)中任一项的方法,其中水的量为相对于式(I)化合物的量的0.3与5.0之间(尤其0.5与3.0之间)当量。
40)另一实施例涉及如实施例1)至37)中任一项的方法,其中水的量为相对于式(I)化合物的量的0.5与2.0之间(尤其0.8与2.0之间)当量。
41)另一实施例涉及如实施例1)或2)中任一项的方法,该方法包括式(I)化合物
其中R
与盐酸于包含有机溶剂及水的混合物中的反应,
其中将该盐酸以氯化氢气体以相对于式(I)化合物的量的1.0当量与3.0当量之间(尤其1.0当量与2.0当量之间)的量添加至反应混合物中;
其中该有机溶剂为乙酸、乙酸甲酯(CH
其中该水的量为相对于式(I)化合物的量的0.3当量与5.0当量之间(尤其0.5当量与3.0当量之间)。
42)另一实施例涉及如实施例1)或2)中任一项的方法,该方法包括式(I)化合物
其中R
与盐酸于包含有机溶剂及水的混合物中的反应,
其中该盐酸通过(C
其中该有机溶剂为乙酸、乙酸甲酯(CH
其中该水的量为相对于式(I)化合物的量介于0.3当量与5.0当量之间(尤其介于0.5当量与3.0当量之间)。
43)另一实施例涉及如实施例41)或42)中任一项的方法,其中R
44)另一实施例涉及如实施例41)至43)中任一项的方法,其中该有机溶剂为乙酸,且其中该式(I)化合物于有机溶剂中的浓度介于20%w/w与30%w/w之间。
45)另一实施例涉及如实施例41)至44)中任一项的方法,其中该式(I)化合物于有机溶剂中的浓度为约25%w/w。
46)另一实施例涉及如实施例41)至45)中任一项的方法,其中该水的量为相对于式(I)化合物的量介于0.8当量与2.0当量之间。
47)另一实施例涉及如实施例1)至46)中任一项的方法,其中该反应在介于20℃与40℃之间的温度下进行。反应温度的下限为20℃、23℃、25℃及27℃,上限为40℃、37℃、35℃及33℃。应了解,可将各下限与各上限组合。因此,本文应公开所有组合。
48)另一实施例涉及如实施例1)至46)中任一项的方法,其中该反应在介于25℃与35℃之间(尤其介于27℃与33℃之间)的温度下进行。
49)另一实施例涉及如实施例1)至48)中任一项的方法,其中于搅拌至少3小时(尤其约4小时)的搅拌时间后将该反应混合物用化合物·HCl的晶种处理。
术语“搅拌时间”意指于将最后试剂/反应物添加至反应混合物后的时间直至添加晶种的时间。优选地,该搅拌时间介于3小时与8小时之间,更佳地介于3.5小时与5小时之间及最佳地约4小时。
优选地,化合物·HCl的晶种呈如WO 2018/055016中所述的晶型2或如本文中所述的晶型(I)。
50)另一实施例涉及如实施例49)的方法,其中化合物·HCl的晶种呈如WO 2018/055016中所述的晶型2。
晶型2的晶种可(例如)自WO 2018/055016中所述的方法或自本文中所述的方法获得。
晶种亦可通过内部接晶种,即,通过自反应混合物移除样品,添加反溶剂(尤其乙酸乙酯)至样品中及再次添加所获得的悬浮液至反应混合物中来获得。优选地,将2.0至4.0mL(最佳地约2.5mL)乙酸乙酯/克样品添加至样品中。
51)另一实施例涉及如实施例49)或50)中任一项的方法,其中晶种的量为相对于式(I)化合物的量的0.1%w/w与2.0%w/w之间。晶种的量的下限为0.1、0.2及0.3%w/w,上限为2.0、1.0及0.6%w/w。应了解,可将各下限与各上限组合。因此,本文应公开所有组合。
52)另一实施例涉及如实施例49)或50)中任一项的方法,其中晶种的量为相对于式(I)化合物的量的0.1%w/w与1.0%w/w之间(尤其0.1%w/w与0.6%w/w之间)。
53)另一实施例涉及如实施例49)或50)中任一项的方法,其中晶种的量为相对于式(I)化合物的量的0.2%w/w与0.6%w/w之间。
54)另一实施例涉及如实施例1)至53)中任一项的方法,其中将反溶剂添加至反应混合物中,其中该反溶剂选自甲苯、丙酮、乙酸乙酯(尤其丙酮或乙酸乙酯)或其任何混合物。
55)另一实施例涉及如实施例54)的方法,其中该反溶剂为乙酸乙酯。
56)另一实施例涉及如实施例54)或55)中任一项的方法,其中添加的反溶剂(尤其乙酸乙酯)的体积为3.0与15之间升/千克式(I)化合物。反溶剂的体积的下限为3.0、3.5、4.0及4.5升/千克式(I)化合物,上限为15、10、7.0及6.0升/千克式(I)化合物。应了解,可将各下限与各上限组合。因此,本文应公开所有组合。
57)另一实施例涉及如实施例54)或55)中任一项的方法,其中添加的反溶剂(尤其乙酸乙酯)的体积为3.5与7.0之间升/千克式(I)化合物。
58)另一实施例涉及如实施例54)或55)中任一项的方法,其中添加的乙酸乙酯(反溶剂)的体积为4.0与6.0之间(尤其约5.0)升/千克式(I)化合物。
59)另一实施例涉及如实施例1)至58)中任一项的方法,其中将反溶剂于1小时至5小时内(尤其于1.5小时至3小时内)添加至反应混合物中。
60)另一实施例涉及如实施例1)至59)中任一项的方法,其中将所获得的沉淀过滤及干燥(或过滤,用反溶剂(尤其乙酸乙酯)洗涤及干燥)以得到呈固体形式的化合物·HCl(下文中亦称作“经沉淀的化合物·HCl”)。
式(I)化合物与乙酰氯(CH
61)另一实施例涉及如实施例1)至60)中任一项的方法,其中该方法进一步包括将经沉淀的化合物·HCl(尤其呈晶型(I)的化合物·HCl)自丙酮及水的混合物再结晶的步骤。
62)另一实施例涉及如实施例61)的方法,其中将该经沉淀的化合物·HCl(尤其呈晶型(I)的化合物·HCl)在35℃与65℃之间的温度下溶解于3.0与25之间升体积的丙酮及水的混合物/千克经沉淀的化合物·HCl中。丙酮/水混合物的体积的下限为3.0、3.2、3.4及3.5升/千克经沉淀的化合物·HCl,上限为25、10、7.0及5.0升/千克经沉淀的化合物·HCl。应了解,可将各下限与各上限组合。因此,本文应公开所有组合。温度的下限为35℃、40℃及45℃,上限为65℃、60℃及55℃。应了解,可将各下限与各上限组合。因此,本文应公开所有组合。
优选地,将经沉淀的化合物·HCl(尤其呈晶型(I)的化合物·HCl)添加至预升温至各自温度(诸如介于35℃与65℃之间的温度)的丙酮及水的混合物中。
63)另一实施例涉及如实施例62)的方法,其中丙酮及水的混合物的体积为3.2与7.0之间升/千克经沉淀的化合物·HCl(尤其呈晶型(I)的化合物·HCl)。
64)另一实施例涉及如实施例62)的方法,其中丙酮及水的混合物的体积为3.4与5.0之间(尤其3.5±0.1)升/千克经沉淀的化合物·HCl(尤其呈晶型(I)的化合物·HCl)。
65)另一实施例涉及如实施例62)至64)中任一项的方法,其中该温度介于40℃与60℃之间。
66)另一实施例涉及如实施例62)至64)中任一项的方法,其中该温度介于45℃与55℃之间(尤其50℃±2℃)。
67)另一实施例涉及如实施例61)至66)中任一项的方法,其中丙酮与水之间的比率介于2:1v/v与20:1v/v之间。丙酮与水之间的比率的下限为2:1v/v、5:2v/v、3:1v/v及7:2v/v,上限为20:1v/v、10:1v/v、7:1v/v及5:1v/v。应了解,可将各下限与各上限组合。因此,本文应公开所有组合。
67)另一实施例涉及如实施例61)至66)中任一项的方法,其中丙酮与水之间的比率介于3:1v/v与7:1v/v之间。
68)另一实施例涉及如实施例61)至66)中任一项的方法,其中丙酮与水之间的比率介于7:2v/v与7:1v/v之间(尤其介于7:2v/v与5:1v/v之间)。
69)另一实施例涉及如实施例61)至68)中任一项的方法,其中在介于25℃与55℃之间的温度下将含于丙酮及水的混合物中的经沉淀的化合物·HCl(尤其呈晶型(I)的化合物·HCl)的溶液用丙酮稀释及/或用化合物·HCl的晶种处理。该温度的下限为25℃及30℃,上限为55℃、45℃及35℃。应了解,可将各下限与各上限组合。因此,本文应公开所有组合。
应了解,假使将溶液用丙酮稀释及用晶种处理,可在将溶液用丙酮稀释之前添加晶种或可在添加晶种之前将溶液用丙酮稀释。优选地,在用丙酮稀释之前添加晶种。
70)另一实施例涉及如实施例69)的方法,其中在用丙酮稀释及/或用化合物·HCl的晶种处理期间,经沉淀的化合物·HCl(尤其呈晶型(I)的化合物·HCl)的溶液的温度介于25℃与35℃之间(尤其30℃±2℃)。
71)另一实施例涉及如实施例69)或70)中任一项的方法,其中将含于丙酮及水的混合物中的经沉淀的化合物·HCl(尤其呈晶型(I)的化合物·HCl)的溶液用丙酮稀释及用化合物·HCl的晶种处理。
72)另一实施例涉及如实施例69)至71)中任一项的方法,其中将含于丙酮及水的混合物中的经沉淀的化合物·HCl(尤其呈晶型(I)的化合物·HCl)的溶液首先用丙酮稀释及随后用化合物·HCl的晶种处理。
73)另一实施例涉及如实施例69)至71)中任一项的方法,其中将含于丙酮及水的混合物中的经沉淀的化合物·HCl(尤其呈晶型(I)的化合物·HCl)的溶液首先用化合物·HCl的晶种处理及随后用丙酮稀释。
74)另一实施例涉及如实施例69)至73)中任一项的方法,其中将含于丙酮及水的混合物中的经沉淀的化合物·HCl(尤其呈晶型(I)的化合物·HCl)的溶液用6.0与20之间升/千克经沉淀的化合物·HCl的量的丙酮稀释。丙酮的量的下限为6.0、8.0及10升/千克经沉淀的化合物·HCl,上限为20、17及14升/千克经沉淀的化合物·HCl。应了解,可将各下限与各上限组合。因此,本文应公开所有组合。
75)另一实施例涉及如实施例69)至73)中任一项的方法,其中将含于丙酮及水的混合物中的经沉淀的化合物·HCl(尤其呈晶型(I)的化合物·HCl)的溶液用8.0与17之间(尤其10与14之间)升/千克经沉淀的化合物·HCl的量的丙酮稀释。
76)另一实施例涉及如实施例69)至75)中任一项的方法,其中丙酮的总体积(即,用于溶解经沉淀的化合物·HCl的丙酮/水混合物中的丙酮的体积连同用于将含于丙酮及水的混合物中的经沉淀的化合物·HCl的溶液稀释的丙酮的体积)为12与35之间升/千克经沉淀的化合物·HCl(尤其呈晶型(I)的化合物·HCl)。丙酮的总体积的下限为12、13.5及15升/千克经沉淀的化合物·HCl,上限为35、25、20及17升/千克经沉淀的化合物·HCl。应了解,可将各下限与各上限组合。因此,本文应公开所有组合。
77)另一实施例涉及如实施例69)至75)中任一项的方法,其中丙酮的总体积为13.5与20之间(尤其15与20之间)升/千克经沉淀的化合物·HCl(尤其呈晶型(I)的化合物·HCl)。
78)另一实施例涉及如实施例69)至75)中任一项的方法,其中丙酮的总体积为15与17之间升/千克经沉淀的化合物·HCl(尤其呈晶型(I)的化合物·HCl)。
79)另一实施例涉及如实施例69)至78)中任一项的方法,其中含于丙酮及水的混合物中的经沉淀的化合物·HCl(尤其呈晶型(I)的化合物·HCl)的溶液的稀释是于1小时至10小时内(尤其于3小时至6小时内及尤其在3小时至4小时之间)进行。
80)另一实施例涉及如实施例69)至79)中任一项的方法,其中化合物·HCl的晶种呈化合物·HCl的晶型2,如WO 2018/055016中所述。
81)另一实施例涉及如实施例69)至80)中任一项的方法,其中化合物·HCl的晶种(尤其呈晶型2的化合物·HCl,如WO 2018/055016中所述)的量为相对于经沉淀的化合物·HCl(尤其呈晶型(I)的化合物·HCl)的量的0.05%w/w与5.0%w/w之间。晶种的量的下限为0.05、0.1、0.2及0.4%w/w,上限为5.0、2.0、1.0及0.6%w/w。应了解,可将各下限与各上限组合。因此,本文应公开所有组合。
呈晶型2的晶种可(例如)自WO 2018/055016中所述的方法或自本文中所述的方法获得。晶种亦可通过内部引晶,即,通过自含于丙酮及水的混合物中的经沉淀的化合物·HCl(尤其呈晶型(I)的化合物·HCl)的溶液移除样品,添加溶液至丙酮中及将所获得的悬浮液再添加至含于丙酮及水的混合物中的经沉淀的化合物·HCl的溶液中获得。优选地,在RT下,将5至40mL(更佳地10至20mL及最优选地约15mL)丙酮/毫升样品用于内部引晶。
82)另一实施例涉及如实施例81)的方法,其中晶种的量为相对于经沉淀的化合物·HCl(尤其呈晶型(I)的化合物·HCl)的量的0.1%w/w与2.0%w/w之间(尤其0.4%w/w与2.0%w/w之间)。
83)另一实施例涉及如实施例81)的方法,其中晶种的量为相对于经沉淀的化合物·HCl(尤其呈晶型(I)的化合物·HCl)的量的0.2%w/w与1.0%w/w之间(尤其0.4%w/w与0.6%w/w之间)。
84)另一实施例涉及如实施例61)至83)中任一项的方法,其中将混合物(尤其于用丙酮稀释及/或用化合物·HCl的晶种处理后,自含于丙酮及水的混合物中的经沉淀的化合物·HCl(尤其呈晶型(I)的化合物·HCl)的溶液获得的悬浮液)冷却至0℃与20℃之间(尤其0℃与10℃之间)的温度及将沉淀分离及视情况用丙酮洗涤。沉淀自母液的分离可通过适用于自液体分离固体的任何方式,诸如过滤(优选)或离心进行。
如“将沉淀分离及视情况用丙酮洗涤”的上下文中所用,术语“视情况”意指将沉淀用丙酮洗涤的步骤可存在或不存在于该方法中。
85)另一实施例涉及如实施例84)的方法,其中将经分离的沉淀于真空中干燥直至经分离的沉淀中的含水量介于4.0%w/w与8.2%w/w之间(优选地介于5.0%w/w与7.0%w/w之间)。含水量可通过卡尔费休(Karl Fischer)滴定测量。所获得的产物为呈晶型2的化合物·HCl,如WO 2018/055016中所述。
86)另一实施例涉及如实施例1)至85)中任一项的方法,该方法进一步包括式(II)化合物
其中R
与式(III)化合物的反应
其中R
87)另一实施例涉及如实施例86)的方法,其中R
88)另一实施例涉及如实施例86)或87)中任一项的方法,其中R
优选地,该式(III)化合物以水合物形式(诸如三水合物形式)的钠盐使用(R
89)另一实施例涉及如实施例86)至88)中任一项的方法,其中该反应在存在EDC及HOBt的混合物下进行。
90)另一实施例涉及如实施例86)至89)中任一项的方法,其中该反应于选自THF、甲苯及水中的两者或三者(尤其THF及水)的溶剂混合物中进行。
91)另一实施例涉及如实施例86)至90)中任一项的方法,其中该反应在4.5与6.0之间(尤其4.8与5.5之间)的pH值下进行。
式(III)化合物可通过式(IV)化合物与氢氧化钠的水溶液于2-丙醇中的反应获得。
该反应可在升高的温度下(例如,在约80℃下)进行及可将式(III)化合物通过结晶(例如,通过自约80℃冷却至约20℃)分离。优选地,可将反应混合物自约80℃至约20℃缓慢冷却(至少4小时),以提高所获得晶体的可过滤性。例如,可将反应混合物于2小时内自约80℃冷却至约50℃,保持在约50℃下另外30分钟及于4小时内进一步冷却至约20℃。可将所获得的晶体用甲苯洗涤及干燥。
91)另一实施例涉及如实施例1)至90)中任一项的方法,该方法进一步包括式(V)化合物
其中R
与TFA的反应,以得到式(II)化合物。
92)另一实施例涉及如实施例91)的方法,其中R
93)另一实施例涉及如实施例91)或92)中任一项的方法,其中在30℃与60℃之间的温度下将含于甲苯中的式(V)化合物的溶液添加至TFA中。温度的下限为30℃、35℃及40℃,上限为60℃、55℃及50℃。应了解,可将各下限与各上限组合。因此,本文应公开所有组合。
94)另一实施例涉及如实施例91)或92)中任一项的方法,其中在40℃与50℃之间的温度(尤其约45℃)下将含于甲苯中的式(V)化合物的溶液添加至TFA中。
95)另一实施例涉及如实施例93)或94)中任一项的方法,其中甲苯溶液中的式(V)化合物的量介于50%w/w与85%w/w之间。甲苯溶液中的式(V)化合物的量的下限为50%w/w、60%w/w及70%w/w,上限为85%w/w及80%w/w。应了解,可将各下限与各上限组合。因此,本文应公开所有组合。
96)另一实施例涉及如实施例93)或94)中任一项的方法,其中甲苯溶液中的式(V)化合物的量介于60%w/w与85%w/w之间(尤其约70%w/w)。
97)另一实施例涉及如实施例91)至96)中任一项的方法,其中TFA的体积为0.5与1.5之间升/千克式(V)化合物。TFA的体积的下限为0.5、0.7及0.9升/千克式(V)化合物,上限为1.5、1.3及1.1升/千克式(V)化合物。应了解,可将各下限与各上限组合。因此,本文应公开所有组合。
98)另一实施例涉及如实施例91)至96)中任一项的方法,其中TFA的体积为0.7与1.3之间(尤其约1.0)升/千克式(V)化合物。
99)另一实施例涉及如实施例1)至98)中任一项的方法,该方法进一步包括式(VI)化合物
/>
其中R
与式(VII)化合物的反应
以得到式(V)化合物。
100)另一实施例涉及如实施例99)的方法,其中R
101)另一实施例涉及如实施例99)或100)中任一项的方法,其中该反应在存在2,4,6-三丙基-1,3,5,2
102)另一实施例涉及如实施例99)至101)中任一项的方法,其中该反应于选自乙酸乙酯、甲苯及THF及水的混合物的溶剂中进行。
103)另一实施例涉及如实施例99)或100)中任一项的方法,其中该反应在存在2,4,6-三丙基-1,3,5,2
104)另一实施例涉及如实施例102)或103)中任一项的方法,其中该溶剂的体积为3.5升与7.5升之间(尤其约3.9升)/千克式(VI)化合物。
105)另一实施例涉及如实施例99)至104)中任一项的方法,其中将含于甲苯中的T3P(尤其以相对于式(VI)化合物的量的约1.03当量的量的T3P)的溶液添加至含于甲苯中的式(VI)化合物、式(VII)化合物(尤其以相对于式(VI)化合物的量的约1.03当量的量的式(VII)化合物)及三乙胺(尤其以相对于式(VI)化合物的量的约3.5当量的量的三乙胺)的混合物中。
106)另一实施例涉及如实施例99)至105)中任一项的方法,其中该反应在-5℃与25℃之间(尤其在10℃与20℃之间)的温度下进行。
107)本发明的另一实施例涉及4-((R)-2-{[6-((S)-3-甲氧基-吡咯烷-1-基)-2-苯基-嘧啶-4-羰基]-氨基}-3-膦酰基-丙酰基)-哌嗪-1-羧酸丁酯盐酸盐(化合物·HCl)的晶型(I),其通过X-射线粉末衍射图中在下列绕射角2θ:5.7°、5.9°及12.9°处的峰的存在表征。
应了解,如实施例107)的晶型包含呈盐酸(氯化氢)盐形式的化合物·HCl。此外,该晶型可包含非配位及/或配位溶剂(尤其非配位及/或配位水)。本文中以用于结晶溶剂化物(尤其结晶水合物)的术语使用配位溶剂(尤其配位水)。为了避免怀疑,于本申请案中,术语“结晶水合物”包含非化学计量水合物。同样,本文中使用非配位溶剂作为用于物理吸附或物理陷留的溶剂的术语(根据Polymorphismin the Pharmaceutical Industry(编辑R.Hilfiker,VCH,2006),第8章:U.J.Griesser:The Importance of Solvates定义)。
108)另一实施例涉及如实施例107)的化合物·HCl的晶型,其通过X-射线粉末衍射图中在下列绕射角2θ:5.1°、5.7°、5.9°、11.0°及12.9°处的峰的存在表征。
109)另一实施例涉及如实施例107)的化合物·HCl的晶型,其通过X-射线粉末衍射图中在下列绕射角2θ:3.7°、5.1°、5.7°、5.9°、11.0°、12.9°、15.2°、18.3°、20.2°及21.0°处的峰的存在表征。
110)另一实施例涉及如实施例107)的化合物·HCl的晶型,其基本上显示如图1中所述的X-射线粉末衍射图。
用于此文本中的缩略语及术语
缩略语:
整篇本说明书及实例使用下列缩略语:
Ac 乙酰基
AcCl 乙酰氯
AcOH 乙酸
AcOEt 乙酸乙酯
AcOMe 乙酸甲酯
aq 水溶液
Boc 叔丁氧羰基
dba 二亚苄基丙酮
DCM二氯甲烷
EDC N-(3-二甲氨基丙基)-N'-乙基-碳二亚胺盐酸盐
eq. 当量
Et 乙基
h 小时
HOBt羟基苯并三唑
HPLC高效液相层析法
IPA 2-丙醇
IPAc 乙酸异丙酯
IPC 工艺中的控制
JT 夹套温度
M 摩尔浓度
Me 甲基
min 分钟
NMP 1-甲基-2-吡咯烷酮
NMR核磁共振
org. 有机
o.t. 理论
RT 室温
%a/a按面积比率测定的百分比
T3P 2,4,6-三丙基-1,3,5,2
THF 四氢呋喃
TFA 三氟乙酸
TMSBr三甲基硅基溴
vol 1vol意指1L溶剂/1kg参考起始物质
实验部分
X-射线粉末绕射分析(XRPD)
X-射线粉末衍射图在配备有以反射模式(耦合的2θ/θ)操作的Lynxeye检测器的Bruker D8 Advance X-射线绕射仪上收集。通常,在40kV/40mA下运行Cu X-射线管。应用在3至50°2θ的扫描范围上0.02°(2θ)的步长及76.8秒的阶跃时间。将发散狭缝设置为固定样品照明(可变狭缝大小)及将抗散射狭缝设置为0.3°。将粉末轻微压缩至具有0.5mm的深度的硅单晶样品架中及将样品在测量期间于其自身平面中旋转。使用
高效液相层析法(HPLC)
实例1:
合成(S)-6-(3-甲氧基吡咯烷-1-基)-2-苯基嘧啶-4-甲腈:
将100mL舒伦克(Schlenk)管用氮气冲洗三次。向管中放入NMP(30.0ml,1.5vol)及将NMP以三个循环(真空/氮气)脱气及添加Pd
于500mL反应器中放入(S)-4-氯-6-(3-甲氧基吡咯烷-1-基)-2-苯基嘧啶(20.0g,69.02mmol,1.00eq.)、Zn(CN)
于500mL反应器中放入甲苯(200.0mL,10.0vol)、氨水25%(44.0mL,2.2vol)及水(100.0mL,5.0vol)及在30至40℃下搅拌及在30至40℃下将反应混合物添加至此乳液中。于完全添加后,将乳液在30至40℃下搅拌30分钟及随后相分离5分钟。然后在25至35℃下,将有机相用先前制备(在制备期间气体形成!)的N-乙酰基-L-半胱氨酸(5.6g,34.32mmol,0.5eq.)、苏打(8.0g,66.04mmol,0.96eq.)及水(100.0ml,5.0vol)的溶液萃取三次持续30分钟及相分离5分钟。之后在30至35℃下,将有机相用水(100.0mL,5.0vol)萃取两次及相分离5分钟。然后在40至60℃下,在减压(通常:150至300毫巴)下,将有机相浓缩至5.0vol。在40至60℃下,在减压(通常:50至200毫巴)下,继续蒸馏,通过添加IPA(500.0mL,25.0vol)保持恒定体积。在蒸馏过程期间,产物沉淀。添加IPA(40.0mL,2.0vol),将浆液升温至75至82℃及在此温度下后搅拌30分钟。然后将溶液于60分钟内冷却至60至70℃及在60至70℃下后搅拌60分钟。然后将浆液于4小时内冷却至0至10℃及在0至10℃后搅拌2小时。在0至10℃下过滤除去固体及在0至25℃下将滤饼用IPA(40.0mL,2.0vol)洗涤两次(置换)。将湿产物于橱柜中在50℃下干燥直至达成恒定重量,以得到呈固体的产物(16.2g)。
再结晶:
将500mL反应器用氮气冲洗三次及在JT≤40℃下,放入粗制物(S)-6-(3-甲氧基吡咯烷-1-基)-2-苯基嘧啶-4-甲腈(20.0g,71.34mmol)、活性炭(Norit CGPSuper)及IPAc(200mL,10.0vol)。然后将浆液升温至65至75℃及在65至75℃下后搅拌60分钟。将溶液通过回火(约75℃)压力过滤器过滤至第二个500mL反应器中。将第一个反应器及过滤器用IPAc(40.0ml,2.0vol)冲洗。将溶液在正常压力下在85至95℃下浓缩至4至5vol。在85至95℃下添加庚烷(160.0mL,8.0vol)及随后将溶液于60分钟内冷却至65至75℃。将所获得的浆液于3小时内冷却至0至10℃及在0至10℃下后搅拌2小时。在0至10℃下过滤掉固体及在0至25℃下,将滤饼用IPAc/庚烷1:2v/v洗涤(置换)两次。将湿产物于橱柜中在50℃下干燥直至达成恒定重量,以得到呈固体的产物(16.8g)。
实例2:
合成(S)-6-(3-甲氧基吡咯烷-1-基)-2-苯基嘧啶-4-羧酸钠盐:
将500mL反应器用氮气冲洗三次,及放入水(323mL,6.5vol)及升温至20至40℃。添加(S)-6-(3-甲氧基吡咯烷-1-基)-2-苯基嘧啶-4-甲腈(50.0g,178mmol,1.0eq.)及2-丙醇(532mL,10.6vol)及将反应混合物升温至70至85℃。经由添加漏斗历时30分钟添加氢氧化钠水溶液(30%,29mL,0.58vol)及将漏斗用水(5.0mL,0.1vol)冲洗。将反应混合物在75至85℃下搅拌至少6小时,历时至少2小时冷却至45至55℃及在45至55℃下再搅拌30分钟。历时至少4小时将所获得的悬浮液冷却至15至25℃及在15至25℃下搅拌至少30分钟。将产物过滤,及将滤饼首先用2-丙醇(136mL)及水(14mL)的混合物及随后用甲苯(150mL)洗涤。将湿产物于真空中在45至55℃下干燥,以得到呈固体的产物。
实例3:合成呈晶型2的4-((R)-2-{[6-((S)-3-甲氧基-吡咯烷-1-基)-2-苯基-嘧啶-4-羰基]-氨基}-3-膦酰基-丙酰基)-哌嗪-1-羧酸丁酯盐酸盐(如WO 2018/055016中所述):
步骤1:合成4-[(R)-2-叔丁氧羰基氨基-3-(二乙氧基-膦酰基)-丙酰基]-哌嗪-1-羧酸丁酯:
向配备有机械搅拌器及滴液漏斗的2.5L玻璃反应器中放入甲苯(780mL,3.9vol)、(R)-2-叔丁氧羰基氨基-3-(二乙氧基-膦酰基)-丙酸(200g,614.8mmol,1.0eq.)、哌嗪-1-羧酸丁酯盐酸盐(141.0g,633.3mmol,1.03eq.)及三乙胺(217.75g,2152mmol,3.5eq.)及将轻浆液的温度调整至10至20℃。在10至20℃下,将含于甲苯中的T3P 50%w/w(430.4g,676.30mmol,1.10eq.)历时1至2小时直接加剂量至反应混合物中。随后将剂量系统用20mL甲苯(0.1vol)冲洗。将反应混合物老化至少1小时。将反应混合物转移至锥形瓶(Erlenmeyer flask)中及将水(800mL,4vol)放入反应器中。在10至25℃下,将反应混合物基于放入反应器中的水历时至少10分钟淬灭。然后,在10至25℃下,历时至少10分钟放入30%w/w苛性钠(123.0g,922.2mmol,1.5eq.)。最大体积:2.6L,13vol。在15至25℃下,将下水层排空(快速相分离,无中间相)。将水(200mL,1vol)添加至有机层中及在15至25℃下,将pH用30%w/w硫酸(约241g)调整至2.5至3.0。将下水层排空(快速相分离,无中间相)。将水(200mL,1vol)添加至有机层中及将混合物搅拌至少5分钟。分离相持续至少30分钟及将下水层排空。将有机层在40至60℃(p=100至300毫巴)下浓缩至约30%w/w及获得透明黄色溶液。
阶段2:合成4-[(R)-2-氨基-3-(二乙氧基-膦酰基)-丙酰基]-哌嗪-1-羧酸丁酯
该批次以用于制备阶段1的(R)-2-叔丁氧羰基氨基-3-(二乙氧基-膦酰基)-丙酸的量计算。
向配备有机械搅拌器、滴液漏斗及蒸馏适配器的1.0L玻璃反应器中放入含于甲苯中的阶段1(500g,自100g(R)-2-叔丁氧羰基氨基-3-(二乙氧基-膦酰基)-丙酸制备)的溶液。将溶液在40至60℃(50至250毫巴)下浓缩至215g及转移至滴液漏斗中。然后,向反应器中放入TFA(150mL,1.5vol在(R)-2-叔丁氧羰基氨基-3-(二乙氧基-膦酰基)-丙酸上)及将温度调整至45℃。随后,在45℃下,历时2小时逐滴添加将阶段1的溶液(添加控制气体逸出)。将批次老化1小时及然后在150毫巴下在45至50℃下开始蒸馏。将压力逐步降低至100毫巴,同时保持温度在45至50℃。总的后搅拌时间(包含蒸馏)为4小时。将反应混合物在10至25℃下基于水(300mL,3vol)及25%氨水(167.6g,8.0eq.)的混合物淬灭。在10至25℃下添加二氯甲烷(300mL,3vol)。分离下DCM层及将水层用DCM(150ml,1.5vol)再萃取两次。将合并的DCM层用20% KHCO
步骤3:合成4-((R)-3-(二乙氧基-膦酰基)-2-{[6-((S)-3-甲氧基-吡咯烷-1-基)-2-苯基-嘧啶-4-羰基]-氨基}-丙酰基)-哌嗪-1-羧酸丁酯:
放入呈含于甲苯(约226g,44.2%w/w)中的溶液的步骤2(100g,254.18mmol,1.0eq.,基于滴定),及添加水(200mL,2vol)。在15至25℃下,将pH用33% HCl aq.(约28g)调整至4.0至5.0。将含有步骤2的水层分离及遗弃有机层。将步骤2的水溶液用含HOBt一水合物3%的THF(274.8g,7.8g HOBt一水合物+300mL THF)稀释及添加(S)-6-(3-甲氧基吡咯烷-1-基)-2-苯基嘧啶-4-羧酸钠盐(83.3g,259mmol,1.02eq.)。将轻浆液的pH用HCl 33%调整至5.0至5.5(目标:5.2)。在15至25℃下,历时至少1小时以至少10份添加EDC(58.47g,305.02mmol,1.2eq.)。监测反应混合物的pH及通过添加10% K
步骤4:合成呈晶型(I)的4-((R)-2-{[6-((S)-3-甲氧基-吡咯烷-1-基)-2-苯基-嘧啶-4-羰基]-氨基}-3-膦酰基-丙酰基)-哌嗪-1-羧酸丁酯盐酸盐:
向含于AcOH(490mL,2.9vol)中的步骤3(171.5g,254.18mmol)的溶液中添加乙醇(29.3g,635.45mmol,2.5eq.)。然后历时至少20分钟逐滴添加乙酰氯(29.9g,381.27mmol,1.5eq.),保持温度在25至35℃。将反应混合物在30℃下搅拌4至5小时及然后用0.5g 4-((R)-2-{[6-((S)-3-甲氧基-吡咯烷-1-基)-2-苯基-嘧啶-4-羰基]-氨基}-3-膦酰基-丙酰基)-哌嗪-1-羧酸丁酯盐酸盐晶种引晶及在30℃下搅拌30分钟。将所获得的浆液在30℃下再后搅拌14小时。在30℃下,历时至少2小时逐滴添加AcOEt(860mL,5vol)。历时1小时将浆液冷却至20℃及老化至少2小时及然后过滤。将湿产物用AcOEt(345mL,2vol,置换洗涤)及AcOEt(345mL,2vol,浆液洗涤)洗涤。将湿产物于橱柜中在45℃下干燥直至具有载气的恒定重量,以得到呈灰白色结晶固体的产物(159.4g,未校正)。
表1:呈晶型(I)的化合物·HCl的表征数据
/>
阶段5:合成呈晶型2(如WO 2018/055016中所述)的4-((R)-2-{[6-((S)-3-甲氧基-吡咯烷-1-基)-2-苯基-嘧啶-4-羰基]-氨基}-3-膦酰基-丙酰基)-哌嗪-1-羧酸丁酯盐酸盐:
将阶段4(30.0g,91.7%w/w,42.0mmol)放入反应器中及添加丙酮/水4:1v/v(105mL,3.5vol,预升温至50℃)及形成透明溶液。将溶液冷却至30℃,用0.5g 4-((R)-2-{[6-((S)-3-甲氧基-吡咯烷-1-基)-2-苯基-嘧啶-4-羰基]-氨基}-3-膦酰基-丙酰基)-哌嗪-1-羧酸丁酯盐酸盐的晶种引晶及在25至35℃下搅拌30分钟。在25至35℃下,历时3小时将丙酮(360mL,12vol)添加至所获得的浆液中。历时2小时将浆液冷却至0至10℃及在0至10℃后搅拌60分钟及然后过滤。将湿产物用丙酮(2x 75mL,2.5vol)洗涤。将湿产物于具有载气(用水饱和的氮气)的旋转蒸发器中在20至35℃下干燥直至达成恒定重量,以得到呈白色固体的呈晶型2(如WO 2018/055016中所述)的产物,产率92%。
或者,可将湿产物在不存在载气下干燥直至不超过8.2%w/w水的含水量。
参考实例1:4-((R)-3-(二乙氧基-膦酰基)-2-{[6-((S)-3-甲氧基-吡咯烷-1-基)-2-苯基-嘧啶-4-羰基]-氨基}-丙酰基)-哌嗪-1-羧酸丁酯的二乙氧基-膦酰基于DCM及浓盐酸水溶液的混合物中的裂解:
将含于DCM(4.0vol)中的4-((R)-3-(二乙氧基-膦酰基)-2-{[6-((S)-3-甲氧基-吡咯烷-1-基)-2-苯基-嘧啶-4-羰基]-氨基}-丙酰基)-哌嗪-1-羧酸丁酯(1.05g)的溶液平分至两个25mL螺旋盖小瓶中。将溶液用1.8vol或3.6vol 32%w/w HCl水溶液处理及在RT下搅拌。在表2中给定的时间点取样及通过HPLC分析,以测定水解产物(R)-2-(6-((S)-3-甲氧基吡咯烷-1-基)-2-苯基嘧啶-4-甲酰氨基)-3-膦酰基丙酸(水解产物)的相对量:
表2:
参考实例2:4-((R)-3-(二乙氧基-膦酰基)-2-{[6-((S)-3-甲氧基-吡咯烷-1-基)-2-苯基-嘧啶-4-羰基]-氨基}-丙酰基)-哌嗪-1-羧酸丁酯的二乙氧基-膦酰基于浓盐酸水溶液中的裂解:
将含于37%w/w HCl水溶液中的4-((R)-3-(二乙氧基-膦酰基)-2-{[6-((S)-3-甲氧基-吡咯烷-1-基)-2-苯基-嘧啶-4-羰基]-氨基}-丙酰基)-哌嗪-1-羧酸丁酯(200mg,0.30mmol)的混合物在表3中给定的条件下搅拌。在表3中给定的时间点取样及通过HPLC分析,以测定水解产物(R)-2-(6-((S)-3-甲氧基吡咯烷-1-基)-2-苯基嘧啶-4-甲酰氨基)-3-膦酰基丙酸(水解产物)的相对量:
表3:
参考实例3:4-((R)-3-(二乙氧基-膦酰基)-2-{[6-((S)-3-甲氧基-吡咯烷-1-基)-2-苯基-嘧啶-4-羰基]-氨基}-丙酰基)-哌嗪-1-羧酸丁酯的二乙氧基-膦酰基于稀盐酸水溶液中的裂解:
将含于37%w/w HCl水溶液及水的混合物(参见表4)中的4-((R)-3-(二乙氧基-膦酰基)-2-{[6-((S)-3-甲氧基-吡咯烷-1-基)-2-苯基-嘧啶-4-羰基]-氨基}-丙酰基)-哌嗪-1-羧酸丁酯(200mg,0.30mmol)的混合物在RT下在表4中给定的条件下搅拌。在表4中给定的时间点取样及通过HPLC分析,以测定水解产物(R)-2-(6-((S)-3-甲氧基吡咯烷-1-基)-2-苯基嘧啶-4-甲酰氨基)-3-膦酰基丙酸(水解产物)的相对量:
表4:
实例4:4-((R)-3-(二乙氧基-膦酰基)-2-{[6-((S)-3-甲氧基-吡咯烷-1-基)-2-苯基-嘧啶-4-羰基]-氨基}-丙酰基)-哌嗪-1-羧酸丁酯的二乙氧基-膦酰基基团于丙酮中利用HCl的裂解:
将含于丙酮及HCl(参见表5)中的4-((R)-3-(二乙氧基-膦酰基)-2-{[6-((S)-3-甲氧基-吡咯烷-1-基)-2-苯基-嘧啶-4-羰基]-氨基}-丙酰基)-哌嗪-1-羧酸丁酯(200mg,0.30mmol)的混合物在RT下在表5中给定的条件下搅拌。在表5中给定的时间点取样及通过HPLC分析,以测定水解产物(R)-2-(6-((S)-3-甲氧基吡咯烷-1-基)-2-苯基嘧啶-4-甲酰氨基)-3-膦酰基丙酸(水解产物)的相对量:
表5:
实例5:4-((R)-3-(二乙氧基-膦酰基)-2-{[6-((S)-3-甲氧基-吡咯烷-1-基)-2-苯基-嘧啶-4-羰基]-氨基}-丙酰基)-哌嗪-1-羧酸丁酯的二乙氧基-膦酰基基团于不同溶剂中利用HCl的裂解:
将HCl气体轻轻鼓泡透过含于各自溶剂(参见表6)中的4-((R)-3-(二乙氧基-膦酰基)-2-{[6-((S)-3-甲氧基-吡咯烷-1-基)-2-苯基-嘧啶-4-羰基]-氨基}-丙酰基)-哌嗪-1-羧酸丁酯的溶液20分钟及将混合物在RT下在表6中给定的条件下搅拌。在表6中给定的时间点取样及通过HPLC分析,以测定水解产物(R)-2-(6-((S)-3-甲氧基吡咯烷-1-基)-2-苯基嘧啶-4-甲酰氨基)-3-膦酰基丙酸(水解产物)的相对量:
表6:
ML:母液;PPT:沉淀;*):于18小时后,过滤沉淀并分析
实例6:4-((R)-3-(二乙氧基-膦酰基)-2-{[6-((S)-3-甲氧基-吡咯烷-1-基)-2-苯基-嘧啶-4-羰基]-氨基}-丙酰基)-哌嗪-1-羧酸丁酯的二乙氧基-膦酰基基团利用原位产生的HCl的裂解:
将含于AcOH(2.0vol)中的4-((R)-3-(二乙氧基-膦酰基)-2-{[6-((S)-3-甲氧基-吡咯烷-1-基)-2-苯基-嘧啶-4-羰基]-氨基}-丙酰基)-哌嗪-1-羧酸丁酯(1.0g)、Ac
表7:
实例7:4-((R)-3-(二乙氧基-膦酰基)-2-{[6-((S)-3-甲氧基-吡咯烷-1-基)-2-苯基-嘧啶-4-羰基]-氨基}-丙酰基)-哌嗪-1-羧酸丁酯的二乙氧基-膦酰基基团利用原位产生的HCl的裂解:
将含于不同体积AcOH(参见表8)中的4-((R)-3-(二乙氧基-膦酰基)-2-{[6-((S)-3-甲氧基-吡咯烷-1-基)-2-苯基-嘧啶-4-羰基]-氨基}-丙酰基)-哌嗪-1-羧酸丁酯(1.0g)、AcCl(1.5eq.)及乙醇(2.5eq)的混合物在RT下搅拌。在表8中给定的时间点取样,以测定水解产物(R)-2-(6-((S)-3-甲氧基吡咯烷-1-基)-2-苯基嘧啶-4-甲酰氨基)-3-膦酰基丙酸(水解产物)的相对量:
表8:
实例8:4-((R)-3-(二乙氧基-膦酰基)-2-{[6-((S)-3-甲氧基-吡咯烷-1-基)-2-苯基-嘧啶-4-羰基]-氨基}-丙酰基)-哌嗪-1-羧酸丁酯的二乙氧基-膦酰基基团利用原位产生的HCl的裂解:
将含于AcOH(3vol)中的起始物质(SM)4-((R)-3-(二乙氧基-膦酰基)-2-{[6-((S)-3-甲氧基-吡咯烷-1-基)-2-苯基-嘧啶-4-羰基]-氨基}-丙酰基)-哌嗪-1-羧酸丁酯(20g)、AcCl(1.5eq.)及乙醇(2.5eq)的混合物在35℃下搅拌4小时,用4-((R)-2-{[6-((S)-3-甲氧基-吡咯烷-1-基)-2-苯基-嘧啶-4-羰基]-氨基}-3-膦酰基-丙酰基)-哌嗪-1-羧酸丁酯盐酸盐晶体引晶,在35℃下进一步再搅拌4小时,及冷却至RT。在表9中给定的时间点取样,以测定水解产物(R)-2-(6-((S)-3-甲氧基吡咯烷-1-基)-2-苯基嘧啶-4-甲酰氨基)-3-膦酰基丙酸(水解产物)的相对量:
表9:
实例9:合成呈晶型2的4-((R)-2-{[6-((S)-3-甲氧基-吡咯烷-1-基)-2-苯基-嘧啶-4-羰基]-氨基}-3-膦酰基-丙酰基)-哌嗪-1-羧酸丁酯盐酸盐(如WO 2018/055016中所述)的替代程序:
a)在65℃下,将化合物·HCl(2.0g,3.1mmol)溶解于44mL丙酮及2.3mL水中。将溶液冷却降至55℃,用3%呈晶型2的化合物·HCl引晶及搅拌1小时。将混合物以3℃/h冷却至15℃,以得到呈晶型2的化合物·HCl(60%产率)。
b)在RT下,将化合物·HCl(2.0g,3.1mmol)溶解于6mL丙酮及3.5mL水中。将溶液添加至含有50mg呈晶型2的化合物·HCl的晶种的60mL丙酮的以10mL/h的速率冷却的混合物(5℃)中及搅拌过夜,以得到呈晶型2的化合物·HCl(78%产率)。
c)在50℃下,将化合物·HCl(5.0g,7.6mmol)溶解于丙酮及水的混合物(4:1v/v,20mL)中。将溶液用丙酮(32.5mL)稀释及用呈晶型2的化合物·HCl的晶种(100mg)处理。将丙酮(38mL)于1小时内添加至混合物中及将混合物以2.8℃/h冷却至5℃,以得到呈晶型2的化合物·HCl(78%产率)。
d)在65℃下,将化合物·HCl(18g,27.5mmol)溶解于丙酮及水的混合物(4:1v/v,63mL)中。将溶液冷却至30℃,用呈晶型2的化合物·HCl的晶种(90mg)处理及搅拌1小时。将丙酮(216mL)于1.5小时内添加至混合物中。将混合物搅拌1小时,以5℃/h冷却至5℃及再搅拌2小时,以得到呈晶型2的化合物·HCl(87%产率)。