作为受体拮抗剂的工程化纤维细胞生长因子变体
文献发布时间:2023-06-19 12:02:28
相关申请的交叉引用
本申请要求于2018年10月09日提交的名称为“作为受体拮抗剂的工程化纤维细胞生长因子变体”的美国临时专利申请号62/743,414的优先权,其内容特此以引用的方式整体并入本文。
发明领域
本发明涉及多肽变体、特别是成纤维细胞生长因子(FGF)变体的领域。
发明背景
人类生长因子在协调许多复杂的过程诸如伤口愈合、组织再生、血管生成和肿瘤形成中起关键作用
先前已经对各种生长因子进行了改良,以提高其热稳定性和蛋白水解稳定性,并且这在体外功能测定和体内实验中均显示出增强其生物活性
本发明通过提供用于工程化蛋白水解稳定的生长因子的方法来满足此需求。本发明还提供了通过所述方法产生的蛋白水解稳定的FGF肽变体,以及此类FGF肽变体的用途。
发明内容
本发明提供了一种人成纤维细胞生长因子1(FGF1)变体,其包含至少一个选自氨基酸取代、氨基酸缺失、氨基酸添加及其组合的成员,其中所得FGF1变体与SEQ ID NO:1的野生型FGF1相比表现出提高的蛋白水解稳定性。
在一些实施方案中,FGF1变体在β-环中或C末端附近包含氨基酸取代、氨基酸缺失、氨基酸添加及其组合。
在一些实施方案中,FGF1变体是成纤维细胞生长因子受体(FGFR)拮抗剂。
在一些实施方案中,FGF1变体在位置28、40、47、93或131处包含至少一个氨基酸取代。
在一些实施方案中,FGF1变体包含至少一个选自由D28N、Q40P、S47I、H93G、L131R和L131K组成的组的氨基酸取代。
在一些实施方案中,FGF1变体包含氨基酸取代L131R。
在一些实施方案中,FGF1变体包含氨基酸取代L131K。
在一些实施方案中,FGF1变体包含氨基酸取代D28N和L131R。
在一些实施方案中,FGF1变体包含氨基酸取代D28N和L131K。
在一些实施方案中,FGF1变体包含氨基酸取代Q40P、S47I、H93G和L131R。
在一些实施方案中,FGF1变体包含氨基酸取代Q40P、S47I、H93G和L131K。
在一些实施方案中,FGF1变体包含氨基酸取代D28N、Q40P、S47I、H93G和L131R。
在一些实施方案中,FGF1变体包含氨基酸取代D28N、Q40P、S47I、H93G和L131K。
在一些实施方案中,FGF1变体不包含氨基酸取代L131A。
在一些实施方案中,FGF1变体缀合至选自可检测部分、水溶性聚合物、水不溶性聚合物、治疗部分、靶向部分及其组合的成员。
在一些实施方案中,FGF1变体缀合至选自放射性同位素、顺磁体、荧光团及其组合的可检测部分。
在一些实施方案中,FGF1变体是诊断显像剂。
本发明还提供了一种包含根据权利要求1的FGF1变体的药物制剂,其中所述变体与药学上可接受的载剂组合。一种抑制或预防有需要的受试者中的血管生成的方法,其包括向有需要的受试者施用根据权利要求1的变体,从而预防或抑制血管生成。
在一些实施方案中,受试者患有癌症。
在一些实施方案中,对受试者进行治疗以预防眼中的新血管形成。
本发明还提供了一种治疗需要治疗的受试者的癌症的方法,所述方法包括向所述受试者施用治疗有效量的如本文所提供的FGF1变体,从而治疗癌症。
本发明还提供了一种减少受试者中选自肿瘤进展、血管生成、转移及其组合的成员的过程的方法,所述方法包括向所述受试者施用足以减少所述过程的量的根据权利要求1的变体。
在一些实施方案中,癌症是选自结直肠癌、口腔癌、肝细胞癌、肾癌、乳腺癌、肺癌、卵巢癌、胃癌、脑癌、前列腺癌及其组合的成员。
本发明还提供了编码如本文所述的FGF1变体多肽的核酸。
本发明还提供了分离的细胞,其包含编码如本文所述的FGF1变体多肽的核酸,并且能够表达如本文所述的FGF1变体多肽。
本发明还提供了一种筛选蛋白水解稳定的生长因子变体的方法,所述方法包括:
i.在酵母展示系统中表达生长因子变体的文库;
ii.通过测量酵母展示的生长因子变体与相关生长因子受体的结合活性,测试来自i)的酵母展示的生长因子变体的正确折叠;
iii.将来自ii)的酵母展示的生长因子变体与至少一种蛋白酶一起孵育;
iv.确定与野生型生长因子的蛋白酶切割相比,来自iii)的酵母展示的生长因子变体的蛋白酶切割;以及
v.从iv)中选择与相同的蛋白酶对野生型生长因子的蛋白酶切割相比,对至少一种蛋白酶表现出降低的蛋白酶切割和/或提高的蛋白水解稳定性的变体,其中所述选择的生长因子变体是蛋白水解稳定的生长因子变体。
在一些实施方案中,至少一种蛋白酶是能够切割野生型生长因子的蛋白酶。
在一些实施方案中,至少一种蛋白酶能够选择性地切割生长因子,并且表现出最小和/或无酵母展示蛋白的非特异性切割。
在一些实施方案中,至少一种蛋白酶选自由以下组成的组:血清、胰蛋白酶、胰凝乳蛋白酶和纤溶酶。
在一些实施方案中,至少一种蛋白酶是血清。
在一些实施方案中,至少一种蛋白酶是胰蛋白酶。
在一些实施方案中,至少一种蛋白酶是胰凝乳蛋白酶。
在一些实施方案中,至少一种蛋白酶是纤溶酶。
附图说明
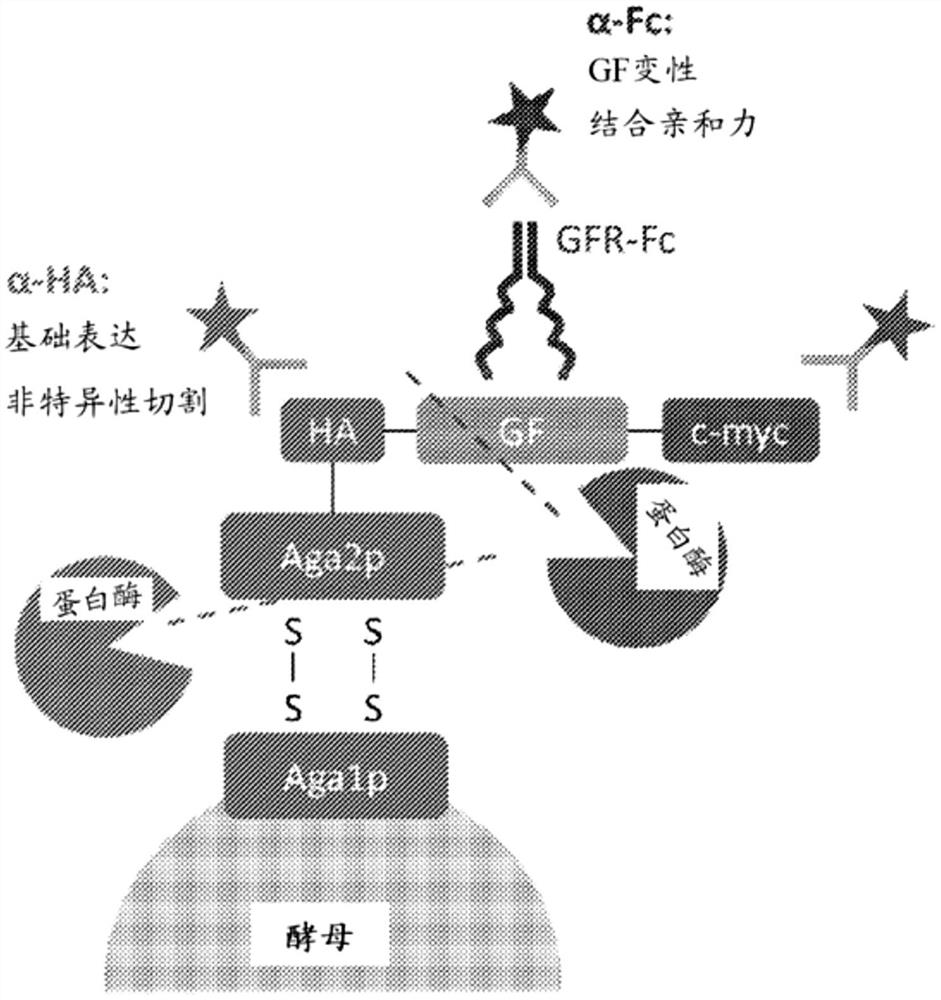
图1.用于工程化蛋白水解稳定性的生长因子的酵母展示。感兴趣的生长因子(GF)表达为与粘附蛋白凝集素Aga2p的融合体,后者通过两个二硫键连接到细胞壁蛋白Aga1p上。与蛋白酶一起孵育后,切割既可以在生长因子内发生(生长因子特异性切割),也可以在酵母展示蛋白Aga1p或Aga2p内发生(非特异性切割)。与生长因子受体的可溶性Fc融合体(GFR-Fc)一起孵育后,可以使用荧光抗体对HA标签、c-myc标签和Fc结构域进行染色。HA信号用于测量生长因子的基础表达水平和蛋白酶的非特异性切割。c-myc信号与HA信号结合起来测量GF特异性切割。Fc信号用于测量GF变性的水平和GF对其受体的结合亲和力。
图2.针对蛋白水解稳定的生长因子突变体的基于FACS的的筛选方法。将生长因子突变体文库转化到EBY100酵母细胞中,并通过酵母展示诱导展示生长因子。将细胞与蛋白酶一起孵育,洗涤,然后与受体的可溶性Fc-融合体孵育。用适当的荧光抗体标记后,使用流激活细胞分选(FACS)来门控和收集表达蛋白水解切割水平低且与可溶性受体的结合水平高的突变体的细胞。将此孵育和细胞分选过程循环多次,以鉴定具有最高蛋白水解稳定性水平的突变体。
图3.FGF1的酵母展示。(A)FGF1表达为与粘附蛋白凝集素Aga2p的融合体,后者通过两个二硫键连接到细胞壁蛋白Aga1p上。FGFR1-Fc是与FGF1结合的相应可溶性受体。(B)c-myc标签的荧光标记显示FGF1在酵母表面成功表达。(C)FGFR1的Fc融合体显示与酵母展示的FGF1的特异性结合。将表达表面展示的FGF1的酵母与可溶性FGFR1-Fc在各种浓度下一起孵育3小时。洗涤细胞,并用抗-Fc AlexaFluor488对可溶性FGFR1-Fc染色。通过流式细胞术测量与酵母细胞结合相关的荧光并作图。
图4.采用胎牛血清的蛋白水解稳定性测定。将展示FGF1突变体文库的酵母细胞与不同浓度的胎牛血清一起孵育。在洗涤细胞并与10nM FGFR1-Fc一起孵育后,将细胞用针对c-myc和可溶性受体的Fc结构域的荧光抗体染色。流式细胞术分析显示,增加FBS的浓度对FGF1特异性切割信号以及FGFR1-Fc结合信号的影响相对较小。
图5.采用胰蛋白酶的蛋白水解稳定性测定。将展示FGF1的酵母细胞与不同浓度的胰蛋白酶一起孵育。在洗涤细胞并与10nM FGFR1-Fc一起孵育后,将细胞用针对c-myc和可溶性受体的Fc结构域的荧光抗体染色。流式细胞术分析显示,增加胰蛋白酶的浓度会导致酵母展示蛋白的切割(c-myc减少)和与FGFR1-Fc的结合丧失。
图6.采用胰凝乳蛋白酶的蛋白水解稳定性测定。将展示FGF1的酵母细胞与不同浓度的胰凝乳蛋白酶一起孵育。在洗涤细胞并与10nM FGFR1-Fc一起孵育后,将细胞用针对c-myc和可溶性受体的Fc结构域的荧光抗体染色。流式细胞术分析显示,增加胰凝乳蛋白酶的浓度会导致酵母展示蛋白的切割(c-myc减少)和与FGFR1-Fc的结合丧失。
图7A-图7B.胰蛋白酶对酵母展示蛋白Aga1和Aga2的非特异性切割。将展示FGF1的酵母细胞与不同浓度的胰蛋白酶一起孵育。洗涤后,将细胞用针对HA和c-myc的荧光抗体染色。流式细胞术分析显示,增加胰蛋白酶的浓度会导致HA信号的丢失,表明酵母展示蛋白Aga1和Aga2的非特异性切割。
图8A-图8B.胰凝乳蛋白酶对FGF1的特异性切割。将展示FGF1的酵母细胞与不同浓度的胰蛋白酶一起孵育。洗涤后,将细胞用针对HA和c-myc的荧光抗体染色。流式细胞术分析显示,增加胰凝乳蛋白酶的浓度会导致c-myc信号的丢失,但不会导致HA信号丢失,表明发生了FGF1特异性切割。
图9.采用纤溶酶的蛋白水解稳定性测定。将展示FGF1的酵母细胞与不同浓度的纤溶酶一起孵育。洗涤后,将细胞用针对HA和c-myc的荧光抗体染色。流式细胞术分析显示存在FGF1的浓度依赖性切割。
图10.纤溶酶对FGF1的特异性切割。将展示FGF1的酵母细胞和仅表达酵母展示蛋白Aga1和Aga2的空白对照与125nM纤溶酶一起孵育。洗涤后,将细胞用针对HA和c-myc的荧光抗体染色。流式细胞术分析显示,对于展示FGF1的酵母细胞,增加纤溶酶的浓度会导致c-myc信号的丢失,而展示空白对照的酵母细胞则不会。这证实了纤溶酶对酵母展示蛋白的切割是FGF1特异性的。
图11.通过区分野生型FGF1与蛋白水解稳定的PM2对蛋白水解稳定性测定的有效性的验证。纤溶酶通过在各种纤溶酶浓度下孵育2天后的酵母表面展示,使野生型FGF1与蛋白水解稳定的突变体(PM2)得以区分。这证明了基于纤溶酶的筛选能够鉴定新的蛋白水解稳定的突变体。
图12.分选1:FGFR1-Fc结合剂的选择。(A)FGFR1-Fc结合剂的筛选方法示意图。随机诱变文库被诱导以在酵母表面上表达FGF突变体。将细胞与10nM FGFR1-Fc一起孵育,洗涤,然后用针对表达(α-c-myc)和FGFR1结合(α-FGFR1-Fc)的荧光抗体染色。荧光激活细胞分选(FACS)用于分析和门控显示高c-myc信号和高FGFR1-Fc信号的细胞。(B)示出了FGF1的FACS点图。在点图上绘制的门旁,示出了从总群体中收集的细胞的百分比。
图13.分选2:对FGF1特异性切割的抗性的选择。(A)分选2的筛选方法的示意图。来自分选1的细胞被诱导以表达并与纤溶酶一起孵育。洗涤细胞,然后用针对表达(α-HA)和对FGF1特异性切割的抗性(α-c-myc)的荧光抗体来染色。荧光激活细胞分选(FACS)用于分析和门控显示出通过HA表达信号归一化的高c-myc信号的细胞。(B)示出了FGF1的FACS点图。将来自每个文库的分选1的细胞在各种浓度的纤溶酶中孵育持续如所详述的不同孵育时间。用红色突出显示用于门控和收集细胞以进行富集的最终条件。对于所有测试条件,绘制相同的门。
图14.肽伪物质的分离。(A)示出了用于FGF1分选2文库的分选的FACS点图。以与分选2相同的方式选择对FGF1特异性切割的抗性。在显示出明显更高的对蛋白水解切割(c-myc)抗性的细胞亚群周围绘制收集门。(B)示出了从门收集的突变体的蛋白质序列。大多数由短肽组成,这些短肽是随机诱变的伪物质,并非衍生自FGF1。
图15肽伪物质不与FGFR1-Fc结合。将在其细胞表面上表达RTTTS或HTTS肽的酵母细胞与10nM FGFR1-Fc一起孵育。用针对表达(α-c-myc)和结合(α-FGFR1-Fc)的荧光抗体对细胞染色。没有检测到明显的结合信号,表明该肽不结合FGFR1-Fc。
图16.分选3和4的示意图。来自先前的细胞被诱导以便表达,并与不同浓度的纤溶酶一起孵育,洗涤,并与FGFR1-Fc一起孵育。最终洗涤后,将细胞用针对表达(α-HA)、对FGF1特异性切割的抗性(α-c-myc)和FGFR1结合(α-FGFR1-Fc)的荧光抗体染色。荧光激活细胞分选(FACS)用于分析和门控显示出通过HA表达信号归一化的高c-myc信号和/或高FGFR1-Fc结合信号的细胞。
图17.分选3:蛋白酶抗性的FGFR1-Fc结合剂的选择。将来自分选2的诱导细胞在指定浓度的纤溶酶中孵育12小时。洗涤后,将细胞与10nM FGFR1-Fc一起孵育。最后一次洗涤后,将细胞用针对表达(α-HA)和FGFR1结合(α-FGFR1-Fc)的荧光抗体染色。荧光激活细胞分选(FACS)用于分析和门控显示出高HA信号和高FGFR1-Fc信号的细胞。示出了FGF1的FACS点图。在点图上绘制的门旁,示出了从总群体中收集的细胞的百分比。下图:与1.25μM纤溶酶一起孵育24小时后,保持与FGFR1-Fc的结合。
图18.分选4:蛋白酶抗性的FGFR1-Fc结合剂的选择。将来自分选3的诱导细胞在各种浓度的纤溶酶中孵育36小时。洗涤后,将细胞与10nM FGFR1-Fc一起孵育。最后一次洗涤后,将细胞用针对表达(α-c-myc)和FGFR1结合(α-FGFR1-Fc)的荧光抗体染色。荧光激活细胞分选(FACS)用于分析和门控显示出高c-myc信号和高FGFR1-Fc信号的细胞。示出了FGF1的FACS点图。用红色突出显示用于门控和收集细胞以进行富集的最终条件。对于给定FGF的所有条件,绘制相同的门。在点图上绘制的门旁,示出了从总群体中收集的细胞的百分比。下图:与3.75μM纤溶酶一起孵育36小时后,保持与FGFR1-Fc的结合。
图19.FGF1结构上的BS4M1突变(PDB代码1E0O)。用蓝色突出显示通过筛选蛋白水解稳定性所鉴定的富集的突变。D28N突变位于使六链β-桶状结构稳定的三个β-发夹之一(红色突出显示)中。L131R突变位于蛋白质的C末端附近,在N末端与C末端之间缺乏稳定的β-发夹。
图20.可溶性野生型FGF1的重组表达。(A)通过非还原考马斯染色凝胶(左)和针对FGF1的蛋白质印迹(右)分析纯化的野生型FGF1。两个明显的条带表明存在FGF1单体(19.7kDa)和二聚体(39.4kDa)。(B)通过观察与酵母展示的FGFR3构建体的特异性结合,证实了FGF1的正确折叠。
图21.FGF2在pBAD载体中的重组表达。(A)通过还原考马斯染色凝胶(左)和针对FGF2的蛋白质印迹(右)分析在pBAD中表达并纯化的野生型FGF2-His。两者均表明通过所表达的FGF2的聚集。(B)在pBAD中表达的FGF2-His不能与酵母展示的FGFR3构建体结合。
图22.FGF2在pET28b载体中的重组表达。野生型FGF2和FGF2突变体(BS5M1、BS5M3、BS5M5)在pET28b载体中表达为与超级折叠物GFP的融合体。通过还原考马斯染色凝胶(左)和针对FGF2的蛋白质印迹(右)分析在pBAD中表达并纯化的野生型FGF2-His。野生型FGF2表达不佳,而FGF2突变体则显示出聚集和/或低聚的迹象。
图23.野生型FGF2在pET32a载体中的重组表达。(A)FGF2在pET32a载体中表达为与硫氧还蛋白的融合体。在用TEV切割并通过Ni-NTA和尺寸排阻色谱法纯化后,通过针对FGF2的蛋白质印迹分析蛋白质。证实了FGF2(19.3kDa)的成功纯化。
图24.纤溶酶中FGF1 WT和BS4M1的蛋白水解稳定性测定。FGF1BS4M1(D28N/L131R)突变体与野生型FGF1相比在纤溶酶中显示出更高的蛋白水解稳定性。将100ng FGF1与600nM纤溶酶在37℃下一起孵育不同的孵育时间。将所孵育的样品在针对FGF1的蛋白质印迹的单独泳道上电泳,以测量在每个时间点下的蛋白质降解程度。将由红色箭头指示的蛋白质条带的条带强度通过图像分析进行定量,以测量剩余蛋白质的量。通过时间点t=0将每种蛋白质的条带强度归一化并作图。
图25.纤溶酶中FGF1 WT、BS4M1、PM2和PM3的蛋白水解稳定性测定。将来自BS4M1(D28N,L131R)的突变与来自PM2(Q40P,S47I,H93G)的突变组合以创建PM3。与BS4M1或PM2相比,PM3在纤溶酶中显示出更高的蛋白水解稳定性。将125ng FGF1与各种浓度的纤溶酶在37℃下一起孵育48小时。将所孵育的样品在针对FGF1的蛋白质印迹的单独泳道上电泳,以测量在每个时间点下的蛋白质降解程度。将由红色箭头指示的蛋白质条带的条带强度通过图像分析进行定量,以测量剩余蛋白质的量。通过与0μM纤溶酶一起孵育时每种构建体的蛋白质的量将条带强度归一化并作图。
图26.胰蛋白酶中FGF1 WT和BS4M1的蛋白水解稳定性测定。FGF1BS4M1(D28N/L131R)突变体与野生型FGF1相比在胰蛋白酶中显示出更高的蛋白水解稳定性。将100ngFGF1同摩尔比1:20的胰蛋白酶与FGF1在37℃下一起孵育不同的孵育时间。将所孵育的样品在针对FGF1的蛋白质印迹的单独泳道上电泳,以测量在每个时间点下的蛋白质降解程度。将由红色箭头指示的蛋白质条带的条带强度通过图像分析进行定量,以测量剩余蛋白质的量。通过在时间点t=0时每种构建体的蛋白质的量对条带强度进行归一化并绘制。
图27.纤溶酶中FGF1 WT、BS4M1、D28N和L131R的蛋白水解稳定性测定。与BS4M1相比,FGF1 L131R单个突变体保留了其大部分的蛋白水解稳定性。甚至与野生型FGF1相比,FGF1 D28N单个突变体的蛋白水解稳定性也较低。将100ng FGF1与各种浓度的纤溶酶在37℃下一起孵育48小时。将所孵育的样品在针对FGF1的蛋白质印迹的单独泳道上电泳,以测量在每个时间点下的蛋白质降解程度。将由红色箭头指示的蛋白质条带的条带强度通过图像分析进行定量,以测量剩余蛋白质的量。通过与0μM纤溶酶一起孵育时每种构建体的蛋白质的量将条带强度归一化并作图。
图28.纤溶酶中FGF1 WT、L131R、L131A和L131K的蛋白水解稳定性测定。与FGF1L131R相比,FGF1 L131K单个突变体保留了其大部分的蛋白水解稳定性。甚至与野生型FGF1相比,FGF1 L131A单个突变体的蛋白水解稳定性也较低。将100ng FGF1与各种浓度的纤溶酶在37℃下一起孵育48小时。将所孵育的样品在针对FGF1的蛋白质印迹的单独泳道上电泳,以测量在每个时间点下的蛋白质降解程度。将由红色箭头指示的蛋白质条带的条带强度通过图像分析进行定量,以测量剩余蛋白质的量。通过与0μM纤溶酶一起孵育时每种构建体的蛋白质的量将条带强度归一化并作图。
图29.FGF1野生型和L131R突变体的ThermoFluor测定。一式三份测量FGF1野生型和L131R突变体的解链温度并作图。两种蛋白质的解链温度之间没有统计学上的显著差异
图30.MDA-MB-231培养物中FGF1野生型和L131R突变体的稳定性。与野生型FGF1相比,FGF1 L131R突变体在MDA-MB-231培养物中显示出更高的稳定性。将500ng FGF1与MDA-Mb-231细胞在37℃下一起孵育不同的孵育时间。将所孵育的样品浓缩并在针对FGF1的蛋白质印迹的单独泳道上电泳,以测量在每个时间点下的蛋白质降解程度。将由红色箭头指示的蛋白质条带的条带强度通过图像分析进行定量,以测量剩余蛋白质的量。通过时间点t=0将每种蛋白质的条带强度归一化并作图。
图31.NIH3T3 ERK磷酸化测定。FGF1 L131R突变体抑制通过野生型FGF1实现的NIH3T3 ERK磷酸化。用FGF1野生型和/或各种浓度的FGF1L131R突变体刺激NIH3T3细胞15小时。将细胞裂解,并在蛋白质印迹上用抗磷酸ERK探测裂解物。通过图像分析对条带强度进行定量,以测量FGF途径激活的程度。下图:用FGF1野生型和/或各种浓度的FGF1 L131R突变体刺激NIH3T3细胞10小时。
图32.NIH3T3细胞中FGF1 L131R突变体抑制FGF1刺激的ERK磷酸化。将NIH3T3细胞与1nM FGF1和各种浓度的FGF1 L131R一起孵育。通过针对磷酸ERK的蛋白质印迹来测量每种条件下ERK磷酸化的程度。通过图像分析对条带强度进行定量并作图以获得IC50值。
图33.FGF1野生型和L131R突变体与NIH3T3细胞的结合。His标记的FGF1 WT和L131R突变体与表达FGFR的NIH3T3细胞的平衡结合滴定。将细胞在4℃下与不同浓度的每种蛋白质一起孵育,并用抗His的荧光抗体染色以定量与细胞的结合。
图34A-图34B.提供IgG1、IgG2、IgG3和IgG4序列的实例。
具体实施方式
I.
成纤维细胞生长因子是参与多种过程的细胞信号传导蛋白家族,最显著地作为正常发育的关键要素。这些生长因子通常用作激活细胞表面受体的细胞外起源的全身或局部循环分子。哺乳动物成纤维细胞生长因子受体家族具有4个成员,即FGFR1、FGFR2、FGFR3和FGFR4。FGFR由三个细胞外免疫球蛋白型结构域(D1-D3)、一个单跨膜结构域和一个细胞内分裂酪氨酸激酶结构域组成。FGF与D2和D3结构域相互作用,其中D3相互作用主要负责配体结合特异性(参见下文)。硫酸乙酰肝素结合通过D3结构域介导。位于D1与D2结构域之间的一小段酸性氨基酸具有自动抑制功能。该‘酸盒’基序与硫酸乙酰肝素结合位点相互作用,以防止在不存在FGF的情况下的受体激活。每个FGFR结合至FGF的特定子集。同样,大多数FGF可以结合几种不同的FGFR亚型。FGF1有时被称为‘通用配体’,因为它能够激活所有7种不同的FGFR。相反,FGF7(角质形成细胞生长因子,KGF)仅与FGFR2b(KGFR)结合。
本发明提供了用于组合方法的方法,所述组合方法利用酵母展示平台和用于筛选的流激活细胞分选(FACS)来工程化蛋白水解稳定的生长因子。描述了使用FGF1作为模型实例建立筛选方法的过程,并提供了工程化示例性蛋白水解稳定的生长因子的方法。本发明还提供了蛋白水解稳定的FGF1突变体的表征。
II.
除非另有定义,否则本文中使用的所有技术和科学术语具有与本发明所属领域的普通技术人员通常理解的含义相同的含义。通常,本文使用的命名法以及细胞培养、分子遗传学、有机化学和核酸化学与杂交中的实验室程序是本领域众所周知的和常用的。标准技术用于核酸和肽合成。所述技术和程序通常根据本领域的常规方法和各种一般参考文献进行(一般参见Sambrook等人Molecular Cloning:A Laboratory Manual,第2版(1989)ColdSpring Harbor Laboratory Press,Cold Spring Harbor,N.Y.,其以引用的方式并入本文),它们在本文档中通篇提供。本文所用的命名法以及下文所述的分析和合成有机化学的实验室程序是本领域众所周知的和常用的。标准技术或其修改用于化学合成和化学分析。
术语“BS4M1”和“PM2”和“PM3”是指具有以下取代的SEQ ID NO:1的变体:(i)BS4M1(D28N和L131R),(ii)PM2(Q40P、S47I、H93G)和(iii)PM3(D28N、Q40P、S47I、H93G、L131R)。FGF1:FNLPPGNYKKPKLLYCSNGGHFLRILPDGTVDGTRDRSDQHIQLQLSAESVGEVYIKSTETGQYLAMDTDGLLYGSQTPNEECLFLERLEENHYNTYISKKHAEKNWFVGLKKNGSCKRGPRTHYGQKAILFLPLPVSSD(SEQ IDNO:1)。SEQ ID NO:1是没有前肽的FGF1序列(https://www.uniprot.org/blast/?about=P05230[16-155]&key=Chain&id=PRO_0000008908)。本文所述的编号是基于以上序列的第一个氨基酸作为位置1(例如:F1,N2等)。FGF1的其它编号可以在编号中包括前肽序列,这会导致编号增加14。然而,本文的编号是基于SEQ ID NO:1并且不包括FGF1前肽。
术语“核酸”或“多核苷酸”是指单链或双链形式的脱氧核糖核酸(DNA)或核糖核酸(RNA)及其聚合物。除非特别限制,该术语涵盖含有天然核苷酸的已知类似物的核酸,所述核酸具有与参考核酸相似的结合特性并且以与天然核苷酸类似的方式进行代谢。除非另有说明,否则特定的核酸序列也隐含涵盖其保守修饰的变体(例如,简并密码子取代)、等位基因、直向同源物、SNP和互补序列以及明确指出的序列。具体而言,简并密码子取代可以通过生成以下序列来实现:其中一个或多个选定的(或全部)密码子的第三位置被混合碱基和/或脱氧肌苷残基取代(Batzer等人,Nucleic Acid Res.19:5081(1991);Ohtsuka等人,J.Biol.Chem.260:2605-2608(1985);以及Rossolini等人,Mol.Cell.Probes8:91-98(1994))。术语核酸可与基因、cDNA和由基因编码的mRNA互换使用。此外,如本文所用,编码本发明的多肽变体的核酸被定义为包括与此核酸序列互补的核酸序列。
术语“基因”是指参与产生多肽链的DNA链段。它可以包括编码区之前和之后的区域(前导区和尾区)以及各个编码链段(外显子)之间的间插序列(内含子)。
当应用于核酸或蛋白质时,术语“分离的”是指该核酸或蛋白质基本上不含在自然状态下与之缔合的其它细胞组分。尽管可以是干燥或水溶液形式,但优选为均质状态。纯度和均质性通常使用诸如聚丙烯酰胺凝胶电泳或高效液相色谱法的分析化学技术测定。蛋白质作为在制剂中存在的主要物质是大体上纯化的。特别地,分离的基因与位于该基因侧翼并编码除目标基因以外的蛋白质的开放阅读框分离。术语“纯化的”表示核酸或蛋白质在电泳凝胶中产生基本上一个条带(band)。具体来说,这意味着核酸或蛋白质的纯度为至少85%、更优选至少95%、最优选至少99%。分离的核酸可以是表达载体的组分。
通常,本发明的分离的多肽具有纯度水平,优选表示为范围。多肽纯度范围的下限为约60%、约70%或约80%,且纯度范围的上限为约70%、约80%、约90%、约95%或大于约95%。当多肽的纯度大于约90%时,它们的纯度也优选表示为范围。纯度范围的下限为约90%、约92%、约94%、约96%或约98%。纯度范围的上限为约92%、约94%、约96%、约98%或约100%。
纯度通过本领域公认的任何分析方法(例如,银染凝胶上的条带强度、聚丙烯酰胺凝胶电泳、HPLC、质谱或类似方法)测定。
术语“氨基酸”是指天然存在的和合成的氨基酸,以及以类似于天然存在的氨基酸的方式起作用的氨基酸类似物和氨基酸模拟物。天然存在的氨基酸是由遗传密码编码的那些,以及后期修饰的那些氨基酸,例如,羟基脯氨酸、γ-羧基谷氨酸和O-磷酸丝氨酸。氨基酸类似物是指与天然存在的氨基酸具有相同的基本化学结构的化合物,即与氢、羧基、氨基和R基团结合的α-碳,例如,高丝氨酸、正亮氨酸、甲硫氨酸亚砜、甲硫氨酸甲基锍。这种类似物具有修饰的R基团(例如,正亮氨酸)或修饰的肽骨架,但保留了与天然存在的氨基酸相同的基本化学结构。“氨基酸模拟物”是指具有这样的结构的化合物,所述结构不同于氨基酸的一般化学结构,但其作用的方式类似于天然存在的氨基酸。
“亲水氨基酸”是指根据Eisenberg等人,1984,J.Mol.Biol.179:125-142的归一化共有疏水性量表,呈现小于零的疏水性的氨基酸。遗传编码的亲水氨基酸包括Thr(T)、Ser(S)、His(H)、Glu(E)、Asn(N)、Gln(Q)、Asp(D)、Lys(K)和Arg(R)。
“酸性氨基酸”是指具有小于7的侧链pK值的亲水氨基酸。由于失去氢离子,所以酸性氨基酸通常在生理pH下具有带负电的侧链。遗传编码的酸性氨基酸包括Glu(E)和Asp(D)。
“碱性氨基酸”是指具有大于7的侧链pK值的亲水氨基酸。由于与水合氢离子缔合,碱性氨基酸通常在生理pH下具有带正电的侧链。遗传编码的碱性氨基酸包括His(H)、Arg(R)和Lys(K)。
“极性氨基酸”是指具有在生理pH下不带电但含有至少一个键的侧链的亲水氨基酸,在所述键中由两个原子共同享有的一对电子被一个原子更紧密地保持。遗传编码的极性氨基酸包括Asn(N)、Gln(Q)、Ser(S)和Thr(T)。
“疏水氨基酸”是指根据Eisenberg,1984,J.Mol.Biol.179:125-142的归一化共有疏水性量表,呈现大于零的疏水性的氨基酸。示例性疏水氨基酸包括Ile(I)、Phe(F)、Val(V)、Leu(L)、Trp(W)、Met(M)、Ala(A)、Gly(G)、Tyr(Y)、Pro(P)和脯氨酸类似物。
“芳族氨基酸”是指具有含有至少一个芳环或杂芳环的侧链的疏水氨基酸。芳环或杂芳环可含有一个或多个取代基,诸如-OH、-SH、-CN、-F、-Cl、-Br、-I、-NO
“非极性氨基酸”是指具有在生理pH下不带电并含有键的侧链的疏水氨基酸,在所述键中由两个原子共同享有的一对电子通常被两个原子中的每个均等地保持(即,侧链不是极性的)。遗传编码的非极性氨基酸包括Leu(L)、Val(V)、Ile(I)、Met(M)、Gly(G)和Ala(A)。
“脂族氨基酸”是指具有脂族烃侧链的疏水氨基酸。遗传编码的脂族氨基酸包括Ala(A)、Val(V)、Leu(L)和Ile(I)。
氨基酸残基Cys(C)不常见,因为它可以与其它Cys(C)残基或其它含磺酰基的氨基酸形成二硫桥。Cys(C)残基(和其它带有含–SH的侧链的氨基酸)以还原的游离SH或氧化的二硫桥接形式存在于肽中的能力会影响Cys(C)残基是否对肽贡献净疏水性或亲水性的特征。尽管Cys(C)根据Eisenberg的归一化共有量表(Eisenberg,1984,同上)呈现出0.29的疏水性,应当理解,尽管上面定义了一般分类,但出于本发明的目的,Cys(C)被归类为极性亲水氨基酸。
术语“接头”是指共价连接两个或更多个多肽的氨基酸多肽间隔子。接头可以是1-15个氨基酸残基。优选地,接头是单个半胱氨酸残基。接头也可以具有氨基酸序列SEQ IDNO:1KESCAKKQRQHMDS。
如本领域技术人员将理解的,以上定义的类别不是相互排斥的。因此,具有表现出两种或更多种物理化学性质的侧链的氨基酸可以被包括在多个类别中。例如,具有进一步被极性取代基诸如Tyr(Y)取代的芳族部分的氨基酸侧链可同时具有芳族疏水性和极性或亲水性,且因此可被同时包括在芳族和极性类别中。任何氨基酸的适当分类对于本领域技术人员将是显而易见的,尤其是根据本文提供的详细公开内容。
当某些氨基酸残基包含在螺旋的内部位置时,它们被称为“螺旋断裂”氨基酸,具有破坏α-螺旋结构的倾向。表现出这种螺旋断裂性质的氨基酸残基是本领域众所周知的(参见,例如,Chou和Fasman,Ann.Rev.Biochem.47:251-276)并且包括Pro(P)、Gly(G)和潜在的所有D-氨基酸(当包含在L-肽中时;相反,当L-氨基酸包含在D-肽中时,它会破坏螺旋结构)以及脯氨酸类似物。尽管这些螺旋断裂氨基酸残基属于上面定义的类别,Gly(G)(下文讨论)除外,但这些残基不应当用于取代螺旋内部位置处的氨基酸残基,而仅应用于取代肽的N末端和/或C末端处的1-3个氨基酸残基。
尽管已经根据遗传编码的氨基酸例示了上面定义的类别,但是氨基酸取代不必是,并且在某些实施方案中优选地不限于遗传编码的氨基酸。实际上,式(I)的许多优选肽包含基因上非编码的氨基酸。因此,除了天然存在的遗传编码的氨基酸之外,式(I)的核心肽中的氨基酸残基也可以被天然存在的非编码氨基酸和合成氨基酸取代。
为式(I)的核心肽提供有用取代的某些常见氨基酸包括但不限于β-丙氨酸(β-Ala)和其它ω-氨基酸,诸如3-氨基丙酸、2,3-二氨基丙酸(Dpr)、4-氨基丁酸等;α-氨基异丁酸(Aib);ε-氨基己酸(Aha);δ-氨基戊酸(Ava);N-甲基甘氨酸或肌氨酸(MeGly);鸟氨酸(Orn);瓜氨酸(Cit);叔丁基丙氨酸(t-BuA);叔丁基甘氨酸(t-BuG);N-甲基异亮氨酸(MeIle);苯甘氨酸(Phg);环己基丙氨酸(Cha);正亮氨酸(Nle);萘丙氨酸(Nal);4-氯苯丙氨酸(Phe(4-Cl));2-氟苯丙氨酸(Phe(2-F));3-氟苯丙氨酸(Phe(3-F));4-氟苯丙氨酸(Phe(4-F));青霉胺(Pen);1/2/3/4-四氢异喹啉-3-甲酸(Tic);β-2-噻吩丙氨酸(Thi);甲硫氨酸亚砜(MSO);高精氨酸(hArg);N-乙酰赖氨酸(AcLys);2,4-二氨基丁酸(Dbu);2,3-二氨基丁酸(Dab);对氨基苯丙氨酸(Phe(pNH2));N-甲基缬氨酸(MeVal);高半胱氨酸(hCys)、高苯丙氨酸(hPhe)和高丝氨酸(hSer);羟脯氨酸(Hyp)、高脯氨酸(hPro)、N-甲基化氨基酸和类肽(N-取代的甘氨酸)。另外,在一些实施方案中,式(I)的核心肽中的氨基酸脯氨酸被脯氨酸类似物取代,所述脯氨酸类似物包括但不限于氮杂环丁烷-2-羧酸盐(A2C)、L-噻唑烷-4-羧酸、顺式-4-羟基-L-脯氨酸(CHP)、3,4-脱氢脯氨酸、硫代脯氨酸和异哌啶酸(Inp)。
氨基酸在本文中可由其通常已知的三字母符号或由IUPAC-IUB生物化学命名委员会推荐的单字母符号来表示。同样地,核苷酸可以由其普遍接受的单字母代码表示。
关于氨基酸序列,本领域技术人员将认识到,对核酸、肽、多肽或蛋白质序列的单个取代、缺失或添加是会改变、添加或缺失所编码序列中的单个氨基酸或一小部分氨基酸序列的“保守修饰的变体”,其中所述改变导致氨基酸被化学上相似的氨基酸取代。提供功能上相似的氨基酸的保守取代表是本领域众所周知的。此类保守修饰的变体是本发明的多态变体、种间同源物和等位基因的补充,并且不排除其。
以下八个组各自包含彼此保守取代的氨基酸:
1)丙氨酸(A),甘氨酸(G);
2)天冬氨酸(D),谷氨酸(E);
3)天冬酰胺(N),谷氨酰胺(Q);
4)精氨酸(R),赖氨酸(K);
5)异亮氨酸(I),亮氨酸(L),甲硫氨酸(M),缬氨酸(V);
6)苯丙氨酸(F),酪氨酸(Y),色氨酸(W);
7)丝氨酸(S),苏氨酸(T);和
8)半胱氨酸(C),甲硫氨酸(M)
(参见,例如,Creighton,Proteins(1984))。
氨基酸取代通常基于氨基酸侧链取代基的相对相似性,例如它们的疏水性、亲水性、电荷、大小等。将一种或多种前述特征考虑在内的示例性取代是本领域技术人员众所周知的,并且包括但不限于(原始残基:示例性取代):(Ala:Gly、Ser),(Arg:Lys),(Asn:Gln、His),(Asp:Glu、Cys、Ser),(Gln:Asn),(Glu:Asp),(Gly:Ala),(His:Asn、Gln),(Ile:Leu、Val),(Leu:Ile、Val),(Lys:Arg),(Met:Leu、Tyr),(Ser:Thr),(Thr:Ser),(Tip:Tyr),(Tyr:Trp、Phe)和(Val:Ile、Leu)。因此,本公开的实施方案考虑如上所述的多肽或蛋白质的功能或生物等效物。具体来说,本发明的实施方案提供了与亲本多肽具有约50%、60%、70%、80%、90%和95%序列同一性的变体。在各种实施方案中,本发明提供了与亲本多肽序列的一部分具有该水平同一性的变体,所述亲本多肽是例如野生型生长因子,包括例如野生型FGF1(SEQ ID NO:1)。在各种实施方案中,变体与亲本多肽或亲本多肽序列的一部分具有至少约95%、96%、97%、98%或99%序列同一性,所述亲本多肽是例如本文所定义的野生型生长因子,包括例如野生型FGF1(SEQ ID NO:1)。
“保守修饰的变体”适用于氨基酸和核酸序列两者。关于特定的核酸序列,“保守修饰的变体”是指编码相同或基本相同的氨基酸序列的那些核酸,或其中核酸不编码氨基酸序列至基本相同的序列。由于遗传密码的简并性,大量功能相同的核酸编码任何给定的蛋白质。例如,密码子GCA、GCC、GCG和GCU都编码氨基酸丙氨酸。因此,在由密码子指定丙氨酸的每个位置处,可以将密码子改变为所描述的任何相应密码子,而不会改变编码的多肽。这样的核酸变异是“沉默变异”,其是保守修饰变异中的一个种类。本文中编码多肽的每个核酸序列还描述了核酸的每个可能的沉默变异。本领域技术人员将认识到,可以修饰核酸中的每个密码子(AUG(其通常是甲硫氨酸的唯一密码子)和TGG(其通常是色氨酸的唯一密码子)除外)以产生功能相同的分子。因此,编码多肽的核酸的每个沉默变异都隐含在每个所述序列中。
如本领域已知的“同一性”是两个或更多个多肽或蛋白质序列之间的关系,如通过比较序列所确定的。在本领域中,“同一性”还指的是多肽或蛋白质之间的序列相关性程度,如通过此类序列串之间的匹配所确定的。“同一性”可易于通过已知的生物信息学方法来计算。
“肽”是指其中单体是氨基酸并且通过酰胺键连接在一起的聚合物。本发明的肽的大小可以变化,例如,从两个氨基酸至数百或数千个氨基酸。较大的肽(例如,至少10个、至少20个、至少30个或至少50个氨基酸残基)可替代地称为“多肽”或“蛋白质”。另外,还包括非天然氨基酸,例如β-丙氨酸、苯甘氨酸、高精氨酸和高苯丙氨酸。非基因编码的氨基酸也可以用于本发明中。此外,已被修饰为包括反应性基团、糖基化序列、聚合物、治疗部分、生物分子等的氨基酸也可以用于本发明中。用于本发明的所有氨基酸可以是d-或l-异构体。l–异构体通常是优选的。另外,其它肽模拟物也可用于本发明中。如本文所用,“肽”或“多肽”是指糖基化和非糖基化的肽或“多肽”。还包括被表达多肽的系统不完全糖基化的多肽。有关一般性综述,参见Spatola,A.F.,Chemistry and Biochemistry of Amino Acids,Peptides and Proteins,B.Weinstein编著,Marcel Dekker,New York,第267页(1983)。
在本申请中,氨基酸残基是根据它们相对于多肽的N末端氨基酸(例如N末端甲硫氨酸)的相对位置来编号的(通常在上标中),编号为“1”。N末端氨基酸可以是甲硫氨酸(M),编号为“1”。如果多肽的N末端在无甲硫氨酸下起始,则可以容易地调节与每个氨基酸残基相关的数目以反映出N末端甲硫氨酸的缺失。应当理解,示例性多肽的N末端可在有或无甲硫氨酸下起始。因此,在向野生型多肽的N末端添加氨基酸接头的情况下,与N末端氨基酸邻接的第一接头氨基酸为-1号,以此类推。例如,如果接头具有氨基酸序列KESCAKKQRQHMDS(SEQ ID NO:2),且S残基与野生型多肽的N末端氨基酸邻接,则最N末端接头氨基酸K为-14,而最C末端接头氨基酸S为-1。以这种方式,保留了野生型多肽和与接头结合的野生型多肽中的氨基酸编号。
术语“亲本多肽”是指野生型多肽,并且该野生型多肽的氨基酸序列或核苷酸序列是公众可访问的蛋白质数据库(例如EMBL Nucleotide Sequence Database、NCBI Entrez、ExPasy、Protein Data Bank等)的一部分。
术语“突变多肽”或“多肽变体”或“突变蛋白”或“变体多肽”是指多肽的一种形式,其中其氨基酸序列不同于其相应野生型(亲本)形式、天然存在的形式或任何其它亲本形式的氨基酸序列。突变多肽可以包含一个或多个突变,例如替换、插入、缺失等,它们产生突变多肽。
术语“对应于亲本多肽”(或此术语的语法变型)用于描述本发明的多肽,其中多肽的氨基酸序列与相应的亲本多肽的氨基酸序列的区别仅在于至少存在氨基酸变异。通常,变体多肽与亲本多肽的氨基酸序列表现出高同一性百分比。在一个实例中,“对应于亲本多肽”意味着变体多肽的氨基酸序列与亲本多肽的氨基酸序列具有至少约50%同一性、至少约60%、至少约70%、至少约80%、至少约90%、至少约约95%或至少约98%同一性。在另一个实例中,编码变体多肽的核酸序列与编码亲本多肽的核酸序列具有至少约50%同一性、至少约60%、至少约70%、至少约80%、至少约90%、至少约约95%或至少约98%同一性。在一些实施方案中,亲本多肽对应于SEQ ID NO:1的FGF1。
术语“向亲本多肽中引入(或添加等)变异”(或其语法变型)或“修饰亲本多肽”以包括变异(或其语法变型)不一定意味着亲本多肽是用于这种转化的物理起始材料,但是亲本多肽提供了用于制备变体多肽的指导氨基酸序列。在一个实例中,“向亲本多肽中引入变体”是指通过适当的突变修饰亲本多肽的基因以产生编码变体多肽的核苷酸序列。在另一个实例中,“向亲本多肽中引入变体”是指使用亲本多肽序列作为指导在理论上设计所得多肽。然后可以通过化学或其它方式生成所设计的多肽。
术语“文库”是指不同多肽的集合,每个多肽对应于共同的亲本多肽。文库中的每个多肽种类被称为文库的成员。优选地,本发明的文库代表具有足以提供从中鉴定先导多肽的群体的数量和多样性的多肽的集合。文库包含至少两种不同多肽。在一个实施方案中,文库包含约2至约100,000,000个成员。在另一个实施方案中,文库包含约10,000至约100,000,000个成员。在仍另一个实施方案中,文库包含约100,000至约100,000,000个成员。在又一个实施方案中,文库包含约1,000,000至约100,000,000个成员。在另一个实施方案中,文库包含约10,000,000至约100,000,000个成员。在仍另一个实施方案中,文库包含多于100个成员。
文库的成员可以是混合物的一部分,或者可以彼此分离。在一个实例中,文库的成员是混合物的一部分,该混合物任选地包含其它组分。例如,一定体积的细胞培养肉汤中存在至少两种多肽。在另一个实例中,文库的成员各自单独表达,并且任选地分离。分离的多肽可任选地包含在多孔容器中,其中每个孔含有不同类型的多肽。在另一个实例中,文库的成员各自表达为与酵母或细菌细胞或噬菌体或病毒颗粒的融合体。
如本文所用,术语“聚合修饰基团”是包括至少一个聚合部分(聚合物)的修饰基团。添加到多肽上的聚合修饰基团可以改变这种多肽的性质,例如其生物利用度、生物活性或其在体内的半衰期。示例性聚合物包括水溶性和水不溶性聚合物。聚合修饰基团可以是直链或支链的,并且可以包括一个或多个独立选择的聚合部分,诸如聚(烷二醇)及其衍生物。在一个实例中,聚合物是非天然存在的。在一个示例性实施方案中,聚合修饰基团包括水溶性聚合物,例如聚(乙二醇)及其衍生物(PEG,m-PEG)、聚(丙二醇)及其衍生物(PPG,m-PPG)等。在一个优选实施方案中,聚(乙二醇)或聚(丙二醇)的分子量基本上是均分散的。在一个实施方案中,聚合修饰基团不是天然存在的多糖。
如本文所用,术语“靶向部分”是指将选择性地定位在身体的特定组织或区域中的种类。通过分子决定簇的特异性识别、靶向剂或缀合物的分子大小、离子相互作用、疏水相互作用等来介导定位。使剂靶向特定组织或区域的其它机制是本领域技术人员已知的。示例性靶向部分包括抗体、抗体片段、转铁蛋白、HS-糖蛋白、凝血因子、血清蛋白、β-糖蛋白、G-CSF、GM-CSF、M-CSF、EPO等。
如本文所用,术语“Fc融合蛋白”意在包括蛋白质,特别是治疗性蛋白质,其包含免疫球蛋白衍生的部分(在本文中将被称为“Fc部分”)和衍生自第二非免疫球蛋白蛋白质的部分,在本文中将被称为“治疗部分”,不管是否打算治疗疾病。
如本文所用,“治疗部分”是指可用于疗法的任何剂,包括但不限于抗生素、抗炎剂、抗肿瘤药、细胞毒素和放射性剂。“治疗部分”包括生物活性剂的前药,其中多于一个治疗部分结合至载剂(例如多价剂)的构建体。
治疗部分还包括蛋白质和包含蛋白质的构建体。
如本文所用,“抗肿瘤药”是指可用于对抗癌症的任何剂,包括。
如本文所用,“细胞毒素或细胞毒性剂”是指对细胞有害的任何剂。实例包括紫杉醇、细胞松弛素B、短杆菌肽D、溴化乙锭、依米丁、丝裂霉素、依托泊苷、替诺泊苷、长春新碱、长春碱、秋水仙碱、阿霉素、柔红霉素、二羟基蒽醌二酮、米托蒽醌、光神霉素、放线菌素D、1-去氢睾酮、糖皮质激素、普鲁卡因、丁卡因、利多卡因、普萘洛尔和嘌呤霉素及其类似物或同系物。其它毒素包括例如蓖麻毒素、CC-1065和类似物、杜卡霉素。其它毒素还包括白喉毒素和蛇毒(例如眼镜蛇毒)。
如本文所用,“放射性剂”包括在诊断或破坏肿瘤方面有效的任何放射性同位素。实例包括但不限于铟-111、钴-60、氟-18、铜-64、铜-67、镥-177或technicium-99m。另外,通常代表放射性同位素的混合物的天然存在的放射性元素诸如铀、镭和钍是放射性剂的合适实例。金属离子通常与有机螯合部分螯合。放射性剂或放射性核素可以是显像剂的组分。
对于光学成像应用,也可以使用标准化学方法对近红外染料进行缀合。“近红外”是指在电磁光谱中与可见光相关的部分附近的部分中的辐射,例如,从约0.7μm至约1μm。近红外染料可包括例如花青或吲哚花青衍生物,诸如Cy5.5。红外染料还可以包括亚磷酰胺染料,例如
许多有用的螯合基团、冠醚、穴状化合物等在本领域中是已知的,并且可以掺入本发明的化合物中(例如,EDTA、DTPA、DOTA、NTA、HDTA 等,以及它们的膦酸酯类似物,诸如DTPP、EDTP、HDTP、NTP等)。参见,例如,Pitt等人,“The Design of Chelating Agents forthe Treatment of Iron Overload,”Inorganic Chemistry in Biology and Medicine;Martell编著;American Chemical Society,Washington,D.C.,1980,第279-312页;Lindoy,The Chemistry of Macrocyclic Ligand Complexes;Cambridge UniversityPress,Cambridge,1989;Dugas,Bioorganic Chemistry;Springer-Verlag,New York,1989,以及其中包含的参考文献。另外,本领域技术人员可以使用多种途径将螯合剂、冠醚和环糊精连接到其它分子上。参见,例如,Meares等人,“Properties of In Vivo Chelate-Tagged Proteins and Polypeptides.”Modification of Proteins:Food,Nutritional,and Pharmacological Aspects;”Feeney,等人,编著,American Chemical Society,Washington,D.C.,1982,第370-387页;Kasina等人,Bioconjugate Chem.,9:108-117(1998);Song等人,Bioconjugate Chem.,8:249-255(1997)。这些金属结合剂可用于结合可在成像模态中检测到的金属离子。
如本文所用,“药学上可接受的载剂”包括当与缀合物组合时保留缀合物的活性并且对受试者的免疫系统无反应的任何材料。“药学上可接受的载剂”包括固体和液体,诸如媒介物、稀释剂和溶剂。实例包括但不限于任何标准药物载剂诸如磷酸盐缓冲盐水溶液、水、乳液诸如油/水乳液和各种类型的润湿剂。其它载剂还可以包括无菌溶液、包括包衣片剂的片剂和胶囊。通常,此类载剂含有赋形剂,诸如淀粉、牛奶、糖、某些类型的粘土、明胶、硬脂酸或其盐、硬脂酸镁或硬脂酸钙、滑石粉、植物脂肪或油、树胶、乙二醇或其它已知的赋形剂。这样的载剂还可以包括调味剂和颜色添加剂或其它成分。通过众所周知的常规方法配制包含此类载剂的组合物。
如本文所用,“施用”意指向受试者经口施用、作为栓剂施用、局部接触、静脉内、腹膜内、肌内、鞘内、病灶内或皮下施用、通过吸入施用或植入缓释装置,例如微型渗透泵。通过任何途径进行施用,包括肠胃外和经粘膜(例如,经口、鼻、阴道、直肠或透皮),特别是通过吸入。肠胃外施用包括例如静脉内、肌内、小动脉内、皮内、皮下、腹膜内、心室内和颅内。此外,在注射用于治疗肿瘤(例如诱导细胞凋亡)的情况下,施用可以直接对肿瘤和/或肿瘤周围的组织进行。其它递送方式包括但不限于使用脂质体制剂、静脉内输注、透皮贴剂等。
术语“改善(ameliorating/ameliorate)”指的是在病理或病症治疗方面取得成功的任何标志,包括任何客观或主观参数,诸如减轻、缓解或消除症状或改善患者的身体或精神健康。症状的改善可基于客观或主观参数;包括身体检查和/或精神病学评估的结果。
术语“疗法”是指疾病或病症的“治疗(treating)”或“治疗(treatment)”,包括预防可能易患疾病但尚未经历或表现出疾病症状的受试者(例如,人)中发生该疾病或病症(预防性治疗);抑制疾病(延缓或阻止其发展);缓解疾病的症状或副作用(包括姑息治疗)以及缓解疾病(导致疾病消退)。
术语“有效量”或“有效……的量”或“治疗有效量”或任何语法等价术语是指当施用于动物或人以治疗疾病时足以实现对该疾病的治疗的量。有效量还可以指的是引起包括例如细胞凋亡、细胞周期起始和/或信号转导在内的细胞应答所必需的量。
术语“药学上可接受的盐”包括活性化合物用相对无毒的酸或碱制备的盐,这取决于本文所述的化合物上所见的具体取代基。当本发明化合物含有相对酸性官能团时,可通过纯净地或在合适的惰性溶剂中使中性形式的所述化合物与足量的所需碱接触来获得碱加成盐。药学上可接受的碱加成盐的实例包括钠、钾、钙、铵、有机氨基或镁盐或类似盐。当本发明化合物含有相对碱性官能团时,可通过纯净地或在合适的惰性溶剂中使中性形式的所述化合物与足量的所需酸接触来获得酸加成盐。药学上可接受的酸加成盐的实例包括衍生自无机酸如盐酸、氢溴酸、硝酸、碳酸、一氢碳酸、磷酸、一氢磷酸、二氢磷酸、硫酸、一氢硫酸、氢碘酸或亚磷酸等的那些;以及衍生自相对无毒的有机酸如乙酸、丙酸、异丁酸、马来酸、丙二酸、苯甲酸、琥珀酸、辛二酸、富马酸、乳酸、扁桃酸、邻苯二甲酸、苯磺酸、对甲苯磺酸、柠檬酸、酒石酸、甲磺酸等的盐。还包括的是氨基酸诸如精氨酸等的盐以及有机酸如葡萄糖醛酸或半乳糖醛酸等的盐(参见,例如,Berge等,Journal of PharmaceuticalScience 66:1-19(1977))。本发明的某些特定化合物既含有碱性官能团也含有酸性官能团,允许化合物转化成碱加成盐或酸加成盐。
中性形式的化合物优选地通过使盐与碱或酸接触并以常规方式分离母体化合物而再生。化合物的母体形式在某些物理特性(诸如在极性溶剂中的溶解度)上不同于各种盐形式,但在其它方面,盐出于本发明的目的等效于化合物的母体形式。
本发明化合物也可在构成所述化合物的一个或多个原子处含有非天然比例的原子同位素。例如,化合物可用放射性同位素(例如像氚(
如本文所用,“反应性官能团”是指包括但不限于以下的基团:烯烃、乙炔、醇、酚、醚、氧化物、卤化物、醛、酮、羧酸、酯、酰胺、氰酸酯、异氰酸酯、硫氰酸酯、异硫氰酸酯、胺、肼、腙、酰肼、重氮、重氮鎓、硝基、腈、硫醇、硫化物、二硫化物、亚砜、砜、磺酸、亚磺酸、缩醛、缩酮、酸酐、硫酸盐、次磺酸异腈、脒、酰亚胺、亚氨酸酯、硝酮、羟胺、肟、异羟肟酸、硫代羟肟酸、丙二烯、原酸酯、亚硫酸盐、烯胺、炔胺、脲、假脲、氨基脲、碳二亚胺、氨基甲酸酯、亚胺、叠氮化物、偶氮化合物、叠氮化合物和亚硝基化合物。反应性官能团还包括用于制备生物缀合物的那些,例如,N-羟基琥珀酰亚胺酯、马来酰亚胺等。制备这些官能团中的每个的方法是本领域众所周知的,并且其对于特定目的的应用或修改在本领域技术人员的能力范围内(参见,例如Sandler和Karo编著Organic Functional Group Preparations,AcademicPress,San Diego,1989)。
III.
在一些实施方案中,与野生型生长因子相比,变体是蛋白水解稳定的变体。在一个示例性实施方案中,变体与野生型相比表现出提高的蛋白水解稳定性。在一些实施方案中,变体是野生型生长因子的任何变体。在一些实施方案中,变体是野生型生长因子结合至的生长因子受体的拮抗剂。
在一些实施方案中,变体是FGF1的变体。在一些实施方案中,提供了一种人成纤维细胞生长因子1(FGF1)变体,其包含至少一个选自氨基酸取代、氨基酸缺失、氨基酸添加及其组合的成员。在一些实施方案中,提供了一种人成纤维细胞生长因子1(FGF1)变体,其包含至少一个选自氨基酸取代、氨基酸缺失、氨基酸添加及其组合的成员,其中所得FGF1变体与SEQ ID NO:1的野生型FGF1相比表现出提高的蛋白水解稳定性。在一些实施方案中,FGF1变体在β-环中或C末端附近包含氨基酸取代、氨基酸缺失、氨基酸添加及其组合。在一些实施方案中,FGF1变体是成纤维细胞生长因子受体(FGFR)拮抗剂。本发明提供了一种FGF1多肽,所述FGF1多肽在至少一个位置上具有至少一个氨基酸,其中在亲本FGF1多肽(野生型,SEQ ID NO:1)中未见到此氨基酸。
FNLPPGNYKKPKLLYCSNGGHFLRILPDGTVDGTRDRSDQHIQLQLSAESVGEVYIKSTETGQYLAMDTDGLLYGSQTPNEECLFLERLEENHYNTYISKKHAEKNWFVGLKKNGSCKRGPRTHYGQKAILFLPLPVSSD(SEQID NO:1)。
在一些实施方案中,SEQ ID NO:1的FGF1变体具有至少一个氨基酸取代。在一些实施方案中,FGF1变体在位置28、40、47、93或131处包含至少一个氨基酸取代。在一些实施方案中,FGF1变体包含至少一个选自由D28N、Q40P、S47I、H93G、L131R和L131K组成的组的氨基酸取代。在一些实施方案中,FGF1变体包含氨基酸取代L131R。在一些实施方案中,FGF1变体包含氨基酸取代L131K。在一些实施方案中,变体包含氨基酸取代D28N和L131R。在一些实施方案中,变体包含氨基酸取代D28N和L131K。在一些实施方案中,变体包含氨基酸取代Q40P、S47I、H93G和L131R。在一些实施方案中,变体包含氨基酸取代Q40P、S47I、H93G和L131K。在一些实施方案中,变体包含氨基酸取代D28N、Q40P、S47I、H93G和L131R。在一些实施方案中,变体包含氨基酸取代D28N、Q40P、S47I、H93G和L131K。在一些实施方案中,FGF1变体不包含氨基酸取代L131A。
在一些实施方案中,变体FGF1是被称为BS4M1(D28N和L131R)变体的变体。在一些实施方案中,BS4M1包含序列FNLPPGNYKKPKLLYCSNGGHFLRILPNGTVDGTRDRSDQHIQLQLSAESVGEVYIKSTETGQYLAMDTDGLLYGSQTPNEECLFLERLEENHYNTYISKKHAEKNWFVGLKKNGSCKRGPRTHYGQKAIRFLPLPVSSD(SEQ ID NO:2)。
在一些实施方案中,变体FGF1是被称为PM2(Q40P、S47I、H93G)的变体。在一些实施方案中,PM2包含序列FNLPPGNYKKPKLLYCSNGGHFLRILPDGTVDGTRDRSDPHIQLQLIAESVGEVYIKSTETGQYLAMDTDGLLYGSQTPNEECLFLERLEENGYNTYISKKHAEKNWFVGLKKNGSCKRGPRTHYGQKAILFLPLPVSSD(SEQ ID NO:3)。
在一些实施方案中,变体FGF1是被称为PM3(D28N、Q40P、S47I、H93G、L131R)的变体。在一些实施方案中,PM3包含序列FNLPPGNYKKPKLLYCSNGGHFLRILPNGTVDGTRDRSDPHIQLQLIAESVGEVYIKSTETGQYLAMDTDGLLYGSQTPNEECLFLERLEENGYNTYISKKHAEKNWFVGLKKNGSCKRGPRTHYGQKAIRFLPLPVSSD(SEQ ID NO:4)。
在一些实施方案中,变体FGF1包含序列FNLPPGNYKKPKLLYCSNGGHFLRILPDGTVDGTRDRSDQHIQLQLSAESVGEVYIKSTETGQYLAMDTDGLLYGSQTPNEECLFLERLEENHYNTYISKKHAEKNWFVGLKKNGSCKRGPRTHYGQKAIRFLPLPVSSD(SEQ ID NO:5)。
在一些实施方案中,变体FGF1包含序列FNLPPGNYKKPKLLYCSNGGHFLRILPDGTVDGTRDRSDQHIQLQLSAESVGEVYIKSTETGQYLAMDTDGLLYGSQTPNEECLFLERLEENHYNTYISKKHAEKNWFVGLKKNGSCKRGPRTHYGQKAIKFLPLPVSSD(SEQ ID NO:6)。
在一些实施方案中,变体是分离的变体。在一些实施方案中,变体表现出本多肽中不存在的至少一种所需特征。示例性特征包括但不限于蛋白水解稳定性的提高、热稳定性的提高、构象柔韧性的提高或降低以及拮抗活性的提高。如本领域技术人员将理解的,变体可以表现出这些改善特征中的两个或更多个的任何组合。
在一些实施方案中,变体FGF1是FGFR受体的拮抗剂。在一些实施方案中,FGF1变体具有选自由以下组成的组的序列:SEQ ID NO:2、SEQ ID NO:3、SEQ ID NO:4、SEQ ID NO:5、SEQ ID NO:6。
在一些实施方案中,生长因子变体与亲本多肽具有至少为约80%、至少约85%、至少约90%、至少约95%或至少约96%、97%、98%或99%的序列同一性。在一些实施方案中,本发明的生长因子变体与亲本多肽具有至少约99.2%、至少约99.4%、至少约99.6%或至少约99.8%的序列同一性。
在一些实施方案中,FGF1变体与亲本多肽具有至少为约80%、至少约85%、至少约90%、至少约95%或至少约96%、97%、98%或99%的序列同一性。在一些实施方案中,本发明的FGF1变体与亲本多肽具有至少约99.2%、至少约99.4%、至少约99.6%或至少约99.8%的序列同一性。
在一些实施方案中,SEQ ID NO:1中突变的位置包括28、40、47、93或131中的一个或多个。如本领域技术人员将意识到的,这些位置的任何组合都可以突变。
在一些实施方案中,与野生型FGF1(例如,SEQ ID NO:相比,亲本多肽在位置28处的氨基酸改变为N
在一些实施方案中,亲本多肽在位置40处的氨基酸改变为P。
在一些实施方案中,亲本多肽在位置47处的氨基酸改变为I。
在一些实施方案中,亲本多肽在位置93处的氨基酸改变为G。
在一些实施方案中,亲本多肽在位置131处的氨基酸改变为R。在一些实施方案中,亲本多肽在位置131处的氨基酸改变为K。
a.
本发明提供了本发明的变体与一种或多种缀合配偶体的缀合物。示例性缀合配偶体包括聚合物、靶向剂、治疗剂、细胞毒性剂、螯合剂和可检测剂。本领域技术人员将认识到,这些非限制性剂类别之间存在重叠。
缀合配偶体或“修饰基团”可以是任何可缀合部分。示例性修饰基团在下面讨论。可以根据其改变给定多肽的性质(例如,生物或物理化学性质)的能力来选择修饰基团。可以通过使用修饰基团来改变的示例性多肽性质包括但不限于药代动力学、药效学、代谢稳定性、生物分布、水溶性、亲脂性、组织靶向能力和治疗活性谱。修饰基团可用于修饰在诊断应用或体外生物测定系统中使用的多肽。
在一些实施方案中,包括例如本文所述的FGF1变体的生长因子变体与Fc部分组合。Fc部分可衍生自人或动物免疫球蛋白(Ig),优选为IgG。IgG可以是IgG1、IgG2、IgG3或IgG4(参见例如图34)。还优选地,Fc部分衍生自免疫球蛋白优选IgG的重链。更优选地,Fc部分包含免疫球蛋白重链恒定区的一部分,诸如结构域。这种Ig恒定区优选包含至少一个选自铰链、CH2、CH3结构域中的任一个或其任何组合的Ig恒定结构域。在一些实施方案中,Fc部分至少包含CH2和CH3结构域。进一步优选地,Fc部分包含IgG铰链区、CH2和CH3结构域。
表1:示例性IgG序列:
IgG1亚类的Fc结构域通常用作Fc部分,因为IgG1具有任何血清蛋白中最长的血清半衰期。较长的血清半衰期可能是动物研究和潜在的人类治疗用途所需的蛋白质特征。另外,IgG1亚类具有最强的执行抗体介导的效应子功能的能力。
在融合蛋白中最有用的主要效应子功能是IgG1抗体介导抗体依赖性细胞毒性的能力。另一方面,对于主要起拮抗剂作用的融合蛋白,这可能是不希望的功能。已经鉴定了几个对IgG1亚类中抗体恒定区介导的活性重要的特定氨基酸残基。因此,包含或排除这些特定氨基酸允许包含或排除特定免疫球蛋白恒定区介导的活性。
根据本发明,还可以修饰Fc部分以调节效应子功能。例如,如果Fc部分来源于IgG1,则根据EU索引位置(Kabat等人,1991)可以引入以下Fc突变:T250Q/M428L;M252Y/S254T/T256E+H433K/N434F;E233P/L234V/L235A/AΔ236+A327G/A330S/P331S;E333A;K322A。
其它Fc突变可以是例如在选自330、331 234或235或其组合的EU索引位置处的取代。在本发明的上下文中,也可以将位于CH2结构域中EU索引位置297处的氨基酸取代引入Fc部分,从而消除N-连接的碳水化合物连接的潜在位点。EU索引位置220处的半胱氨酸残基也可被替换。
本发明的Fc融合蛋白可以是单体或二聚体。Fc融合蛋白也可以是“伪二聚体”,其含有二聚体Fc部分(例如,两个二硫桥接的铰链-CH2-CH3构建体的二聚体),其中仅一个与治疗部分融合。
Fc融合蛋白可以是含有两个不同治疗部分的异二聚体,或含有单个治疗部分的两个拷贝的同二聚体。
在一些实施方案中,如本文所述的生长因子变体(包括例如FGF1变体)的体内半衰期可以用聚乙二醇(PEG)部分延长。用PEG对多肽进行化学修饰(PEG化)会增加其分子大小,且通常降低表面和官能团的可及性,其各自取决于与多肽连接的PEG部分的数量和大小。此修饰经常导致血浆半衰期和蛋白水解稳定性的改善,以及免疫原性和肝吸收的降低(Chaffee等人J.Clin.Invest.89:1643-1651(1992);Pyatak等人Res.Commun.Chem.PatholPharmacol.29:113-127(1980))。例如,据报道,白介素-2的PEG化增强其体内抗肿瘤能力(Katre等人Proc.Natl.Acad.Sci.USA.84:1487-1491(1987)),并且来源于单克隆抗体A7的F(ab’)2的PEG化改善了其肿瘤定位(Kitamura等人Biochem.Biophys.Res.Commun.28:1387-1394(1990))。因此,在另一个实施方案中,通过本发明方法用PEG部分衍生的多肽的体内半衰期相对于非衍生化的亲本多肽的体内半衰期是增加的。
多肽体内半衰期的增加最好表示为相对于亲本多肽的百分比增加范围。百分比增加范围的下限为约40%、约60%、约80%、约100%、约150%或约200%。范围的上限为约60%、约80%、约100%、约150%或大于约250%。
许多水溶性聚合物是本领域技术人员已知的,并且可用于实施本发明。术语水溶性聚合物包括以下种类诸如糖类(例如葡聚糖、直链淀粉、透明质酸、聚(唾液酸)、乙酰肝素、肝素等);聚(氨基酸),例如聚(天冬氨酸)和聚(谷氨酸);核酸;合成聚合物(例如,聚(丙烯酸)、聚(醚),例如聚(乙二醇);肽、蛋白质等。本发明可以用任何水溶性聚合物实施,唯一的限制是该聚合物必须包括可以连接缀合物的其余部分的点。参见,例如Harris,Macronol.Chem.Phys.C25:325-373(1985);Scouten,Methods in Enzymology 135:30-65(1987);Wong等人,Enzyme Microb.Technol.14:866-874(1992);Delgado等人,CriticalReviews in Therapeutic Drug Carrier Systems 9:249-304(1992);Zalipsky,Bioconjugate Chem.6:150-165(1995);以及Bhadra,等人,Pharmazie,57:5-29(2002)。
在另一个实施方案中,类似于以上讨论的那些,改性糖包括水不溶性聚合物,而不是水溶性聚合物。本发明的缀合物还可以包括一种或多种水不溶性聚合物。通过使用缀合物作为媒介物与治疗性多肽一起以受控方式递送来说明本发明的此实施方案。聚合药物递送系统是本领域已知的。参见,例如Dunn等人,编著Polymeric Drugs And Drug DeliverySystems,ACS Symposium Series第469卷,American Chemical Society,Washington,D.C.1991。本领域技术人员将理解,大体上任何已知的药物递送系统均适用于本发明的缀合物。
代表性水不溶性聚合物包括但不限于聚膦嗪、聚(乙烯醇)、聚酰胺、聚碳酸酯、聚亚烷基、聚丙烯酰胺、聚烷二醇、聚环氧烷、聚对苯二甲酸亚烷基酯、聚乙烯醚、聚乙烯酯、聚卤乙烯、聚乙烯吡咯烷酮、聚乙交酯、聚硅氧烷、聚氨酯、聚(甲基丙烯酸甲酯)、聚(甲基丙烯酸乙酯)、聚(甲基丙烯酸丁酯)、聚(甲基丙烯酸异丁酯)、聚(甲基丙烯酸己酯)、聚(甲基丙烯酸异癸酯)、聚(甲基丙烯酸月桂酯)、聚(甲基丙烯酸苯酯)、聚(丙烯酸甲酯)、聚(丙烯酸异丙酯)、聚(丙烯酸异丁酯)、聚(丙烯酸十八烷酯)、聚乙烯、聚丙烯、聚(乙二醇)、聚(环氧乙烷)、聚(对苯二甲酸乙二酯)、聚(乙酸乙烯酯)、聚氯乙烯、聚苯乙烯、聚乙烯吡咯烷酮、普朗尼克和聚乙烯苯酚及其共聚物。
用于本发明的缀合物中的代表性可生物降解的聚合物包括但不限于聚乳酸、聚乙交酯及其共聚物、聚(对苯二甲酸乙二酯)、聚(丁酸)、聚(戊酸)、聚(丙交酯-共-己内酯)、聚(丙交酯-共-乙交酯)、聚酸酐、聚原酸酯、其共混物和共聚物。形成凝胶的组合物特别有用,诸如包括胶原蛋白、普朗尼克等的那些凝胶。
示例性可吸收聚合物包括例如合成产生的聚(α-羟基-羧酸)/聚(氧化烯的可吸收嵌段共聚物(参见,Cohn等人,美国专利号4,826,945)。这些共聚物没有交联并且是水溶性的,因此主体可以排泄降解的嵌段共聚物组合物。参见,Younes等人.,JBiomed.Mater.Res.21:1301-1316(1987);以及Cohn等人,J Biomed.Mater.Res.22:993-1009(1988)。
作为水凝胶组分的聚合物也可用于本发明中。水凝胶是能够吸收相对大量水的聚合材料。形成水凝胶的化合物的实例包括但不限于聚丙烯酸、羧甲基纤维素钠、聚乙烯醇、聚乙烯吡咯烷、明胶、角叉菜胶和其它多糖、羟乙烯甲基丙烯酸(HEMA)及其衍生物等。可以产生稳定、可生物降解和可生物吸收的水凝胶。此外,水凝胶组合物可包含表现出这些性质中的一种或多种的亚基。
在另一个实施方案中,凝胶是热可逆凝胶。目前优选的是包括以下组分的热可逆凝胶:诸如普朗尼克、胶原蛋白、明胶、透明质酸、多糖、聚氨酯水凝胶、聚氨酯-脲水凝胶及其组合。
在仍另一个示例性实施方案中,本发明的缀合物包括脂质体的组分。脂质体可以根据例如如1985年6月11日颁布的Eppstein等人的美国专利4,522,811中所述的本领域技术人员已知的方法来制备。例如,脂质体制剂可通过将适当的脂质(诸如硬脂酰磷脂酰乙醇胺、硬脂酰磷脂酰胆碱、花生四烯酰磷脂酰胆碱和胆固醇)溶解于无机溶剂中来制备,然后将所述无机溶剂蒸发,留下干脂质薄膜在容器的表面上。随后将活性化合物或其药学上可接受的盐的水溶液引入容器中。然后用手旋转容器以从容器的侧面上释放脂质物质并分散脂质聚集体,从而形成脂质体悬浮液。
本发明还提供了类似于上述缀合物的缀合物,其中将多肽缀合至治疗部分、诊断部分、靶向部分、毒素部分等。上述每个部分可以是小分子、天然聚合物(例如多肽)或合成聚合物。
在各种实施方案中,变体缀合至基质的组分以用于组织再生。示例性基质是本领域已知的,并且本领域技术人员能够选择和修饰与本发明的生长因子变体(包括例如FGF1变体)一起使用的合适基质。本发明的生长因子变体(包括例如FGF1变体)通常用于再生医学应用,包括例如眼、肝、肌肉、神经和心脏组织的再生。
在一些实施方案中,本发明提供了由于存在靶向剂作为缀合物的组分而选择性地定位在特定组织中的缀合物。在一个示例性实施方案中,靶向剂是蛋白质。示例性蛋白质包括转铁蛋白(脑、血池)、HS-糖蛋白(骨、脑、血池)、抗体(脑、具有抗体特异性抗原的组织、血池)、凝血因子V-XII(受损的组织、血块、癌症、血池)、血清蛋白例如α-酸性糖蛋白、胎球蛋白、α-胎蛋白(脑、血池)、β2-糖蛋白(肝、动脉粥样硬化斑块、脑、血池)、G-CSF、GM-CSF、M-CSF和EPO(免疫刺激、癌症、血池、红细胞生产过剩、神经保护)、白蛋白(半衰期延长)、IL-2和IFN-α。
在另一个实施方案中,本发明提供了介于本发明的生长因子变体(包括例如FGF1变体)与治疗部分之间的缀合物。可用于实施本发明的治疗部分包括来自具有多种药理活性的广泛药物类别的药物。将治疗剂和诊断剂缀合到各种其它物质上的方法是本领域技术人员众所周知的。参见,例如,Hermanson,Bioconjugate Techniques,Academic Press,SanDiego,1996;以及Dunn等人,编著Polymeric Drugs And Drug Delivery Systems,ACSSymposium Series第469卷,American Chemical Society,Washington,D.C.1991。
有用的治疗部分的类别包括例如抗肿瘤药(例如抗雄激素(例如亮丙瑞林或氟他胺)、杀细胞剂(例如多柔比星、阿霉素、紫杉醇、环磷酰胺、白消安、顺铂、β-2-干扰素)抗雌激素(例如他莫昔芬)、抗代谢物(例如氟尿嘧啶、甲氨蝶呤、巯基嘌呤、硫代鸟嘌呤)。在此类别中还包括用于诊断和治疗的基于放射性同位素的剂,以及缀合毒素,诸如蓖麻毒素、格尔德霉素、丹参素(mytansin)、CC-1065、杜卡霉素、奇霉素(Chlicheamycin)及其相关结构和类似物。
治疗部分也可以是激素(例如,甲羟孕酮、雌二醇、亮丙瑞林、孕酮、奥曲肽或生长抑素);内分泌调节药物(例如,避孕药(例如,乙二醇、乙炔雌二醇、炔诺酮、美雌醇、去氧孕烯、甲羟孕酮)。在本发明的各种实施方案中使用的是与以下各物的缀合物:雌激素(例如,己烯雌酚)、糖皮质激素(例如,曲安西龙、倍他米松等)和孕激素诸如炔诺酮、炔诺醇、炔诺酮、左炔诺孕酮;甲状腺剂(例如碘塞罗宁或左甲状腺素)或抗甲状腺剂(例如甲巯咪唑);抗高泌乳素血症药(例如卡麦角林);激素抑制剂(例如达那唑或戈舍瑞林);催产药(例如甲基麦角新碱或催产素)和前列腺素诸如米洛前列醇、前列地尔或地诺前列酮。
其它有用的修饰基团包括免疫调节药(例如抗组胺药、肥大细胞稳定剂诸如洛多沙胺和/或色甘酸、类固醇(例如曲安西龙、倍氯米松、可的松、地塞米松、泼尼松龙、甲基泼尼松龙、倍氯米松或氯倍他索)、组胺H2拮抗剂(例如法莫替丁、西咪替丁、雷尼替丁)、免疫抑制剂(例如硫唑嘌呤、环孢菌素)等。也可以使用具有抗炎活性的基团,诸如舒林酸、依托度酸、酮洛芬和酮咯酸。与本发明结合使用的其它药物对于本领域技术人员将是显而易见的。
在一些实施方案中,通过反应性氨基酸与反应性氨基酸的反应性缀合配偶体之间的反应形成缀合物。反应性氨基酸和反应性缀合配偶体在它们的框架内都包括一个或多个反应性官能团。两个结合物质之一可以包括“离去基团”(或活化基团),指的是那些在酶调节的亲核取代反应中容易置换的部分,或者在利用亲核反应配偶体的化学反应中被替换的部分(例如,带有巯基的氨基酸部分)。为每种类型的反应选择合适的离去基团在本领域技术人员的能力范围内。许多活化的糖是本领域已知的。参见,例如Vocadlo等人,Carbohydrate Chemistry and Biology,第2卷,Ernst等人编著,Wiley-VCH Verlag:Weinheim,Germany,2000;Kodama等人,Tetrahedron Lett.34:6419(1993);Lougheed,等人,J.Biol.Chem.274:37717(1999))。
在各种实施方案中,氨基酸取代是天然存在的FGF1的变体(或变体),它是缀合配偶体例如侧链氨基酸(例如半胱氨酸、赖氨酸、丝氨酸等)的连接点。
在实施本发明中有用的反应性基团和反应类别通常是生物缀合物化学领域中众所周知的那些。目前优选的具有反应性糖部分的反应类别是那些在相对温和条件下进行的反应。这些包括但不限于亲核取代(例如,胺和醇与酰基卤、活性酯的反应)、亲电子取代(例如,烯胺反应)以及碳-碳和碳-杂原子多键的加成(例如,迈克尔(Michael)反应、狄尔斯-阿尔德(Diels-Alder)加成)。在例如March,Advanced Organic Chemistry,第3版,JohnWiley&Sons,New York,1985;Hermanson,Bioconjugate Techniques,Academic Press,SanDiego,1996;以及Feeney等人,Modification of Proteins;Advances in ChemistrySeries,第198卷,American Chemical Society,Washington,D.C.,1982中讨论了这些和其它有用的反应。
b.
反应性氨基酸或反应性缀合配偶体上有用的反应性官能团包括但不限于:
(a)羧基及其各种衍生物,包括但不限于N-羟基琥珀酰亚胺酯、N-羟基苯并三唑酯、酰基卤、酰基咪唑、硫代酯、对硝基苯基酯、烷基、烯基、炔基和芳族酯;
(b)羟基,其可被转化为例如酯、醚、醛等
(c)卤代烷基,其中卤化物稍后可被亲核基团置换,所述亲核基团例如为胺、羧酸根阴离子、硫醇阴离子、碳负离子或烷氧基离子,从而造成新基团在卤素原子的官能团处的共价连接;
(d)能够参与狄尔斯-阿尔德反应的亲双烯基团,例如马来酰亚胺基;
(e)醛基或酮基,这样可能通过形成羰基衍生物(例如亚胺、腙、缩氨基脲或肟)或通过诸如格氏加成或烷基锂加成的机制进行后续衍生化;
(f)磺酰卤基团,其随后与胺反应,例如形成磺酰胺;
(g)硫醇基团,其可以例如被转化为二硫化物或与酰基卤反应;
(h)胺基或巯基,其可以例如被酰化、烷基化或氧化;
(i)烯烃,其可以经历例如环加成、酰化、迈克尔加成等;以及
(j)环氧化物,其可以与例如胺和羟基化合物反应。
可以选择反应性官能团,使得它们不参与或干扰组装反应性糖核或修饰基团所必需的反应。或者,可以通过存在保护基来保护反应性官能团免于参与反应。本领域技术人员理解如何保护特定的官能团,以使其不干扰所选的一组反应条件。对于有用的保护基的实例,参见,例如Greene等人,Protective Groups in Organic Synthesis,John Wiley&Sons,New York,1991。
连接多肽与缀合配偶体的基团也可以是交联基团,例如零阶或更高阶的交联基团(关于交联试剂和交联程序的综述,参见:Wold,F.,Meth.Enzymol.25:623-651,1972;Weetall,H.H.和Cooney,D.A.,Enzymes as Drugs.(Holcenberg和Roberts编著)第395-442页,Wiley,New York,1981;Ji,T.H.,Meth.Enzymol.91:580-609,1983;Mattson等人,Mol.Biol.Rep.17:167-183,1993,这些文献都以引用的方式并入本文)。优选的交联试剂衍生自各种零长度、均双功能和杂双功能的交联试剂。零长度的交联试剂包括两个内在化学基团的直接缀合,而无需引入外在材料。催化二硫键形成的剂属于这一类。另一个实例是诱导羧基与伯氨基缩合以形成酰胺键的试剂,诸如碳二亚胺、氯甲酸乙酯、伍德沃德试剂K(2-乙基-5-苯基异噁唑-3'-磺酸盐)和羰基二咪唑。除这些化学试剂外,还可以使用转谷氨酰胺酶(谷氨酰肽γ-谷氨酰转移酶;EC 2.3.2.13)作为零长度的交联试剂。此酶催化与蛋白质结合的谷氨酰胺残基的羧酰胺基团上的酰基转移反应,通常以伯氨基为底物。优选的均双功能和异双功能试剂分别含有两个相同或两个不同的位点,它们可对氨基、巯基、胍基、吲哚或非特异性基团有反应性。
与本发明的多肽连接的示例性缀合配偶体包括但不限于PEG衍生物(例如,烷基-PEG、酰基-PEG、酰基-烷基-PEG、烷基-酰基-PEG氨基甲酰基-PEG、芳基-PEG)、PPG衍生物(例如,烷基-PPG、酰基-PPG、酰基-烷基-PPG、烷基-酰基-PPG氨基甲酰基-PPG、芳基-PPG)、治疗部分、诊断部分、甘露糖-6-磷酸、肝素、乙酰肝素、Sle
除共价连接外,本发明的生长因子变体(包括例如FGF1变体)可通过非共价相互作用连接到生物材料的表面上。非共价蛋白掺入可以例如通过包囊或吸收来完成。本发明的多肽与生物材料的连接可以通过肝素介导。在一些实施方案中,本发明的多肽连接到肝素-藻酸盐聚合物和藻酸盐上,如Harada等人,J.Clin.Invest.(1994)94:623-630;Laham等人,Circulation(1999)1865-1871及其中引用的参考文献中所述。在其它实施方案中,本发明的多肽连接到基于胶原蛋白的生物材料上。
c.
本发明的示例性缀合物是包含本发明的变体和可检测部分的显像剂,所述可检测部分在成像模式中可检测。迫切需要分子成像探针,该探针将特异性靶向活体受试者中的Met受体,并允许对患者定制的癌症治疗和疾病管理的肿瘤非侵入性表征。通过非侵入性成像检测表达Met的肿瘤的能力也可以作为转移风险的指标。
本发明的缀合物可利用的示例性成像模式包括但不限于正电子发射断层扫描(PET),其中本发明的变体被正电子发射同位素标记。典型的同位素包括
在一个示例性实施方案中,缀合配偶体通过在选定条件下切割的键被连接到本发明的多肽变体上。示例性条件包括但不限于选定的pH(例如,胃、肠、胞吞液泡)、活性酶(例如,酯酶、还原酶、氧化酶)的存在、光、热等。许多可切割基团是本领域已知的。参见,例如,Jung等人,Biochem.Biophys.Acta,761:152-162(1983);Joshi等人,J.Biol.Chem.,265:14518-14525(1990);Zarling等人,J.Immunol.,124:913-920(1980);Bouizar等人,Eur.J.Biochem.,155:141-147(1986);Park等人,J.Biol.Chem.,261:205-210(1986);Browning等人,J.Immunol.,143:1859-1867(1989)。
IV.
本发明的生长因子变体(包括例如FGF1变体)及其缀合物具有广泛的药物应用。
因此,另一方面,本发明提供了一种药物组合物,其包含至少一种本发明的多肽或多肽缀合物以及药学上可接受的稀释剂、载剂、媒介物、添加剂或其组合。本发明的药物组合物适用于多种药物递送系统。用于本发明的合适的制剂见于Remington’sPharmaceutical Sciences,Mace Publishing Company,Philadelphia,PA,第17版(1985)中。关于药物递送方法的简要综述,参见Langer,Science 249:1527-1533(1990)。
可以将药物组合物配制成用于任何合适的施用方式,包括例如局部、经口、鼻、静脉内、颅内、腹膜内、皮下或肌内施用。对于肠胃外施用(诸如皮下注射),载剂优选包含水、盐水、乙醇、脂肪、蜡或缓冲剂。对于经口施用,可以使用任何上述载剂或固体载剂,诸如甘露醇、乳糖、淀粉、硬脂酸镁、糖精钠、滑石粉、纤维素、葡萄糖、蔗糖和碳酸镁。也可将生物可降解的基质诸如微球(例如聚乳酸聚乙醇酸)用作本发明的药物组合物的载剂。合适的生物可降解的微球公开于例如美国专利号4,897,268和5,075,109中。
通常,药物组合物被皮下或肠胃外例如静脉内施用。因此,本发明提供了用于肠胃外施用的组合物,其包含溶解或悬浮在可接受的载剂中的化合物,所述载剂优选为水性载剂,例如水、缓冲水、盐水、PBS等。组合物还可包含去污剂,诸如Tween 20和Tween 80;稳定剂,诸如甘露醇、山梨醇、蔗糖和海藻糖;以及防腐剂,诸如EDTA和间甲酚。组合物可含有接近生理条件所需的药学上可接受的辅助物质,诸如pH调节剂和缓冲剂、张力调节剂、润湿剂、去污剂等。
这些组合物可以通过常规的灭菌技术进行灭菌,或可被无菌过滤。所得水溶液可按原样或以冻干形式被包装使用,冻干的制剂在施用之前与无菌水性载剂合并。制剂的pH通常介于3与11之间,更优选5至9且最优选7至8。
在一些实施方案中,可以将本发明的糖肽掺入由标准囊泡形成脂质形成的脂质体中。多种方法可用于制备脂质体,如在例如Szoka等人,Ann.Rev.Biophys.Bioeng.9:467(1980);美国专利号4,235,871、4,501,728和4,837,028中所述。使用多种靶向剂(例如,本发明的唾液酸半乳糖苷)靶向脂质体是本领域众所周知的(参见,例如,美国专利号4,957,773和4,603,044)。
可以使用将靶向剂偶联至脂质体的标准方法。这些方法通常涉及将脂质组分诸如磷脂酰乙醇胺(其可被活化以连接靶向剂)或衍生的亲脂性化合物诸如本发明的脂质衍生的糖肽掺入到脂质体中。
靶向机制通常需要将靶向剂以这样的方式放置在脂质体的表面上,使得靶部分可用于与靶(例如细胞表面受体)相互作用。在使用本领域技术人员已知的方法形成脂质体之前,本发明的碳水化合物可以连接到脂质分子上(例如,碳水化合物上存在的羟基分别被长链烷基卤或脂肪酸烷基化或酰化)。
或者,脂质体可以这样的方式形成,使得在形成膜时首先将连接部分掺入到膜中。连接部分必须具有亲脂性部分,该部分被牢固地嵌入和锚定在膜中。它还必须具有反应性部分,该部分在脂质体的水性表面上化学可及。选择反应性部分,使得其在化学上适合与稍后被添加的靶向剂或碳水化合物形成稳定的化学键。在一些实施方案中,可能将靶剂直接连接到连接分子上,但是在大多数情况下,更适合使用第三分子充当化学桥,从而将膜中的连接分子与靶剂或碳水化合物连接,这些靶剂或碳水化合物在三维方向上从囊泡表面延伸出来。
通过本发明的方法制备的生长因子变体(包括例如FGF1变体)也可以用作诊断试剂。例如,标记的化合物可用于在疑似患有炎症的患者中定位炎症或肿瘤转移的区域。为此用途,可以用
V.
在一些实施方案中,本发明提供了一种分离的核酸,其编码根据上文阐述的任何实施方案的生长因子变体,包括例如FGF1变体。在一些实施方案中,本发明提供了一种与此核酸互补的核酸。
在一些实施方案中,本发明提供了一种表达载体,其包含与启动子可操作连接的编码根据上文阐述的任何实施方案的多肽变体的核酸。
VI.
在各种实施方案中,还提供了生长因子变体多肽(包括例如FGF1变体多肽)的文库,其包含多个不同成员,其中文库的每个成员对应于共同的亲本长因子多肽或FGF1亲本多肽,且其中文库的每个成员在某位置处包含氨基酸,在亲本多肽的该位置处未见到所述氨基酸。
a.
为了生成FGF1或其它生长因子的随机文库,制备编码各种FGF1或其它生长因子序列的寡核苷酸。以合成方式或通过标准重组技术制备用于在酵母中表达生长因子变体多肽(包括例如FGF1变体多肽)的DNA。在要改变氨基酸的情况下,针对给定位置合成了二十个不同的密码子,每个密码子编码一个不同的氨基酸。已经使用随机化的寡核苷酸合成来创建其中约5至约15个氨基酸被随机化的编码盒(参见例如Burritt等人,(1996)Anal.Biochem.238:113;Lowman(1997)Annu.Rev.Biophys.Biomol.Struct.26:410 24;Wilson(1998)Can.J.Microbiol.44:313 329)。
通常用于改良突变体的进化的酵母展示载体被称为“pCT”。在Wittrup等人于2004年7月29日公开的名称为“Yeast cell surface display of proteins and usesthereof”的US 2004/0146976中进一步描述了载体。如其中所述,载体提供了目标多肽的N末端与酵母Aga2p细胞壁蛋白的C末端的遗传融合体。每个酵母细胞的外壁可展示约10
在本发明的一些实施方案中,通常用于工程化高亲和力结合物的酵母展示平台也被用于工程化具有更高蛋白水解稳定性的蛋白质(参见例如图1)。在一些实施方案中,单个生长因子变体的数千个拷贝作为束缚融合体展示在酵母表面上。在一些实施方案中,血凝素(HA)标签在生长因子的上游表达,而c-myc标签在生长因子的下游表达。在一些实施方案中,细胞可以与相应受体的可溶性Fc融合体一起孵育,所述融合体可以与酵母展示的生长因子结合。
在一些实施方案中,将酵母展示平台与流激活细胞分选(FACS)组合以工程化具有更高蛋白水解稳定性的生长因子(参见,例如图2)。在一些实施方案中,可以通过随机诱变、定向诱变或DNA改组或如上所述或本领域已知的其它重组技术来生成生长因子突变体的文库。在一些实施方案中,将酵母细胞文库与目标蛋白酶一起孵育,在此期间发生酵母表面展示的蛋白质的切割。在一些实施方案中,具有更高蛋白水解稳定性的生长因子突变体对酵母细胞表面上的切割更具抗性。在一些实施方案中,在蛋白酶孵育之后,将细胞洗涤并与功能性受体的可溶性Fc融合体一起孵育,所述功能性受体与保留有受体结合亲和力的正确折叠的生长因子突变体结合。在一些实施方案中,FACS用于分选正确折叠的、未切割的生长因子突变体,将其扩增并诱导用于下一轮分选。
在一些实施方案中,针对Fc结构域、c-myc结构域和HA标签的荧光抗体标记物被用于测量受体结合、生长因子特异性切割和非特异性切割(参见,例如,下表2)。在一些实施方案中,结合的Fc融合受体的检测可以确认生长因子的突变不会严重降低对该受体的结合亲和力或导致不正确的蛋白质折叠。在一些实施方案中,生长因子特异性切割是生长因子蛋白水解稳定性的直接量度。在一些实施方案中,通过c-myc信号检测生长因子特异性切割,因为切割的生长因子将去除C-末端c-myc标签。在一些实施方案中,当蛋白酶在酵母表面展示蛋白(例如酵母展示蛋白Aga1p和Aga2p)内切割时,发生非特异性切割。在一些实施方案中,在非特异性切割期间,所有三种标记物的荧光信号均降低。在一些实施方案中,这是不希望出现的,因为用于检测生长因子切割和结合活性的动态范围缩小。在一些实施方案中,HA信号被用于确保目标蛋白酶的非特异性切割最小。
表2:不同事件对由荧光抗体标记物观察到的信号的影响。
在一些实施方案中,可以将野生型生长因子及其变体克隆到pCT载体中。在一些实施方案中,野生型生长因子及其变体可以作为与Aga2p交配蛋白的融合体在酿酒酵母的酵母细胞表面上表达。在一些实施方案中,野生型生长因子及其变体在酵母细胞表面上的成功表达可以通过检测蛋白质的C-末端上的c-myc标签来证实。在一些实施方案中,可以通过测量对野生型生长因子-Fc的特异性结合活性来确认酵母展示的野生型生长因子及其变体的正确折叠。
在一些实施方案中,SEQ ID NO:1的FGF1多肽被用作模型来说明蛋白水解稳定性筛选的建立。在一些实施方案中,将野生型FGF1克隆到pCT载体中。在一些实施方案中,此FGF1多肽及其FGF1变体可以作为与Aga2p交配蛋白的融合体在酿酒酵母的酵母细胞表面上表达(参见,例如图3A)。在一些实施方案中,可以通过检测蛋白质的C末端上的c-myc标签来确认FGF1在酵母细胞表面上的成功表达(参见,例如图3B)。在一些实施方案中,可以通过测量对FGFR1-Fc的特异性结合活性来确认酵母展示的FGF的正确折叠(参见,例如图3C)。
在一些实施方案中,血清、胰蛋白酶、胰凝乳蛋白酶和纤溶酶可用于开发针对生长因子变体多肽(包括例如FGF1变体多肽)的蛋白水解稳定性筛选。在一些实施方案中,这些蛋白酶基于它们与生长因子变体多肽(包括例如FGF1变体多肽)的科学和生物学相关性来选择。在一些实施方案中,蛋白酶对筛选的适用性是通过其以合理的速率切割生长因子的能力以及酵母展示蛋白的最小非特异性切割来确定的。在一些实施方案中,血清可用于开发针对生长因子变体多肽(包括例如FGF1变体多肽)的蛋白水解稳定性筛选。在一些实施方案中,胰蛋白酶可用于开发针对生长因子变体多肽(包括例如FGF1变体多肽)的蛋白水解稳定性筛选。在一些实施方案中,胰凝乳蛋白酶可用于开发针对生长因子变体多肽(包括例如FGF1变体多肽)的蛋白水解稳定性筛选。在一些实施方案中,纤溶酶可用于开发针对生长因子变体多肽(包括例如FGF1变体多肽)的蛋白水解稳定性筛选。
在一些实施方案中,通过比较野生型生长因子的蛋白水解切割与变体生长因子的蛋白水解切割来确定稳定性。在一些实施方案中,通过比较野生型FGF1的蛋白水解切割与FGF1变体的蛋白水解切割来确定稳定性。
在一些实施方案中,生长因子变体的稳定性与野生型生长因子相比提高了至少5%至至少95%。在一些实施方案中,生长因子变体的稳定性与野生型生长因子相比提高了至少10%至至少90%。在一些实施方案中,生长因子变体的稳定性与野生型生长因子相比提高了至少5%至至少90%。在一些实施方案中,生长因子变体的稳定性与野生型生长因子相比提高了至少5%至至少85%。在一些实施方案中,生长因子变体的稳定性与野生型生长因子相比提高了至少5%至至少80%。在一些实施方案中,生长因子变体的稳定性与野生型生长因子相比提高了至少5%至至少75%。在一些实施方案中,生长因子变体的稳定性与野生型生长因子相比提高了至少5%至至少70%。在一些实施方案中,生长因子变体的稳定性与野生型生长因子相比提高了至少10%至至少70%。在一些实施方案中,生长因子变体的稳定性与野生型生长因子相比提高了至少5%。在一些实施方案中,生长因子变体的稳定性与野生型生长因子相比提高了至少10%。在一些实施方案中,生长因子变体的稳定性与野生型生长因子相比提高了至少15%。在一些实施方案中,生长因子变体的稳定性与野生型生长因子相比提高了至少20%。在一些实施方案中,生长因子变体的稳定性与野生型生长因子相比提高了至少25%。在一些实施方案中,生长因子变体的稳定性与野生型生长因子相比提高了至少30%。在一些实施方案中,生长因子变体的稳定性与野生型生长因子相比提高了至少35%。在一些实施方案中,生长因子变体的稳定性与野生型生长因子相比提高了至少40%。在一些实施方案中,生长因子变体的稳定性与野生型生长因子相比提高了至少45%。在一些实施方案中,生长因子变体的稳定性与野生型生长因子相比提高了至少50%。在一些实施方案中,生长因子变体的稳定性与野生型生长因子相比提高了至少5%。在一些实施方案中,生长因子变体的稳定性与野生型生长因子相比提高了至少60%。在一些实施方案中,生长因子变体的稳定性与野生型生长因子相比提高了至少65%。在一些实施方案中,生长因子变体的稳定性与野生型生长因子相比提高了至少70%。在一些实施方案中,生长因子变体的稳定性与野生型生长因子相比提高了至少75%。在一些实施方案中,生长因子变体的稳定性与野生型生长因子相比提高了至少80%。在一些实施方案中,生长因子变体的稳定性与野生型生长因子相比提高了至少85%。在一些实施方案中,生长因子变体的稳定性与野生型生长因子相比提高了至少90%。在一些实施方案中,生长因子变体的稳定性与野生型生长因子相比提高了至少95%。
在一些实施方案中,FGF1变体的稳定性与野生型FGF1相比提高了至少5%至至少95%。在一些实施方案中,FGF1变体的稳定性与野生型FGF1相比提高了至少10%至至少90%。在一些实施方案中,FGF1变体的稳定性与野生型FGF1相比提高了至少5%至至少90%。在一些实施方案中,FGF1变体的稳定性与野生型FGF1相比提高了至少5%至至少85%。在一些实施方案中,FGF1变体的稳定性与野生型FGF1相比提高了至少5%至至少80%。在一些实施方案中,FGF1变体的稳定性与野生型FGF1相比提高了至少5%至至少75%。在一些实施方案中,FGF1变体的稳定性与野生型FGF1相比提高了至少5%至至少70%。在一些实施方案中,FGF1变体的稳定性与野生型FGF1相比提高了至少10%至至少70%。在一些实施方案中,FGF1变体的稳定性与野生型FGF1相比提高了至少5%。在一些实施方案中,FGF1变体的稳定性与野生型FGF1相比提高了至少10%。在一些实施方案中,FGF1变体的稳定性与野生型FGF1相比提高了至少15%。在一些实施方案中,FGF1变体的稳定性与野生型FGF1相比提高了至少20%。在一些实施方案中,FGF1变体的稳定性与野生型FGF1相比提高了至少25%。在一些实施方案中,FGF1变体的稳定性与野生型FGF1相比提高了至少30%。在一些实施方案中,FGF1变体的稳定性与野生型FGF1相比提高了至少35%。在一些实施方案中,FGF1变体的稳定性与野生型FGF1相比提高了至少40%。在一些实施方案中,FGF1变体的稳定性与野生型FGF1相比提高了至少45%。在一些实施方案中,FGF1变体的稳定性与野生型FGF1相比提高了至少50%。在一些实施方案中,FGF1变体的稳定性与野生型FGF1相比提高了至少5%。在一些实施方案中,FGF1变体的稳定性与野生型FGF1相比提高了至少60%。在一些实施方案中,FGF1变体的稳定性与野生型FGF1相比提高了至少65%。在一些实施方案中,FGF1变体的稳定性与野生型FGF1相比提高了至少70%。在一些实施方案中,FGF1变体的稳定性与野生型FGF1相比提高了至少75%。在一些实施方案中,FGF1变体的稳定性与野生型FGF1相比提高了至少80%。在一些实施方案中,FGF1变体的稳定性与野生型FGF1相比提高了至少85%。在一些实施方案中,FGF1变体的稳定性与野生型FGF1相比提高了至少90%。在一些实施方案中,FGF1变体的稳定性与野生型FGF1相比提高了至少95%。
在一些实施方案中,生长因子变体的稳定性与野生型生长因子相比提高了至少1倍至至少10倍。在一些实施方案中,生长因子变体的稳定性与野生型生长因子相比提高了至少1倍。在一些实施方案中,生长因子变体的稳定性与野生型生长因子相比提高了至少2倍。在一些实施方案中,生长因子变体的稳定性与野生型生长因子相比提高了至少3倍。在一些实施方案中,生长因子变体的稳定性与野生型生长因子相比提高了至少4倍。在一些实施方案中,生长因子变体的稳定性与野生型生长因子相比提高了至少5倍。在一些实施方案中,生长因子变体的稳定性与野生型生长因子相比提高了至少6倍。在一些实施方案中,生长因子变体的稳定性与野生型生长因子相比提高了至少7倍。在一些实施方案中,生长因子变体的稳定性与野生型生长因子相比提高了至少8倍。在一些实施方案中,生长因子变体的稳定性与野生型生长因子相比提高了至少9倍。在一些实施方案中,生长因子变体的稳定性与野生型生长因子相比提高了至少10倍。
在一些实施方案中,FGF1变体的稳定性与野生型FGF1相比提高了至少1倍至至少10倍。在一些实施方案中,FGF1变体的稳定性与野生型FGF1相比提高了至少1倍。在一些实施方案中,FGF1变体的稳定性与野生型FGF1相比提高了至少2倍。在一些实施方案中,FGF1变体的稳定性与野生型FGF1相比提高了至少3倍。在一些实施方案中,FGF1变体的稳定性与野生型FGF1相比提高了至少4倍。在一些实施方案中,FGF1变体的稳定性与野生型FGF1相比提高了至少5倍。在一些实施方案中,FGF1变体的稳定性与野生型FGF1相比提高了至少6倍。在一些实施方案中,FGF1变体的稳定性与野生型FGF1相比提高了至少7倍。在一些实施方案中,FGF1变体的稳定性与野生型FGF1相比提高了至少8倍。在一些实施方案中,FGF1变体的稳定性与野生型FGF1相比提高了至少9倍。在一些实施方案中,FGF1变体的稳定性与野生型FGF1相比提高了至少10倍。
b.
在一些实施方案中,筛选可包括使用细胞分选仪。可商购获得的流式细胞仪可以每秒大约50,000个细胞的速率测量在四个或更多个波长下单细胞水平的荧光发射(Ashcroft和Lopez,2000)。可以示出典型的流式细胞仪数据,其中已用两种不同颜色的荧光探针标记酵母以测量蛋白质表达水平和结合的可溶性配体(例如,生长因子受体)。由于每个细胞上蛋白质表达水平的变化,出现了“对角线”细胞群:表达更多蛋白质的细胞将结合更多配体。平衡结合常数(K
可以在FACS Vantage(BD Biosciences)多参数激光流式细胞仪和细胞分选仪上进行细胞分选。在分选之前,如上所述进行荧光染色,从而如上所述检测各种多肽水平的分析。
VII.
a.
可以使用常规的逐步溶液或固相合成来制备本发明的多肽变体(参见,例如,Chemical Approaches to the Synthesis of Peptides and Proteins,Williams等人,编著,1997,CRC Press,Boca Raton Florida及其中引用的参考文献;Solid Phase PeptideSynthesis:A Practical Approach,Atherton&Sheppard编著,1989,IRL Press,Oxford,England及其中引用的参考文献)。
或者,本发明的肽可通过链段缩合来制备,如在例如Liu等人,1996,TetrahedronLett.37(7)933 936;Baca等人,1995,J.Am.Chem.Soc.117:1881-1887;Tam等人,1995,Int.J.Peptide Protein Res.45:209-216;
含有N-和/或C-末端封闭基团的多肽变体可以使用有机化学的标准技术来制备。例如,用于酰化肽的N-末端或酰胺化或酯化肽的C-末端的方法是本领域众所周知的。在N-和/或C-末端进行其它修饰的方式对本领域技术人员将是显而易见的,保护任何侧链官能团的方式对于连接末端封闭基团可为必需的。药学上可接受的盐(抗衡离子)可以通过离子交换色谱法或本领域众所周知的其它方法方便地制备。
呈串联多聚体形式的本发明化合物可通过在合成的适当步骤将接头添加到肽链上来方便地合成。或者,可以合成螺旋链段,并且每个链段都与接头反应。当然,实际的合成方法将取决于接头的组成。合适的保护方案和化学方法是众所周知的,并且对于本领域技术人员而言将是显而易见的。
可以使用在Tam,1988,Proc.Natl.Acad.Sci.USA 85:5409-5413及Demoor等人,1996,Eur.J.Biochem.239:74-84中描述的三聚和四聚树脂及化学方法方便地合成呈支链网络形式的本发明化合物。修饰合成树脂及合成更高或更低阶或包含不同核心肽螺旋链段的组合的分支网络的策略完全在肽化学和/或有机化学领域技术人员的能力范围内。如果需要,则通常在温和的氧化剂存在下进行二硫键的形成。
可以使用化学氧化剂,或者可以将化合物简单地暴露于大气氧中以实现这些键联。各种方法是本领域已知的,包括例如由Tam等人,1979,Synthesis955-957;Stewart等人,1984,Solid Phase Peptide Synthesis,第2版,Pierce Chemical Company Rockford,IL;Ahmed等人,1975,J.Biol.Chem.250:8477-8482;以及Pennington等人,1991Peptides1990 164-166,Giralt和Andreu编著,ESCOM Leiden,The Netherlands所述的那些方法。另一替代方案由Kamber等人,1980,Helv.Chim.Acta 63:899-915描述。在固体支持物上进行的方法由Albericio,1985,Int.J.Peptide Protein Res.26:92-97描述。任何这些方法均可用于在本发明的肽中形成二硫键。
VIII.
a.
掺入有本发明的O-连接的糖基化序列的变体和/或突变多肽的生成可以通过改变相应亲本多肽的氨基酸序列,通过多肽的突变或完全化学合成来实现。多肽氨基酸序列优选通过DNA水平的改变而改变,特别是通过在预选碱基处使编码多肽的DNA序列突变以产生密码子,该密码子将翻译成所需氨基酸。DNA突变优选地使用本领域已知的方法进行。
本发明依赖于重组遗传学领域的常规技术。公开本发明中使用的一般方法的基础教科书包括Sambrook和Russell,Molecular Cloning,A Laboratory Manual(第3版2001);Kriegler,Gene Transfer and Expression:A Laboratory Manual(1990);以及Ausubel等人,编著,Current Protocols in Molecular Biology(1994)。
核酸大小以千碱基(kb)或碱基对(bp)给出。这些是衍生自琼脂糖或丙烯酰胺凝胶电泳、测序的核酸或公开的DNA序列的估计值。对于蛋白质,大小以千道尔顿(kDa)或氨基酸残基数量给出。根据凝胶电泳、测序的蛋白质、衍生的氨基酸序列或公开的蛋白质序列估算蛋白质大小。
可以例如根据首先由Beaucage&Caruthers,Tetrahedron Lett.22:1859-1862(1981)描述的固相亚磷酰胺三酯方法,使用如Van Devanter等人,Nucleic Acids Res.12:6159-6168(1984)所述的自动合成仪以化学方式合成不可商购获得的寡核苷酸。整个基因也可以用化学方式合成。寡核苷酸的纯化使用任何本领域公认的策略进行,这些策略是例如Pearson&Reanier,J.Chrom.255:137-149(1983)中所述的天然丙烯酰胺凝胶电泳或阴离子交换HPLC。
克隆的野生型多肽基因、编码突变多肽的多核苷酸和合成寡核苷酸的序列可以在克隆之后使用例如Wallace等人,Gene 16:21-26(1981)的双链模板测序的链终止方法进行验证。
在一个示例性实施方案中,通过改组多核苷酸添加糖基化序列。编码候选多肽的多核苷酸可以用DNA改组方案进行调节。DNA改组是递归重组和突变的过程,其是通过相关基因库的随机片段化来进行,然后通过聚合酶链反应样过程来重新组装片段。参见,例如Stemmer,Proc.Natl.Acad.Sci.USA 91:10747-10751(1994);Stemmer,Nature 370:389-391(1994);以及美国专利号5,605,793、5,837,458、5,830,721和5,811,238。
b.
已经确定了编码野生型多肽的许多多核苷酸序列,并且可以从商业供应商获得,例如人生长激素,例如GenBank登录号NM 000515、NM 002059、NM 022556、NM 022557、NM022558、NM 022559、NM 022560、NM 022561和NM 022562。
人类基因组研究的迅速发展使得克隆方法成为可能,其中可以在人DNA序列数据库中搜索与已知核苷酸序列具有一定百分比序列同源性的任何基因链段,诸如编码先前鉴定的多肽的片段。如此鉴定出的任何DNA序列都可以随后通过化学合成和/或聚合酶链反应(PCR)技术诸如重叠延伸法获得。对于短序列,完全从头合成可能就足够了;然而,为了获得更大的基因,可能有必要使用合成探针从人cDNA或基因组文库中进一步分离出全长编码序列。
或者,可以使用标准克隆技术(诸如聚合酶链反应(PCR))从人cDNA或基因组DNA文库中分离编码多肽的核酸序列,其中基于同源性的引物通常可以从编码多肽的已知核酸序列中衍生而来。在标准教科书例如Sambrook和Russell,同上中描述了用于该目的的最常用技术。
适用于获得野生型多肽的编码序列的cDNA文库可以商购获得或可以构建。分离mRNA、通过反转录制备cDNA、将cDNA连接到重组载体中、转染到重组宿主中进行繁殖、筛选和克隆的一般方法是众所周知的(参见,例如Gubler和Hoffman,Gene,25:263-269(1983);Ausubel等人,同上)。通过PCR获得核苷酸序列的扩增链段后,该链段可进一步用作探针以便从cDNA文库中分离编码野生型多肽的全长多核苷酸序列。适当程序的一般描述可见于Sambrook和Russell,同上中。
可以遵循相似的程序从人类基因组文库中获得编码野生型多肽,例如上述GenBank登录号中的任一个的全长序列。人类基因组文库是可商购获得的,或者可以根据各种本领域公认的方法来构建。通常,为了构建基因组文库,首先从可能发现多肽的组织中提取DNA。然后对DNA进行机械剪切或酶消化,以产生长度约12-20kb的片段。随后通过梯度离心将片段与大小不合适的多核苷酸片段分离,并插入噬菌体λ载体中。这些载体和噬菌体在体外被包装。重组噬菌体通过如Benton和Davis,Science,196:180-182(1977)中所述的噬菌斑杂交来分析。菌落杂交如由Grunstein等人,Proc.Natl.Acad.Sci.USA,72:3961-3965(1975)所述进行。
基于序列同源性,简并寡核苷酸可被设计为引物集,并且PCR可以在合适的条件下进行(参见,例如White等人,PCR Protocols:Current Methods and Applications,1993;Griffin和Griffin,PCR Technology,CRC Press Inc.1994),以便从cDNA或基因组文库中扩增核苷酸序列的链段。使用扩增的链段作为探针,获得编码野生型多肽的全长核酸。
获得编码野生型多肽的核酸序列后,可将编码序列亚克隆到载体例如表达载体中,使得可以从所得构建体产生重组野生型多肽。随后可以对野生型多肽编码序列进行进一步的修饰,例如核苷酸取代,以改变分子特征。
c.
从编码的多核苷酸序列,可以确定野生型多肽的氨基酸序列。随后,可以通过在氨基酸序列的各个位置处引入额外的糖基化序列来修饰此氨基酸序列以改变蛋白质的糖基化模式。
建立了多种突变产生方案并在本领域中进行了描述。参见,例如Zhang等人,Proc.Natl.Acad.Sci.USA,94:4504-4509(1997);以及Stemmer,Nature,370:389-391(1994)。这些程序可以单独使用或组合使用以产生一组核酸的变体,从而产生编码多肽的变体。用于诱变、文库构建和其它多样性产生方法的试剂盒可商购获得。
产生多样性的突变方法包括例如定点诱变(Botstein和Shortle,Science,229:1193-1201(1985));使用含尿嘧啶的模板的诱变(Kunkel,Proc.Natl.Acad.Sci.USA,82:488-492(1985));寡核苷酸定向诱变(Zoller和Smith,Nucl.Acids Res.,10:6487-6500(1982));硫代磷酸酯修饰的DNA诱变(Taylor等人,Nucl.Acids Res.,13:8749-8764和8765-8787(1985));以及使用缺口双链DNA的诱变(Kramer等人,Nucl.Acids Res.,12:9441-9456(1984))。
产生突变的其它方法包括点错配修复(Kramer等人,Cell,38:879-887(1984));使用修复缺陷型宿主菌株的诱变(Carter等人,Nucl.Acids Res.,13:4431-4443(1985));缺失诱变(Eghtedarzadeh和Henikoff,Nucl.Acids Res.,14:5115(1986));限制选择和限制纯化(Wells等人,Phil.Trans.R.Soc.Lond.A,317:415-423(1986));通过全基因合成的诱变(Nambiar等人,Science,223:1299-1301(1984));双链断裂修复(Mandecki,Proc.Natl.Acad.Sci.USA,83:7177-7181(1986));通过多核苷酸链终止方法的诱变(美国专利号5,965,408)和易错PCR(Leung等人,Biotechniques,1:11-15(1989))。
d.
可以进一步改变编码多肽变体的多核苷酸序列,以与特定宿主的优选密码子使用一致。例如,一种细菌细胞菌株的优选密码子使用可用于衍生编码本发明的多肽变体并包括该菌株所偏好的密码子的多核苷酸。可以通过对由宿主细胞表达的大量基因中的优选密码子使用频率取平均值来计算由宿主细胞表现出的优选密码子使用频率(例如,计算服务可从Kazusa DNA Research Institute,Japan的网站获得)。此分析优选地限于由宿主细胞高度表达的基因。例如,美国专利号5,824,864提供了由双子叶植物和单子叶植物展示的高表达基因的密码子使用频率。
修饰完成后,通过测序验证多肽变体编码序列,然后将其亚克隆到合适的表达载体中,以与野生型多肽相同的方式进行重组产生。
IX.
在序列验证之后,可以使用重组遗传学领域中的常规技术,依靠编码本文公开的多肽的多核苷酸序列来产生本发明的多肽变体。
a.
为了获得编码本发明的突变多肽的核酸的高水平表达,通常将编码突变多肽的多核苷酸亚克隆到表达载体中,所述表达载体含有指导转录的强启动子、转录/翻译终止子和用于翻译起始的核糖体结合位点。合适的细菌启动子是本领域众所周知的并且描述于例如Sambrook和Russell,同上,及Ausubel等人,同上中。用于表达野生型或突变多肽的细菌表达系统可在例如大肠杆菌、芽孢杆菌属、沙门氏菌属和柄杆菌属中获得。用于这种表达系统的试剂盒可商购获得。用于哺乳动物细胞、酵母和昆虫细胞的真核表达系统是本领域众所周知的,并且也可商购获得。在一个实施方案中,真核表达载体是腺病毒载体、腺相关载体或逆转录病毒载体。
用于指导异源核酸表达的启动子取决于特定的应用。启动子任选地位于与异源转录起始位点大致相同的距离,与在其天然环境中距转录起始位点的距离相同。然而,如本领域中已知的,在不损失启动子功能的情况下,可以适应该距离的一些变化。
除了启动子之外,表达载体通常还包括转录单元或表达盒,其含有宿主细胞中表达突变多肽所需的所有另外的元件。典型的表达盒因此含有与编码突变多肽的核酸序列可操作连接的启动子以及转录物的有效聚腺苷酸化、核糖体结合位点和翻译终止所需的信号。编码多肽的核酸序列通常连接至可切割的信号肽序列,以促进转化细胞分泌多肽。此类信号肽尤其包括来自以下各物的信号肽:组织纤溶酶原激活物、胰岛素和神经元生长因子、以及烟芽夜蛾的少年激素酯酶。盒的另外的元件可以包括增强子,并且如果基因组DNA用作结构基因,则包括具有功能性剪接供体和受体位点的内含子。
除了启动子序列之外,表达盒还应该在结构基因的下游含有转录终止区,以提供有效的终止。终止区可以从与启动子序列相同的基因获得,或者可以从不同的基因获得。
用于将遗传信息运输到细胞中的特定表达载体不是特别关键。可以使用用于在真核或原核细胞中表达的任何常规载体。标准细菌表达载体包括质粒,诸如基于pBR322的质粒、pSKF、pET23D以及融合表达系统诸如GST和LacZ。也可以将表位标签添加到重组蛋白中,以提供方便的分离方法,例如c-myc。
含有来自真核病毒的调控元件的表达载体通常用于真核表达载体例如SV40载体、乳头瘤病毒载体和源自爱泼斯坦-巴尔(Epstein-Barr)病毒的载体中。其它示例性真核载体包括pMSG、pAV009/A
在一些示例性实施方案中,表达载体选自pCWin1、pCWin2、pCWin2/MBP、pCWin2-MBP-SBD(pMS
一些表达系统具有提供基因扩增的标记物,诸如胸苷激酶、潮霉素B磷酸转移酶和二氢叶酸还原酶。或者,不涉及基因扩增的高产量表达系统也是合适的,诸如昆虫细胞中的杆状病毒载体,其具有在多角体蛋白启动子或其它强的杆状病毒启动子的指导下编码突变多肽的多核苷酸序列。
表达载体中通常包含的元件还包括在大肠杆菌中起作用的复制子、编码抗生素抗性以允许选择带有重组质粒的细菌的基因以及在质粒的非必需区中允许插入真核序列的独特限制位点。所选的特定抗生素抗性基因不是关键的,本领域已知的许多抗性基因中的任一种都是合适的。
原核序列被任选地选择,以使它们在必要时不干扰DNA在真核细胞中的复制。
当需要重组蛋白(例如,本发明的hgh突变体)的周质表达时,表达载体还包含编码分泌信号的序列,诸如大肠杆菌OppA(周质寡肽结合蛋白)分泌信号或其修饰形式,其直接连接至要表达的蛋白质的编码序列的5'。此信号序列引导细胞质中产生的重组蛋白通过细胞膜进入周质空间。表达载体可以进一步包含信号肽酶1的编码序列,其在重组蛋白进入周质空间时能够酶促切割信号序列。关于重组蛋白的周质产生的更详细描述可见于例如Gray等人,Gene 39:247-254(1985);美国专利号6,160,089和6,436,674中。
如上所述,本领域技术人员将认识到,可以对任何野生型或突变多肽或其编码序列进行各种保守取代,同时仍保留该多肽的生物活性。此外,还可以进行多核苷酸编码序列的修饰以适应在特定表达宿主中的优选密码子使用,而不改变所得的氨基酸序列。
b.
使用标准转染方法来产生表达大量突变多肽的细菌、哺乳动物、酵母或昆虫细胞系,然后使用标准技术(参见,例如Colley等人.,J.Biol.Chem.264:17619-17622(1989);Guide to Protein Purification,Methods in Enzymology,第182卷(Deutscher编著,1990))对其进行纯化。真核细胞和原核细胞的转化是根据标准技术(参见,例如Morrison,J.Bact.132:349-351(1977);Clark-Curtiss&Curtiss,Methods in Enzymology 101:347-362(Wu等人,编著,1983)进行的。
可以使用将外来核苷酸序列引入宿主细胞中的任一种众所周知的方法。这些方法包括使用磷酸钙转染、聚凝胺、原生质体融合、电穿孔、脂质体、显微注射、血浆载体、病毒载体及任何其它众所周知的方法来将克隆的基因组DNA、cDNA、合成DNA或其它外来遗传物质引入宿主细胞中(参见,例如,Sambrook和Russell,同上)。仅需要所使用的特定基因工程程序能够将至少一个基因成功地引入能够表达突变多肽的宿主细胞中。
c.
将表达载体引入合适的宿主细胞中之后,在有利于突变多肽表达的条件下培养转染的细胞。然后筛选细胞中重组多肽的表达,随后使用标准技术(参见,例如Scopes,Protein Purification:Principles and Practice(1982);美国专利号4,673,641;Ausubel等人,同上;以及Sambrook和Russell,同上)从培养物中回收重组多肽。
筛选基因表达的几种通用方法是本领域技术人员众所周知的。首先,可以在核酸水平下检测基因表达。使用核酸杂交技术的多种特异性DNA和RNA测量方法是常用的(例如Sambrook和Russell,同上)。一些方法涉及电泳分离(例如,用于检测DNA的Southern印迹和用于检测RNA的Northern印迹),但是也可以在不进行电泳的情况下(诸如通过斑点印迹)进行DNA或RNA的检测。也可以使用序列特异性引物,通过PCR或RT-PCR检测转染细胞中编码突变多肽的核酸的存在。
其次,可以在多肽水平上检测基因表达。本领域技术人员常规地使用各种免疫测定来测量基因产物的水平,特别是使用与本发明的突变多肽特异性反应的多克隆或单克隆抗体(例如Harlow和Lane,Antibodies,A Laboratory Manual,第14章,Cold SpringHarbor,1988;Kohler和Milstein,Nature,256:495-497(1975))。此类技术需要通过选择对突变多肽或其抗原部分具有高特异性的抗体来制备抗体。产生多克隆和单克隆抗体的方法已经充分建立,并且它们的描述可见于以下文献中,参见,例如Harlow和Lane,同上;Kohler和Milstein,Eur.J.Immunol.,6:511-519(1976)。在后续部分中提供了制备针对本发明的突变多肽的抗体和进行检测该突变多肽的免疫测定的更详细描述。
X.
一旦确认了重组突变多肽在转染宿主细胞中的表达,就以适当规模培养宿主细胞,以便纯化重组多肽。
a.
当本发明的突变多肽由转化的细菌大量重组产生(通常在启动子诱导后)时,尽管表达可以是组成型的,但这些蛋白质可形成不溶性聚集体。有几种适用于纯化蛋白质包涵体的方案。例如,聚集蛋白(下文称为包涵体)的纯化通常涉及通过破坏细菌细胞,例如通过在约100-150μg/ml溶菌酶和0.1%Nonidet P40(非离子型去污剂)的缓冲液中孵育来提取、分离和/或纯化包涵体。可以使用Polytron研磨机(Brinkman Instruments,Westbury,NY)研磨细胞悬液。或者,可以在冰上对细胞进行超声处理。溶解细菌的替代方法在Ausubel等人以及Sambrook和Russell,都同上中进行了描述,并且对于本领域技术人员而言将是显而易见的。
对于从细菌包涵体中纯化重组多肽的进一步描述,参见,例如Patra等人,ProteinExpression and Purification 18:182-190(2000)。
可通过本领域技术人员众所周知的标准分离技术将存在于上清液中的重组蛋白与宿主蛋白分离。
b.
为了证实重组突变多肽的产生,免疫学测定可能对检测样品中多肽的表达有用。免疫学测定也可用于定量重组激素的表达水平。针对突变多肽的抗体对于进行这些免疫学测定是必需的。
c.
产生与目标免疫原特异性反应的多克隆和单克隆抗体的方法是本领域技术人员已知的(参见,例如Coligan,Current Protocols in Immunology Wiley/Greene,NY,1991;Harlow和Lane,Antibodies:A Laboratory Manual Cold Spring Harbor Press,NY,1989;Stites等人(编著)Basic and Clinical Immunology(第4版)Lange MedicalPublications,Los Altos,CA及其中引用的参考文献;Goding,Monoclonal Antibodies:Principles and Practice(第2版)Academic Press,New York,NY,1986;以及Kohler和Milstein Nature 256:495-497,1975)。这类技术包括通过从噬菌体或类似载体中的重组抗体文库中选择抗体来制备抗体(参见,Huse等人,Science 246:1275-1281,1989;以及Ward等人,Nature 341:544-546,1989)。
为了产生含有具有所需特异性的抗体的抗血清,可以使用目标多肽(例如,本发明的突变多肽)或其抗原片段来免疫合适的动物,例如小鼠、兔或灵长类动物。可以根据标准免疫方案使用标准佐剂,诸如弗氏佐剂。或者,可以将衍生自该特定多肽的合成抗原肽与载体蛋白缀合,然后用作免疫原。
通过采集试验出血并测定与目标抗原的反应性的效价来监测动物对免疫原制剂的免疫反应。当获得适当高效价的针对抗原的抗体时,从动物中收集血液并制备抗血清。随后可以进行抗血清的进一步分级分离以富集与抗原特异性反应的抗体并纯化抗体,参见,Harlow和Lane,同上,以及上文提供的蛋白质纯化的一般描述。
使用本领域技术人员熟悉的各种技术获得单克隆抗体。通常通过与骨髓瘤细胞融合来使来自用所需抗原免疫的动物的脾细胞永生化(参见,Kohler和Milstein,Eur.J.Immunol.6:511-519,1976)。永生化的替代方法包括例如用爱泼斯坦巴尔病毒、癌基因或逆转录病毒进行转化,或本领域众所周知的其它方法。筛选由单一永生化细胞产生的菌落,以产生具有所需特异性和对抗原的亲和力的抗体,并且可以通过各种技术来提高由此类细胞产生的单克隆抗体的产率,这些技术包括注射到脊椎动物宿主的腹膜腔中。
另外,根据由Huse等人,同上概述的一般方案,通过筛选人B细胞cDNA文库鉴定编码具有所需特异性的抗体或该抗体的结合片段的核酸序列后,也可以重组产生单克隆抗体。以上讨论的重组多肽产生的一般原理和方法适用于通过重组方法的抗体产生。
需要时,可以测试能够特异性识别本发明的突变多肽的抗体与野生型多肽的交叉反应性,由此与针对野生型蛋白的抗体区分开。例如,从用突变多肽免疫的动物获得的抗血清可以穿过固定有野生型多肽的柱。穿过柱的抗血清部分仅识别突变多肽,而不识别野生型多肽。类似地,在仅识别突变体而不识别野生型多肽时,也可以针对突变多肽的单克隆抗体进行排他性筛选。
仅特异性识别本发明的突变多肽而不识别野生型多肽的多克隆或单克隆抗体可用于从野生型蛋白中分离突变蛋白,例如,通过将样品与固定在固体支持物上的突变肽特异性多克隆或单克隆抗体一起孵育来实现。
XI.
在各种实施方案中,本发明提供了一种预防、改善或治疗疾病状态的方法。在这些实施方案中,本发明提供了一种方法,该方法包括向有需要的受试者施用足以预防、改善或治疗疾病状态的量的本发明的多肽变体。示例性疾病状态是癌症。所公开的激动剂变体可用于促进细胞生长,特别是用于血管生成,以及用于治疗心血管、肝脏、肌肉骨骼和神经元疾病。举例来说,本发明的某些多肽变体可用于预防或治疗过度增殖性疾病或病症,例如各种形式的癌症。
在一个示例性实施方案中,本发明提供了一种在需要这种治疗的受试者中治疗癌症的方法。所述方法包括向受试者施用治疗有效量的本发明的多肽变体。
预期本发明的多肽变体可用于治疗多种FGF反应性病症,包括例如各种眼部病症,在以下癌症中的FGF反应性肿瘤细胞:肺癌、乳腺癌、结肠癌、前列腺癌、卵巢癌、头颈癌、卵巢癌、多发性骨髓瘤、肝癌、胃癌、食道癌、肾癌、鼻咽癌、胰腺癌、间皮瘤、黑色素瘤和成胶质细胞瘤。
在示例性实施方案中,癌症是癌,例如,结肠直肠癌、鳞状细胞癌、肝细胞癌、肾癌、乳腺癌或肺癌。
多肽变体可用于抑制或减少肿瘤细胞的增殖。在这种方法中,将肿瘤细胞暴露于治疗有效量的多肽变体,以抑制或减少肿瘤细胞的增殖。在某些实施方案中,多肽变体将肿瘤细胞增殖抑制了至少50%、60%、70%、80%、90%、95%或100%。
在某些实施方案中,将多肽变体用于抑制或减少肿瘤细胞的增殖,其中所述变体降低FGF1结合至FGFR的能力。在某些实施方案中,FGF1多肽变体用于抑制或促进伤口愈合。
另外,多肽变体可用于抑制或减慢哺乳动物中的肿瘤生长或发育。在这种方法中,将有效量的多肽变体施用于哺乳动物,以抑制或减慢哺乳动物中的肿瘤生长。因此,多肽变体可用于治疗例如哺乳动物中的肿瘤。所述方法包括向哺乳动物施用治疗有效量的多肽变体。多肽变体可以单独施用或与另一种药物活性分子组合施用,以治疗肿瘤。
通常,多肽变体的治疗有效量将在约0.1mg/kg至约100mg/kg、任选约1mg/kg至约100mg/kg、任选约1mg/kg至10mg/kg的范围内。所施用的量将取决于变量诸如待治疗的疾病或适应症的类型和程度、特定患者的总体健康状况、所递送的多肽变体的相对生物学功效、多肽变体的制剂、制剂中赋形剂的存在和类型以及施用途径。为了迅速达到所需的血液水平或组织水平,施用的初始剂量可以增加至超过上限,或者初始剂量可以小于最佳剂量,并且在治疗过程中,每日剂量可以视具体情况逐渐增加。人用剂量可被优化,例如,在被设计为0.5mg/kg至20mg/kg的常规I期剂量递增研究中。给药频率可以变化,这取决于诸如施用途径、剂量和所治疗的疾病状况的因素。示例性给药频率是每天一次、每周一次和每两周一次。优选的施用途径是肠胃外,例如静脉内输注。基于蛋白质的药物制剂在本领域普通技术范围内。在本发明的一些实施方案中,在施用时,将多肽变体,例如基于蛋白质的多肽冻干,并在缓冲盐水中重构。
多肽变体可以单独施用或与其它药物活性成分组合施用。其它活性成分(例如免疫调节剂)可以与多肽变体一起施用,或者可以在多肽变体之前或之后施用。
包含用于治疗用途的多肽变体的制剂通常包括与药学上可接受的载剂组合的多肽变体。如本文所用,“药学上可接受的载剂”意指在合理的医学判断范围内;适用于与人类和动物的组织接触而没有过度毒性、刺激性、过敏反应或其它问题或并发症;与合理的利益/风险比相称的缓冲剂、载剂和赋形剂。从与制剂的其它成分相容并且对接受者无害的意义上说,载剂必须为“可接受的”。在这方面,药学上可接受的载剂旨在包括与药物施用相容的任何和所有缓冲剂、溶剂、分散介质、包衣、等渗和吸收延迟剂等。用于药物活性物质的此类介质和剂的用途是本领域已知的。
制剂可以剂量单位形式方便地存在,并且可通过任何合适的方法(包括药学领域中众所周知的任何方法)来制备。Remington’s Pharmaceutical Sciences,第18版(MackPublishing Company,1990)。
在示例性实施方案中,多肽变体在体外或体内用于诊断目的,通常用可检测部分直接或间接标记多肽变体。可检测部分可以是能够直接或间接产生可检测信号的任何部分。例如,可检测部分可以是放射性同位素,诸如
多肽变体可用于本领域可用的多种免疫测定技术中。示例性免疫测定包括例如夹心免疫测定、竞争性免疫测定、免疫组织化学程序。
在夹心免疫测定中,使用两种结合目标分析物或抗原的抗体,例如,一种固定在固相支持物上,而另一种游离在溶液中并用可检测部分标记。当将含有抗原的样品引入此系统中时,抗原既与固定的抗体也与标记的抗体结合,从而在支持物表面形成“夹心”免疫复合物。通过洗去非结合的样品组分和过量的标记抗体,并测量与支持物表面上的蛋白质复合的标记抗体的量,可以检测出复合蛋白。或者,可以通过用结合了游离抗体的可检测部分标记的第三抗体来检测溶液中游离的抗体。免疫学测定设计、理论和方案的详细综述可见于许多教科书中,包括Butt编著,(1984)Practical Immunology,Marcel Dekker,NewYork;Harlow等人编著(1988)Antibodies,A Laboratory Approach,Cold Spring HarborLaboratory;以及Diamandis等人编著(1996)Immunoassay,Academic Press,Boston。
预期标记的多肽变体可用作体内显像剂,由此多肽变体可将显像剂靶向受体中特定的目标组织。用于体内成像的远程可检测部分包括放射性原子
在体内成像中也有用的非放射性部分包括硝基氧自旋标记以及镧系元素和过渡金属离子,所有这些都在原位诱导质子弛豫。除了成像外,复合放射性部分还可用于标准放射免疫疗法方案中以破坏靶细胞。
广泛多种荧光标记是本领域已知的,包括但不限于异硫氰酸荧光素、罗丹明、藻红蛋白、藻蓝蛋白、别藻蓝蛋白、邻苯二甲醛和荧光胺。有用的化学发光标记可包括鲁米诺、异鲁米诺、芳香族吖啶酯、咪唑、吖啶盐或草酸酯。
所公开的多肽变体也可以用荧光标记物标记,以允许在体内检测。在一些实施方案中,荧光标记是Cy5.5(GE Healthcare)。在其它实施方案中,荧光标记是Alexa
高剂量放射疗法的示例性核苷酸包括放射性原子
实施例
实施例1:用于工程化蛋白水解稳定的生长因子的高通量筛选方法
摘要
生长因子是一类重要的调节蛋白,具有开发作为再生医学和癌症治疗的治疗分子的巨大的潜力。然而,生长因子作为治疗分子的活性和功效由于其不良的热稳定性和蛋白水解稳定性而受到极大限制。尽管已经开发出许多方法来工程化具有提高的热稳定性的生长因子,但是缺乏对开发具有提高的蛋白水解稳定性的工程化生长因子的关注和方法开发。蛋白酶(诸如纤溶酶、弹性蛋白酶、uPA、组织蛋白酶和MMP)在细胞外基质降解和信号转导(尤其是伤口愈合和肿瘤形成)中起关键作用。据报道,这些蛋白酶通常也降解生长因子。在这项工作中,描述了一种用于工程化生长因子以提高蛋白水解稳定性的通用方法。利用酵母展示平台和FACS筛选作为组合方法来选择具有提高的蛋白水解稳定性的突变体。通过证明筛选以区分野生型FGF1与文献中报道的蛋白水解稳定的FGF1突变体的能力来验证此方法。
引言
此实施例描述了一种组合方法,该方法使用酵母展示平台和用于筛选的流激活细胞分选(FACS)来工程化蛋白水解稳定的生长因子。说明了设置以FGF1为模型实例的筛选方法的过程。筛选是为FGF1设置的,因为FGF1具有极差的热稳定性和蛋白水解稳定性
结果
工程化蛋白水解稳定的蛋白的组合筛选方法的工作流程
酵母展示平台常用于工程化高亲和力结合物,也可用于工程化具有更高蛋白水解稳定性的蛋白质(图1)。单个生长因子变体的数千个拷贝以束缚融合体的形式展示在酵母表面上。血凝素(HA)标签在生长因子的上游表达,而c-myc标签在生长因子的下游表达。可以将细胞与相应受体的可溶性Fc融合体一起孵育,该融合体可以与酵母展示的生长因子结合。
酵母展示平台与流激活细胞分选(FACS)结合使用,以便工程化具有更高蛋白水解稳定性的生长因子(图2)。生长因子突变体的文库是通过随机诱变、定向诱变或DNA改组生成的。将酵母细胞文库与目标蛋白酶一起孵育,在此过程中发生酵母表面展示蛋白的切割。具有更高蛋白水解稳定性的生长因子突变体对酵母细胞表面的切割有更高的抗性。蛋白酶孵育后,将细胞洗涤并与功能性受体的可溶性Fc融合体一起孵育,所述Fc融合体结合至保留有受体结合亲和力的正确折叠的生长因子突变体。FACS被用于分选正确折叠的、未切割的生长因子突变体,将其扩增并诱导用于下一轮分选。
针对Fc结构域、c-myc结构域和HA标签的荧光抗体标记物被用于测量受体结合、生长因子特异性切割和非特异性切割(表2.1)。结合的Fc融合受体的检测对于确保生长因子的突变不会严重降低对该受体的结合亲和力或导致蛋白质的不正确折叠来说至关重要。生长因子特异性切割是生长因子蛋白水解稳定性的直接量度。它被c-myc信号检测到,因为切割的生长因子将去除C末端c-myc标签。当蛋白酶在酵母表面展示蛋白Aga1p和Aga2p内切割时,发生非特异性切割。在非特异性切割过程中,所有三个标记物的荧光信号均降低。这是不希望的,因为用于检测生长因子切割和结合活性的动态范围缩小。因此,HA信号被用于确保目标蛋白酶的非特异性切割最小。
FGF1的酵母展示
选择FGF1作为模型来说明蛋白水解稳定性筛选的设置。将野生型FGF1克隆到pCT载体中,以便作为与Aga2p交配蛋白的融合体在酿酒酵母的酵母细胞表面上表达(图3A)。通过检测蛋白质C末端上的c-myc标签确认了FGF1在酵母细胞表面上的成功表达(图3B)。最后,通过测量对FGFR1-Fc的特异性结合活性,证实了酵母展示的FGF的正确折叠(图3C)。
用于工程化蛋白水解稳定的FGF1的蛋白酶的选择
测试了使用血清、胰蛋白酶、胰凝乳蛋白酶和纤溶酶来开发FGF1的蛋白水解稳定性筛选。选择这些蛋白酶是基于它们与FGF1的科学和生物学相关性。蛋白酶对筛选的适用性是由其以合理的速率切割生长因子的能力确定的,而酵母展示蛋白的非特异性切割最少。
首先尝试开发使用血清(由多种蛋白酶组成的天然血液产品)的筛选,这些蛋白酶可能会在人体内遇到生长因子
接下来,使用胰蛋白酶和胰凝乳蛋白酶(这两种蛋白酶通常用于测量和报告蛋白质的蛋白水解稳定性)测试了筛选的开发。用各种浓度的胰蛋白酶(图5)和胰凝乳蛋白酶(图6)孵育酵母展示的野生型FGF1,然后测量蛋白切割的程度(α-c-myc)和与FGFR1-Fc的结合。发现胰蛋白酶和胰凝乳蛋白酶都对观察到的蛋白质切割程度和与FGFR1-Fc的结合有浓度依赖性效应。然后,确定观察到的蛋白质切割是由于非特异性切割(α-HA)还是FGF1特异性切割(α-c-myc)引起的。发现与更高浓度的胰蛋白酶一起孵育后,HA信号显著降低,表明许多所观察到的胰蛋白酶对蛋白质的切割是由于非特异性切割所致(图7)。因此,得出结论,胰蛋白酶不能用于蛋白水解稳定性筛选。同时,发现只有在与更高浓度的胰凝乳蛋白酶孵育下HA信号相对不受影响时,c-myc信号才会降低,表明胰凝乳蛋白酶对蛋白质的切割主要归因于FGF1内的切割(图8)。因此,得出结论,胰凝乳蛋白酶是用于蛋白水解稳定性筛选的合理候选物。
最后,评估了使用纤溶酶的蛋白水解稳定性筛选的开发,纤溶酶是一种降解细胞外基质蛋白的蛋白酶,并且据报道可降解FGF1
通过区分野生型FGF1与蛋白水解稳定的FGF1突变体进行的筛选方法的验证
为了测试基于纤溶酶的筛选区分具有不同蛋白水解稳定性的FGF的能力,比较野生型(WT)FGF1与文献中通过合理设计开发的热稳定化的FGF1突变体(PM2)
将展示PM2或WT FGF1的酵母细胞与不同浓度的纤溶酶一起孵育48小时,并针对非特异性切割(抗HA)和FGF1特异性切割(抗c-myc)进行染色(图11)。发现通过c-myc信号的差异可获得群体的干净分离,而对HA信号的影响相对较小。切割信号的这种差异证实了使用纤溶酶可以使筛选正确鉴别具有更高蛋白水解稳定性的新FGF突变体,并通过FACS对这些群体进行分选。
讨论
在此实施例中,描述了使用酵母展示平台和流激活细胞分选对蛋白水解稳定的生长因子进行工程化的高通量、通用筛选方法的开发。举例来说,提供了针对FGF1(即高度不稳定的生长因子)的筛选的设置。
在建立目标生长因子的筛选时,第一步是确保生长因子可以在酵母表面上表达,并且能够结合至其受体的可溶形式。证实了FGF1可以作为与Aga2酵母展示蛋白的C末端融合体在pCT载体中表达,并且它特异性地结合至FGFR1-Fc。过去,VEGF、EGF和HGF已通过酵母展示成功地表达
第二步是为了确定用于蛋白水解筛选的蛋白酶。测试了将血清、胰蛋白酶、胰凝乳蛋白酶和纤溶酶用于工程化酵母展示的FGF1。发现胎牛血清(FBS)即使在高浓度下也无法提供足够的选择性压力。尽管蛋白酶见于FBS中,但见于FBS中的蛋白酶抑制剂诸如α-1-抗蛋白酶和α-1-抗胰凝乳蛋白酶会使它们的活性降低
筛选设置中的最后一步是确定是否可以区分具有不同蛋白水解稳定性的生长因子突变体。这个任选步骤提供了一个重要的基准,使得人们对筛选以选择蛋白水解稳定的突变体的能力充满信心。对于FGF1,证实PM2(一种具有提高的热稳定性和蛋白水解稳定性的FGF1突变体)在酵母表面上展示时可与野生型FGF1区分开。在不存在可用的蛋白水解稳定化的生长因子突变体的情况下,只要蛋白酶显示出酵母展示蛋白的高生长因子特异性切割和低非特异性切割能力,筛选便可进行。在实施例2中,报告了使用我们开发的方法对FGF1就蛋白水解稳定性进行的工程化。
材料和方法
酵母展示构建体的克隆
将FGF1从人FGF1 cDNA(MGC克隆:9218,图像:3896359,残基:Phe16至Asp155)克隆到pCT载体(限制性位点:NheI,BamHI)中进行酵母展示。对于蛋白水解稳定的FGF1突变体PM2,使用定点诱变对FGF1进行Q40P(CAA至CCA)、S47I(TCC至ATC)和H93G(CAT至GGT)突变。
酵母展示的FGF1的结合测定
将50,000个诱导的酵母细胞与不同浓度的人FGFR1β(IIIc)-Fc(R&D Systems)在含1g/L BSA(PBSA)的磷酸盐缓冲盐水中在室温下一起孵育。将细胞以足够大的体积孵育以避免配体耗尽,并进行足够长的时间(通常3至24小时)以达到平衡。在孵育的最后30分钟期间,将酵母细胞与鸡抗c-Myc(Invitrogen)的1:2500稀释液在PBSA中一起孵育。将酵母沉淀,洗涤,然后与二抗的1:200稀释液在冰上一起孵育10分钟:针对抗c-myc的抗人IgG-FITC(Sigma Aldrich)和抗鸡IgY-PE(Santa Cruz Biotechnology)。将酵母洗涤,沉淀并重悬于PBSA中,之后立即使用EMD Millipore Guava EasyCyte,通过流式细胞仪进行分析。使用FlowJo(v7.6.1)分析流式细胞仪数据。绘制结合曲线,并且使用GraphPad Prism 6获得K
用于筛选的蛋白水解稳定性测定
胎牛血清(Gibco)、来自牛胰腺的胰蛋白酶(Sigma Aldrich)、来自牛胰腺的胰凝乳蛋白酶VII型(Sigma Aldrich)或来自人血浆的纤溶酶(Sigma Aldrich)被用作蛋白酶或蛋白酶混合物以进行孵育。在Dulbecco改良的Eagle培养基(Gibco)中稀释胎牛血清。将胰蛋白酶和胰凝乳蛋白酶在胰蛋白酶缓冲液(100mM Tris-HCl(pH 8)、1mM CaCl
在适当的缓冲液中,将一百万个诱导的酵母细胞与各种浓度的蛋白酶一起孵育。孵育结束时,将细胞用PBSA(PBS+0.1%BSA)洗涤一次,并重悬于缓冲蛋白酶抑制剂混合物(Sigma Aldrich)中,以淬灭残留的蛋白酶活性。5分钟后,用PBSA再次洗涤细胞。对于仅测量FGFR结合活性的实验,将细胞在10nM人FGFR1β(IIIc)-Fc(R&D Systems)在pBSA中孵育1小时。最后一次洗涤后,将细胞与适当的荧光抗体一起孵育。
对于测量FGFR1结合活性和c-myc信号的实验,将细胞与鸡抗c-Myc(Invitrogen)的1∶2000稀释液在PBSA中一起孵育30分钟。洗涤后,将细胞在二抗中于冰上孵育10分钟:针对抗c-myc的抗人IgG-FITC(Sigma Aldrich)和抗鸡IgY-PE(Santa Cruz Biotechnology)。
对于测量HA和c-myc信号的实验,将细胞与抗HA-Tag(6E2)小鼠mAb(CellSignaling)的1∶1000稀释液和鸡抗c-Myc(Invitrogen)的1∶2000稀释液一起孵育30分钟。洗涤后,将细胞在二抗中在冰上孵育10分钟:山羊抗小鼠PE(Invitrogen)和山羊抗鸡IgY-AlexaFluor488(Santa Cruz Biotechnology)。
将酵母洗涤,沉淀并重悬于PBSA中,之后立即使用EMD Millipore GuavaEasyCyte,通过流式细胞仪进行分析。使用FlowJo(v7.6.1)分析流式细胞仪数据。绘制结合曲线,并且使用GraphPad Prism 6获得K
表3.不同事件对由荧光抗体标记物观察到的信号的影响。
实施例2:工程化蛋白水解稳定化的成纤维细胞生长因子
摘要
FGF1在伤口愈合、组织再生、肿瘤形成和其它血管生成依赖性疾病过程中,在细胞分化和诱导血管生成中起重要作用。因此,基于FGF1的激动剂和拮抗剂可以在细胞培养和蛋白质治疗中具有重要的应用。然而,据报道当暴露于培养物中的蛋白酶时,FGF1表现出对降解的敏感性。它的不良蛋白水解稳定性可能会阻碍它们在细胞培养中或作为治疗性分子开发时的活性和功效。在此实施例中,使用实施例1中所述的基于酵母展示的筛选方法对FGF1肽就蛋白水解稳定性进行工程化。探索了用于选择蛋白水解稳定的FGF并成功鉴定出表征候选物的门控策略。
引言
成纤维细胞生长因子(FGF)是重要的调节生物活性的生长因子家族的一部分,所述生物活性包括胚胎发育、细胞分化、细胞增殖、细胞迁移、血管生成、新陈代谢和伤口愈合
据报道,FGF1保护功能性血管免于退化、诱导动脉生长和促进毛细血管增生
尽管FGF1在诱导用于伤口愈合和组织再生的血管生成方面具有潜力,但使用FGF1作为治疗剂的努力在很大程度上没有成功。I期和II期临床研究显示,呈可注射的肌内编码FGF1质粒形式的基因疗法改善晚期下肢缺血患者的灌注并减少截肢的需要
重组FGF1无法在许多临床应用中有效的部分原因可能是其稳定性差。野生型FGF1在条件培养基或培养物中于37℃下孵育后会迅速降解,降解半衰期为约25分钟
此实施例描述了使用实施例1中所述开发的筛选方法对FGF1就提高针对纤溶酶的蛋白水解稳定性进行工程化。使用酵母表面展示平台对FGF1进行工程化以建立基因与蛋白质的连锁。筛选每种生长因子的随机诱变文库的FGFR1结合物,然后筛选与蛋白酶一起孵育后保持未切割的突变体,最后筛选与蛋白酶一起孵育后保持未切割并保留FGFR1结合的突变体。鉴定了几个有希望的突变,这些突变似乎提高每种生长因子的蛋白水解稳定性,并如实施例3中所述生成表征候选物。
结果
野生型FGF的酵母展示和随机诱变文库的产生
正确折叠的野生型FGF1的成功酵母展示如下所述。易错PCR与核苷酸类似物一起用于在野生型FGF1中随机产生突变。生成了具有3.3×10
分选1:FGFR1-Fc结合物的选择
对于第一分选,针对保留对FGFR1-Fc的结合亲和力,对FGF1突变体进行分选(图12A)。假设大多数随机突变将导致结合亲和力的丧失。与FGFR1-Fc一起孵育后,门控并收集显示出高表达(α-c-myc)和高结合信号(α-FGFR1-Fc)的细胞。对于FGF1文库,观察到非结合突变体与保留FGFR结合亲和力的突变体之间的清晰分离(图12B)。
分选2:对FGF1特异性切割的抗性的选择
对于第二分选,扩增来自分选1的细胞,并针对在与纤溶酶一起孵育后保留切割抗性的FGF1突变体进行分选(图13A)。为了获得有效的动态范围并区分具有不同蛋白水解稳定性的突变体,用不同的浓度和孵育时间测试细胞的孵育。发现对于FGF1文库来说,与400nM纤溶酶一起孵育36小时对于实现切割的与未切割的突变体群体之间的清晰分离是必要的(图13B)。收集表现出最高水平的蛋白酶抗性的前1-2%的细胞(高α-c-myc),通过表达水平(α-HA)归一化。
筛选过程中肽伪物质的分离
扩增FGF1文库的第二分选物,并使用HA和c-myc信号进行另一轮针对FGF1特异性切割的抗性的选择。与200nM纤溶酶一起孵育36小时后,可以清楚地观察到,与文库的其余部分相比,细胞群显示出高得多的c-myc信号,表明酵母展示的蛋白对纤溶酶切割的抗性明显更高(图14A)。收集此群体并对单个克隆测序以进行分析(图14B)。发现大多数细胞在其细胞表面上不表达FGF2突变体,而是在随机诱变过程中可能已经生成的短肽伪物质。
证实这些肽没有表现出对FGFR1-Fc的任何特异性结合,表明对FGF1特异性切割的抗性的分选导致表达这些肽伪物质的非常小的细胞群体的快速富集(图15)。因此,对于所有后续分选,继续进行以包括保留FGFR1结合亲和力的选择性压力。
分选3-4:对蛋白酶抗性的FGFR1-Fc结合物的选择
对于其余的分选3和4,将文库与纤溶酶一起孵育,然后与FGFR1-Fc一起孵育,然后进行选择(图16)。使用α-HA、α-c-myc和α-FGFR1-Fc的不同组合选择保持未切割并保留对FGFR1的结合亲和力的FGF1突变体
对于FGF1的第三分选,扩增来自分选2的细胞,并分选与纤溶酶一起孵育后保留FGFR1结合的FGF突变体(图17)。用更高浓度的纤溶酶进行12小时孵育。最终将1.5μM纤溶酶用于FGF1文库的分选。门控并收集显示出高结合信号(α-FGFR1-Fc)和高表达(α-HA)的细胞。
对于FGF1的第四分选,扩增来自分选3的细胞,并将纤溶酶的孵育时间从12小时增加到36小时。将来自分选3的细胞在1.5μM纤溶酶或500nM纤溶酶中孵育。最终分选与500nM纤溶酶一起孵育的细胞(图18)。门控并收集对切割具有高抗性(α-c-myc)和高结合信号(α-FGFR1-Fc)的细胞。通过分选4,已就FGF1文库达成了完全一致,并被用于进行另外轮次的分选。
分选的FGF1突变体的序列分析
对于最后的分选4,随机选择单个克隆并进行测序分析。能够在四轮分选中达成完全一致。突变体(BS4M1)包含两个突变:D28N和L131R(图19)。天冬氨酸28是三个关键β-发夹中的第一个(LPDG)的一部分,这些发夹封闭FGF1的六链β-桶状结构。亮氨酸131见于FGF1的N末端与C末端之间的β链对中。
讨论
在此实施例中,描述了就针对纤溶酶的蛋白水解稳定性对FGF1进行的工程化。第一步,能够成功表达野生型FGF1和野生型FGF2。通过检测针对FGFR1-Fc或sFGFR3-D2D3-Fc的特异性结合而测试的c-myc标签,可以证明蛋白质的成功表达和正确折叠。观察到FGF1表现出相对高的表达信号,这很有趣,因为FGF1被认为是不稳定的、具有短的半衰期和低的解链温度
酵母展示的FGF1高表达导致在FGF1随机诱变文库的分选和后续分选过程中产生良好的信号动态范围。FGF1分选物可以经受高浓度的纤溶酶和较长的孵育时间,以提高严格性。这在分选2中特别有价值,其中切割的FGF1突变体与未切割的FGF1突变体之间实现清晰的分离。
尽管完成了第一轮分选以选择分选物1中的FGFR结合物,但发现仅使用HA和c-myc信号来测量切割会导致非FGFR结合肽在分选物中的迅速富集。仅在两种这样的分选中,就鉴定出表达肽的群体与文库其余部分的清晰分离。尽管文库是通过以野生型FGF作为支架的随机诱变构建的,但随机诱变过程中的罕见错误可能导致表达肽伪物质的细胞群体极小。虽然这些伪物质对于亲和力成熟的更传统的酵母展示筛选影响不大,但它们迅速变得显著,而没有受体结合的选择性压力。因此,得出结论,用于测量结合亲和力的选择性压力是必不可少的,并且仅基于切割活性来选择突变体能完成不超过一次分选。
通过筛选过程,鉴定出几种富集的突变,这些突变对于改善蛋白水解稳定性可能是重要的。有趣的是,突变见于蛋白质的β环区域中或C末端附近,这被认为是确定蛋白质稳定性的关键区域。对于FGF1文库,已在四轮分选中达成了完全一致。FGF1 BS4M1突变体包含两个突变:D28N和L131R。天冬氨酸28是三个关键β-发夹中的第一个(LPDG)的一部分,这些发夹封闭FGF1的六链β-桶状结构。先前已经研究过Asx-Pro-Asx-Gly基序在其对FGF1稳定性的贡献中的重要性,并且已经证明用丙氨酸取代Asx残基会极大地破坏FGF1的稳定性
总之,已表明蛋白水解稳定性的筛选能够成功地富集在文献中据报道对FGF1蛋白的稳定性重要的位置处的突变。在实施例3中,表征了在最终的FGF1 BS4M1突变体中鉴定出的突变对可溶性表达的FGF1的稳定性和配体调节FGF途径的能力的影响。
材料和方法
蛋白质的酵母表面展示
YPD培养基由20g/L右旋糖、20g/L蛋白胨和10g/L酵母抽提物组成。选择性SD-CAA培养基由以下各物组成:20g/L右旋糖、6.7g/L无氨基酸的酵母含氮碱基(Difco)、5g/L酪蛋白氨基酸(Bacto)、5.4g/L Na
通过电穿孔将pCT酵母展示质粒转化到啤酒酵母EBY100菌株中,并在30℃下于YPD中回收1小时,然后铺于SD-CAA板上。3天后,将酵母菌落在SD-CAA中接种过夜。根据已建立的程序,在SG-CAA中于30℃下诱导蛋白质的表达和酵母展示持续24小时
文库创建
将FGF1从人FGF1 cDNA(MGC克隆:9218,图像:3896359,残基:Phe16至Asp155)克隆到pCT载体(限制性位点:NheI,BamHI)中进行酵母展示。FGF1随机诱变文库是使用易错PCR生成的,如先前所述
文库筛选
将展示FGF1突变体的诱导的EBY100酵母细胞与纤溶酶在纤溶酶消化缓冲液(100mM Tris-HCl,0.01%BSA,pH 8.5)中在37℃下和/或FGFR1-Fc在PBS+0.1%BSA(PBSA)中在室温下一起孵育,如针对每次分选所述。蛋白酶消化步骤后,将细胞用PBSA洗涤,与蛋白酶抑制剂混合物(Sigma)的1:100稀释液在PBSA中一起孵育5分钟,然后再次用PBSA洗涤。FGFR孵育步骤后,将细胞用PBSA洗涤。为每次分选而孵育的酵母细胞数量是前一次分选中收集的细胞数量的~10x。将细胞以每毫升200万个细胞的密度孵育。在所有孵育步骤之后,将细胞用一抗和二抗染色。为了进行初次染色,将细胞适当地与抗HA-Tag(6E2)小鼠mAb(Cell Signaling)的1:1000稀释液和/或鸡抗c-Myc(Invitrogen)的1:2000稀释液一起孵育30分钟。初次染色之后,用PBSA洗涤细胞。在冰上进行二次染色10分钟。为了进行二次染色,针对每次分选使用以下抗体:分选1、3、4-针对抗c-myc的抗鸡IgY-PE(Santa CruzBiotechnology)和针对FGFR1-Fc的抗人IgG-FITC(Sigma Aldrich);分选2-山羊抗小鼠PE(Invitrogen)和山羊抗鸡IgY-AlexaFluor488(Santa Cruz Biotechnology)。
使用BD FACS Aria II(Stanford Shared FACS Facility),通过荧光激活细胞分选(FACS)对标记的酵母细胞进行分选。在每次分选中,根据为每次分选设置的标准,收集0.5%至10%的酵母细胞。将每次分选中收集的细胞在SD-CAA(pH 5,以限制细菌污染)中生长几天,直到OD达到5至8。在下一轮分选之前,在30℃下诱导克隆以在SG-CAA中进行酵母展示表达24小时。
为了测序和克隆,使用Zymoprep Yeast Plasmid Miniprep I试剂盒(ZymoResearch)从酵母细胞中提取质粒DNA。将所提取的DNA转化到DH10B电感受态细胞中并铺板。选择单个菌落并在LB培养基(Fisher Scientific)中生长。使用质粒miniprep试剂盒(GeneJet)从单菌落培养物中分离质粒DNA。DNA测序通过MCLAB进行。
酵母展示肽RTTTS和HTTS的结合亲和力测定
将50,000个诱导的酵母细胞与各种浓度的FGFR1-Fc在室温下在含1g/L BSA(PBSA)的磷酸盐缓冲盐水中一起孵育。将细胞以足够大的体积孵育以避免配体耗尽,并进行足够长的时间(通常3至24小时)以达到平衡。在孵育的最后30分钟期间,将酵母细胞与鸡抗c-Myc(Invitrogen)的1:2500稀释液在PBSA中一起孵育。将酵母沉淀,洗涤,然后与二抗的1:200稀释液在冰上一起孵育10分钟:针对FGFR1-Fc的抗人IgG-FITC(Sigma Aldrich)和针对抗c-myc的抗鸡IgY-PE(Santa Cruz Biotechnology)。将酵母洗涤,沉淀并重悬于PBSA中,之后立即使用EMD Millipore Guava EasyCyte,通过流式细胞仪进行分析。使用FlowJo(v7.6.1)分析流式细胞仪数据。绘制结合曲线,并且使用GraphPad Prism 6获得K
实施例3:蛋白水解稳定化的成纤维细胞生长因子的表征
摘要
蛋白水解稳定性可以在提高不稳定生长因子(诸如FGF1)的功效中发挥重要作用。研究显示,FGF1在培养物中迅速降解,部分原因是血清中所见的或由细胞表达的蛋白酶。在实施例2中,描述了为提高蛋白水解稳定性而进行的FGF1工程化。筛选出FGF1随机诱变文库中在表面酵母上表现出提高的蛋白水解稳定性的FGF1突变体。在此实施例中,描述了可溶性FGF1的重组表达和通过高通量筛选鉴定以改善FGF1中的蛋白水解稳定性的突变的表征。FGF1和FGF2在大肠杆菌表达系统中重组表达和纯化。已证实与野生型FGF1相比,FGF1BS4M1(D28N,L131R)和L131R突变体在蛋白水解方面更稳定,并且FGF1 L131R突变体充当有效的FGF途径拮抗剂。
引言
FGF1是诱导血管生成的有效调控分子,但其不良的稳定性限制了其维持蛋白质活性和延长功效的能力。在实施例2中,描述了使用高通量筛选来选择在与纤溶酶孵育时表现出提高的蛋白水解稳定性的FGF1突变体,纤溶酶是在ECM降解的疾病领域发现的一种重要的蛋白酶。在FGF1文库的四次分选之后,达成了完全一致,并鉴定了FGF1 BS4M1(D28N,L131R)突变体。在蛋白质区域中发现了突变,据报道这些突变对于FGF的稳定性很重要。
在此实施例中,描述了在筛选中鉴定的源自FGF1 BS4M1突变体的突变体FGF的可溶性表达和表征。从酵母展示载体克隆野生型和FGF1 BS4M1突变体,并将其插入大肠杆菌表达载体。纯化后,通过检测与酵母展示的FGFR3构建体的特异性结合来测试重组FGF1的正确折叠。
对于FGF1 BS4M1突变体和野生型FGF1,其可溶性稳定性和调节FGF途径的能力得到了进一步表征。测试蛋白质在纤溶酶或胰蛋白酶中的蛋白水解稳定性,并使用蛋白质印迹和条带强度定量测量它们在不同时间点的降解程度。探讨了D28N和L131R突变的意义及其对蛋白质稳定性的影响。测量FGF1的热稳定性以分析它们与蛋白水解稳定性的关系。为了测试FGF1在更多生物学相关条件下的稳定性,对它们在MDA-MB-231乳腺癌细胞培养物中的降解程度进行表征。此外,还进行了NIH3T3细胞中的ERK磷酸化测定,以表征调节FGFR激活下游信号分子(诸如ERK)的激活的能力。这些表征研究结果证明了工程化的FGF1突变体改善的蛋白水解稳定性和拮抗活性,以及它们用于抗血管生成疗法的潜力。
结果
FGF的重组表达
为了表达可溶性的工程化FGF1突变体并将其与野生型蛋白质进行比较,将这些蛋白质在大肠杆菌表达系统中重组表达。将FGF1和FGF1突变体克隆到pBAD载体中,以在细胞内表达重组蛋白。将pBAD FGF1表达载体转化到大肠杆菌菌株Rosetta中,该菌株利用大肠杆菌中很少使用的密码子来增强真核蛋白的表达。使用基于去污剂的溶液裂解细胞。然后使用Ni-NTA柱色谱法和尺寸排阻色谱法纯化蛋白质。通过考马斯染色的蛋白质凝胶和蛋白质印迹证实野生型FGF1的身份和纯度(图20A)。通过观察与酵母展示的FGFR3构建体的特异性结合,证实FGF1的正确折叠(图20B)。
FGF1突变体BS4M1在纤溶酶中的蛋白水解稳定性
为了测量野生型FGF1和FGF1突变体BS4M1(D28N/L131R)的蛋白水解稳定性,将100ng可溶性FGF在纤溶酶中孵育不同的孵育时间。然后,通过在蛋白质印迹上运行样品并对抗FGF染色来评估其降解率。通过测量每种条件下的条带强度,并由无纤溶酶孵育的蛋白质的条带强度的归一化,计算出FGF的残留量。发现与野生型FGF1相比,BS4M1(D28N,L131R)突变体在纤溶酶中在所有孵育时间点均表现出较低的降解水平(图21)。因此,证实了用于提高FGF1针对纤溶酶的蛋白水解稳定性的筛选是成功的。
将来自BS4M1的突变(D28N和L131R)掺入到稳定化的PM2(Q40P、S47I、H93G)突变体中,以生成PM3(D28N、Q40P、S47I、H93G、L131R)。孵育48小时后,在不同纤溶酶浓度下测量每种构建体的降解。发现将来自BS4M1的突变引入到来自PM2的突变中会导致在所有测试浓度下对蛋白水解降解的抗性显著提高(图22)。因此,得出的结论是,当与来自PM2的突变组合时,在BS4M1中新鉴定的突变对蛋白水解稳定性具有累加效应。
工程化的FGF突变体在胰蛋白酶中的蛋白水解稳定性
以类似的方式在胰蛋白酶中测量野生型FGF1和FGF1突变体BS4M1的蛋白水解稳定性。据推测,就针对纤溶酶的蛋白水解稳定性进行的工程化可以提高胰蛋白酶中的蛋白水解稳定性,因为纤溶酶和胰蛋白酶具有赖氨酸和精氨酸相同的主要特异性。另外,证实了对胰蛋白酶的降解更具抗性的FGF1突变体PM2(Q40P、S47I、H93G)对纤溶酶的切割也更有抗性。发现与野生型FGF1相比,BS4M1(D28N、L131R)突变体在胰蛋白酶中的所有孵育时间点均表现出较低的降解水平(图23)。因此,得出结论,针对纤溶酶中FGF1的蛋白水解稳定性进行的工程化也成功地提高了胰蛋白酶的蛋白水解稳定性。
FGF1突变体BS4M1中突变的意义
为了确定D28N和L131R突变对于向BS4M1突变体赋予蛋白水解稳定性是否都重要,创建了仅具有D28N或L131R突变的FGF1版本。通过评估其在纤溶酶中随时间的降解速率,测量了这些突变体的蛋白水解稳定性。发现与BS4M1(D28N/L131R)突变体相比,L131R突变体表现出相当的蛋白水解稳定性,但与野生型FGF1相比,D28N突变体表现出低得多的蛋白水解稳定性(图24)。结论是,当掺入可溶性表达的蛋白中时,D28N突变未转化为显著提高FGF1的蛋白水解稳定性。因此,L131R突变体的进一步表征得以继续。
还想知道在位置131处突变成精氨酸对于赋予蛋白水解稳定性而言是否是独特的,或者远离亮氨酸的突变是否重要。因此,将位置131替代地突变为丙氨酸(L131A)或赖氨酸(L131K),以便看出这些单个突变体是否维持或失去其蛋白水解稳定性的增强。在纤溶酶中评估了它们的降解率;发现与L131R相比,L131K保持相似的降解水平,而与野生型FGF1相比,L131A表现出更高的降解水平(图25)。
野生型FGF1和FGF1 L131R突变体的热稳定性
为了确定FGF1 L131R突变体的蛋白水解稳定性的改善是否归因于热稳定性的改善,测量了野生型FGF1和FGF L131R突变体的解链温度。使用ThermoFluor测定和疏水性染料来测量温度逐渐升高时每种蛋白质的去折叠
FGF1 L131R突变体在细胞培养物中的稳定性
在含有MDA-MB-231(一种乳腺癌细胞系,其表达尿激酶纤溶酶原激活物(uPA)以激活纤溶酶原并将其转化为纤溶酶)的培养物中测试FGF1 L131R突变体的稳定性。将500ng蛋白质与MDA-MB-231在培养物中一起孵育不同的孵育时间。浓缩每种条件下的所有蛋白质,并将每种条件上样到单独的孔中,以便通过蛋白质印迹进行分析。通过测量条带强度并由未在培养物中孵育的500ng蛋白质的归一化来对蛋白质的剩余量进行定量。发现与野生型蛋白相比,FGF1 L131R突变体在培养物中表现出提高的稳定性(图27)。
FGF1 L131R突变体是FGFR拮抗剂
为了表征FGF1 L131R调节FGF途径的能力,评估了其调节ERK(MAPK)磷酸化的能力,ERK是FGFR激活的下游的关键信号分子并且对诱导细胞增殖很重要
FGF1 L131R突变体与NIH3T3细胞的结合
表征了FGF1 L131R突变体的结合亲和力,并将其与野生型FGF1的结合亲和力比较。将表达FGFR的NIH3T3细胞与不同浓度的FGF1在4℃下一起孵育,以防止孵育。用荧光标记的抗His抗体标记细胞,以检测结合的His标记的FGF1。发现FGF1 WT和FGF1 L131R突变体对NIH3T3细胞均表现出10nM的结合亲和力(图30)。
讨论
在此实施例中,对在实施例2的纤溶酶中的蛋白水解稳定性筛选中鉴定的FGF突变体进行可溶表达和表征。重组表达后,发现FGF1易于表达和纯化。然而,发现表达的产量相当低,为1-3mg/L表达。如此低的蛋白质产量可能是由于FGF1的稳定性差所致。
与野生型FGF1相比,成功地证实了可溶性FGF1 BS4M1(D28N,L131R)突变体在纤溶酶中显示出更高的蛋白水解稳定性。当突变D28N和L131R与来自Zakrzewska等人
通过表征FGF1 D28N和L131R单突变体,发现大多数提高的蛋白水解稳定性归因于L131R,而D28N单突变体的蛋白水解稳定性甚至低于野生型。酵母展示的FGF1与可溶性大肠杆菌衍生的FGF1之间的D28N突变显著性的差异可能归因于仅在酵母展示的蛋白质中发生的糖基化。天冬氨酸突变为天冬酰胺导致真核生物引入NGx糖基化位点
L131R突变是违反直觉的突变,因为纤溶酶对精氨酸具有主要特异性。实际上,没有合理的设计策略会涉及将新的潜在切割位点引入蛋白质。然而,如实施例1中所讨论,主要特异性不是决定蛋白切割是否发生在潜在的切割位点的唯一决定因素;该位点周围的多个氨基酸以及该位点对蛋白酶的空间可及性也起很大作用。为了进一步探究远离亮氨酸的突变或位置131处的精氨酸突变对于提高蛋白水解稳定性是否重要,对FGF1 L131A和L131K单个突变体进行表征。已发现,与野生型FGF1相比,通过合理设计将亮氨酸131改变为丙氨酸导致蛋白水解稳定性降低,丙氨酸是常用于取代蛋白酶切割位点的氨基酸。然而,亮氨酸131突变为赖氨酸导致纤溶酶中提高的蛋白水解稳定性的保留,赖氨酸是纤溶酶具有主要特异性的另一种氨基酸。虽然考虑了纤溶酶对FGF1蛋白在位置131处的切割是否导致所得的蛋白片段的蛋白水解稳定性提高,但得出的结论是筛选不能针对这种突变进行选择。酵母展示的FGF1内的任何切割都将导致c-myc信号的丢失以及远离该突变体的选择。由于赖氨酸和精氨酸都是带正电的氨基酸,所以在该位置添加正电荷可能对提高FGF1的蛋白水解稳定性很重要。发现野生型FGF1与FGF1 L131R突变体之间的解链温度没有统计学上的显著差异,这表明热稳定性的提高不能解释蛋白水解稳定性的提高。因此,需要进一步的研究来确定性地发现L131R提高蛋白水解稳定性的机制。
还发现在含MDA-MB-231乳腺癌细胞的细胞培养物中,FGF1 L131R突变体似乎比野生型FGF1更稳定。这些细胞表达尿激酶纤溶酶原激活物(uPA),该酶将纤溶酶原切割并激活为纤溶酶。该结果对于证明FGF1 L131R突变体在更有生物学相关性的背景下显示出更高的稳定性具有重要意义。
最后,在NIH3T3细胞中使用ERK磷酸化测定,发现L131R突变将FGF1转变为FGF途径拮抗剂。鉴于提高蛋白水解稳定性的筛选仅选择与FGFR结合的突变体,而不选择蛋白质用作激动剂抑或拮抗剂,该结果很有趣,但是合理。FGF1 L131R突变体与NIH3T3细胞的结合亲和力与野生型FGF1大致相同,这解释了为什么FGF1 L131R突变体的IC50与抑制测定中使用的野生型FGF1浓度大致等摩尔。FGF1 L131R突变体对ERK磷酸化的抑制作用不完全,因为在野生型FGF1的存在下用最高浓度的FGF L13R突变体处理的样品显示出低的ERK磷酸化水平,但高于未处理的细胞。然而,在FGF1 R50E突变体中也观察到此现象,这是文献
总之,在实施例2和3中描述的结果表明能够成功地利用高通量筛选来提高FGF1在纤溶酶中的蛋白水解稳定性,并且鉴定出FGF2的关键蛋白水解稳定化的候选物。显示FGF1突变体在纤溶酶和胰蛋白酶中显示出更高的蛋白水解稳定性,并在培养物中显示出更高的稳定性。表明FGF1 L131R突变体充当可用于抑制NIH3T3细胞中FGF1诱导的ERK磷酸化的有效的FGF途径拮抗剂。FGF1突变体显示出以下前景:开发出一种蛋白水解稳定化的治疗分子,用于抗血管生成疗法以治疗疾病诸如癌症和眼中不必要的新血管形成。
材料和方法
重组FGF1表达和纯化
使用Rosetta(DE3)感受态细胞(Novagen)表达FGF1。该基因从人FGF1cDNA(Dharmacon)克隆到具有N末端6x His标签和阿拉伯糖诱导型启动子的pBAD/His B载体(Invitrogen)中。限制性位点XhoI和HindIII用于克隆。将pBAD FGF1-His质粒转化到化学感受态Rosetta(DE3)细胞中,在37℃下回收到1mL LB中伴随235rpm的振荡,并铺于含有氨苄青霉素(Amp)选择的LB板上。将菌落接种到5mL LB Amp中,并在37℃下生长过夜。将1mL过夜培养物用于接种100mL LB Amp表达培养物。使细胞在37℃下在235rpm的振荡下生长2至2.5小时。在~0.5的OD600下,用0.2%L-阿拉伯糖(Sigma Aldrich)诱导细胞。使蛋白质表达并维持在细胞质中。在将细胞离心分离并收集之前,使表达培养物在37℃下生长6小时。
用溶菌酶、DNA酶I和硫酸肝素在B-PER细菌蛋白提取试剂(Thermo Scientific)中裂解细胞30分钟。将提取混合物以15,000g离心10分钟,然后收集上清液并通过0.22μm过滤器过滤。将含有FGF1的上清液用结合缓冲液以稀释度1:10稀释用于Ni-NTA亲和纯化,如部分2.5.6.1中所详述。将上清液和结合缓冲液混合物加载到Ni-NTA柱上。浓缩来自Ni-NTA亲和纯化的洗脱液,并使用截断值为10kDa的Amicon Ultra-4 Centrifugal Filter Unit将缓冲液更换为PBS。如部分2.5.6.1中所述,使用采用Superdex 75柱的尺寸排阻色谱法纯化最终的FGF1-His蛋白。
FGF1单突变体的克隆
重叠延伸PCR用于将野生型FGF1突变为单个氨基酸突变体
蛋白水解稳定性测定
对于每种条件,将125ng蛋白质在20μL纤溶酶消化缓冲液(100mM Tris-HCl,0.01%BSA,pH 8.5)中与不同浓度的纤溶酶一起孵育或在37℃下孵育不同的孵育时间。在每个样品的适当孵育时间结束时,通过将样品保存在-20℃下来停止蛋白酶的消化。所有孵育完成后,将样品在冰上解冻以进行分析。将每个20μL样品与5μL NuPAGE LDS样品缓冲液和2μL NuPAGE样品还原剂混合。在运行SDS-PAGE凝胶之前,将样品加热到95℃持续10分钟。将凝胶与20%乙醇一起孵育10分钟,然后使用Invitrogen iBlot Gel Transfer Device(程序0,7分钟)将其印迹到硝酸纤维素膜上。
将蛋白质印迹用5%脱脂奶粉(Bio-Rad)在TBST(137mM NaCl、2.7mM KCl、25mMTris、0.1%Tween 20)中封闭1小时。初次染色是用小鼠抗FGF1(Sigma Aldrich,克隆2E12)的1:1000稀释液在含5%牛奶的TBST中进行1小时。在TBST中洗涤3次持续15分钟后,用山羊抗小鼠HRP(ThermoFisher Scientific)的1:2500稀释液进行二次染色持续2小时。在TBST中再洗涤3次持续15分钟后,用BioRad ChemiDoc XRS系统以Chemi Hi Sensitivity模式对印迹成像。使用ImageJ量化条带强度,并使用GraphPad Prism 6绘制归一化的条带强度。
用于测量解链温度的ThermoFluor测定
将50μL的1.2mg/mL蛋白质加载到96孔薄壁PCR板(Bio-Rad)中。将0.5μL SYPROOrange(Molecular Probes)添加到样品中并充分混合。在用BioRad CFX96 RT SystemC1000 Touch进行板分析之前,用塑料盖密封板。将板冷却至4℃达5分钟,然后将板以每分钟1℃的速率缓慢加热至100℃。在每个℃下监测和测量荧光变化。在Microsoft Excel上绘制荧光随温度变化的曲线,并通过找到荧光等于最大和最小荧光信号平均值的温度来计算解链温度。
细胞培养稳定性测定
将MDA-MB-231细胞以100,000个细胞/孔的密度接种在6孔板(Sigma Aldrich)上含10%胎牛血清(FBS)(Gibco)的Dulbecco改良的Eagle培养基(DMEM)(Gibco)中并于37℃下在5%CO
将蛋白质印迹用5%脱脂奶粉(Bio-Rad)在TBST(137mM NaCl、2.7mM KCl、25mMTris、0.1%Tween 20)中封闭1小时。初次染色是用小鼠抗FGF1(Sigma Aldrich,克隆2E12)的1:1000稀释液在含5%牛奶的TBST中进行1小时。在TBST中洗涤3次持续15分钟后,用山羊抗小鼠HRP(ThermoFisher Scientific)的1:2500稀释液进行二次染色持续2小时。在TBST中再洗涤3次持续15分钟后,用BioRad ChemiDoc XRS系统以Chemi Hi Sensitivity模式对印迹成像。使用ImageJ量化条带强度,并使用GraphPad Prism 6绘制归一化的条带强度。
NIH3T3 ERK磷酸化测定
将MDA-MB-231细胞以100,000个细胞/孔的密度接种在6孔板(Sigma Aldrich)上含10%新生小牛血清(NBCS)(Gibco)的Dulbecco改良的Eagle培养基(DMEM)(Gibco)中并于37℃下在5%CO
将蛋白质印迹用5%脱脂奶粉(Bio-Rad)在TBST(137mM NaCl、2.7mM KCl、25mMTris、0.1%Tween 20)中封闭1小时。初次染色是用兔抗磷酸-ERK1/2(Y202/Y204)抗体(Cell Signaling)或兔抗ERK1/2(Cell Signaling)的1:1000稀释液在含5%牛奶的TBST中进行1小时。在TBST中洗涤3次持续15分钟后,用山羊抗兔HRP(Santa Cruz Biotechnology)的1:2500稀释液进行二次染色2小时。在TBST中再洗涤3次持续15分钟后,用BioRadChemiDoc XRS系统以Chemi Hi Sensitivity模式对印迹成像。条带强度使用ImageJ进行定量,并使用GraphPad Prism 6绘制。
NIH3T3细胞结合测定
将NIH3T3细胞与不同浓度的野生型FGF1或FGF1 BS4M1突变体在结合缓冲液(20mMTris-HCl(pH 7.5),含有1mM MgCl
参考文献
1.Turner,N.&Grose,R.Fibroblast growth factor signalling:Fromdevelopment to cancer.Nat.Rev.Cancer 10,116-129(2010).
2.Behm,B.,Babilas,P.,Landthaler,M.&Schreml,S.Cytokines,chemokines andgrowth factors in wound healing.J.Eur.Acad.Dermatology Venereol.26,812-820(2012).
3.Andrae,J.,Gallini,R.&Betsholtz,C.Role of platelet-derived growthfactors in physiology and medicine.Genes Dev.1276-1312(2008).doi:10.1101/gad.1653708.revealing
4.Carmeliet,P.VEGF as a key mediator of angiogenesis incancer.Oncology 69,4-10(2005).
5.Anitua,E.,Sánchez,M.,Orive,G.&Andia,I.Delivering growth factors fortherapeutics.Trends Pharmacol.Sci.29,37-41(2008).
6.Papo,N.,Silverman,A.P.,Lahti,J.L.&Cochran,J.R.Antagonistic VEGFvariants engineered to simultaneously bind to and inhibit VEGFR2 and v 3integrin.Proc.Natl.Acad.Sci.108,14067-14072(2011).
7.Kapur,S.et al.Engineered ligand-based VEGFR antagonists withincreased receptor binding affinity more effectively inhibit angiogenesis.Bioeng.Transl.Med.2,81-91(2017).
8.Steed,D.L.Clinical evaluation of recombinant human platelet-derivedgrowth factor for the treatment of lower extremity diabeticulcers.J.Vasc.surgeryl 21,71-78(1995).9.
Kitamura,M.et al.Randomized Placebo-Controlled and Controlled Non-Inferiority Phase III Trials Comparing Trafermin,a Recombinant HumanFibroblast Growth Factor 2,and Enamel Matrix Derivative in PeriodontalRegeneration in Intrabony Defects.J.Bone Miner.Res.31,806-814(2016).
10.Boontheekul,T.&Mooney,D.J.Protein-based signaling systems intissue engineering.Curr.Opin.Biotechnol.14,559-565(2003).
11.Werb,Z.,Vu,T.H.,Rinkenberger,J.L.&Coussens,L.M.Matrix-degradingproteases and angiogenesis during development and tumor formation.Apmis 107,11-18(1999).
12.Copeland,R.a et al.The structure of human acidic fibroblast growthfactor and its interaction with heparin.Arch.Biochem.Biophys.289,53-61(1991).
13.Mignatti,P.,Rifkin,D.B.,Welgus,H.G.&Parks,W.C.Proteinases andtissue remodeling.in The Molecular and Cellular Biology of Wound Repair(ed.Clark,R.A.F.)(Springer,1988).
14.Zakrzewska,M.et al.Increased protein stability of FGF1 cancompensate fbr its reduced affinity for heparin.J.Biol.Chem.284,25388-25403(2009).
15.Motomura,K.et al.An FGF1:FGF2 chimeric growth factor exhibitsuniversal FGF receptor specificity,enhanced stability and augmented activityuseful for epithelial proliferation and radioprotection.Biochim.Biophys.Acta-Gen.Subj.1780,1432-1440(2008).
16.Lee,J.&Blaber,M.Increased Functional Half-life of FibroblastGrowth Factor-1 by Recovering a Vestigial Disulfide Bond.Proteins andProteomics 1,37-42(2010).
17.Kobielak,A.et al.Protease Resistant Variants of FGF1 withProlonged Biological Activity.Protein Pept.Lett.434-443(2014).
18.Draghia-Akli,R.et al.Myogenic expression of an injectableprotease-resistant growth hormone-releasing hormone augments long-term growthin pigs.Nat.Biotechnol.17,1179-1183(1999).
19.Cook,a L.et al.Purification and analysis of proteinase-resistantmutants of recombinant platelet-derived growth fsctor-BB exhibiting improvedbiological activity.Biochem.J.281,57-65(1992).
20.Gosalia,D.N.,Salisbury,C.M.,Ellman,J.A.&Diamond,S.L.HighThroughput Substrate Specificity Profiling of Serine and Cysteine ProteasesUsing Solution-phase Fluorogenic Peptide Microarrays.Mol.Cell.Proteomics 4,626-636(2005).
21.Buchtova,M.et al.Instability restricts signaling of multiplefibroblast growth factors.Cell.Mol.Lfie Sci.72,2445-2459(2015).
22.
23.Romero,N.,Tinker,D.,Hyde,D.&Rucker,R.B.Role of plasma and serumproteases in the degradation of elastin.Arch.Biochem.Biophys.244,161-168(1986).
24.Wagner,I.et al.Serum Proteases Potentiate BMP-Induced Cell CycleRe-entry of Dedifferentiating Muscle Cells during Newt LimbRegeneration.Dev.Cell 40,608-617(2017).
25.Rosengart,T.K.,Johnson,W.V.,Friesel,R.,Clark,R.&Maciag,T.Heparinprotects heparin-binding growth factor-I from proteolytic inactivation in vitro.Biochem.Biophys.Res.Commun.152,432-440(1988).
26.Cochran,J.R.,Kim,Y.S.,Lippow,S.M.,Rao,B.&Wittrup,K.D.Improvedmutants from directed evolution are biased to orthologoussubstitutions.Protein Eng.Des.Sel.19,245-253(2006).
27.Jones,D.S.,Tsai,P.-C.&Cochran,J.R.Engineering hepatocyte growthfactor fragments with high stability and activity as Met receptor agonistsand antagonists.Proc.Natl.Acad.Sci.108,13035-13040(2011).
28.Zheng,X.et al.Proteomic analysis for the assessment of differentlots of fetal bovine serum as a raw material for cell culture.PartIV.Application of proteomics to the manufacture of biologicaldrugs.Biotechnol.Prog.22,1294-1300(2006).
29.INAGAMI,T.&STURTEVANT,J.M.Nonspecific catalyses by alpha-chymotrypsin and trypsin.J.Birol.Chem.235,1019-23(1960).
30.Lauer,G.et al.Expression and proteolysis of vascular endothelialgrowth factor is increased in chronic wounds.J.Invest.Dermatol.115,12-18(2000).
31.Korc,M.&Friesel,R.E.The role of fibroblast growth factors in tumorgrowth.Curr.Cancer Drug Targets 9,639-51(2009).
32.Beenken,A.&Mohammadi,M.The FGF family:biology,pathophysiology andtherapy.Nat Rev Drug Discov.8,235-253(2009).
33.Mason,I.Initiation to end point:The multiple roles of fibroblastgrowth factors in neural development.Nat.Rev.Neurosci.8,583-596(2007).
34.Teven,C.M.,Farina,E.M.,Rivas,J.&Reid,R.R.Fibroblast growth factor(FGF)signaling in development and skeletal diseases.Genes Dis,1,199-213(2014).
35.Eswarakumar,V.P.,Lax,I.&Schlessinger,J.Ccllular signaling byfibroblast growth factor receptors.Cytokine Growth Factor Rev.16,139-149(2005).
36.Knights,V.&Cook,S.J.De-regulated FGF receptors as therapeutictargets in cancer.Pharmacol.Ther.125,105-117(2010).
37.Zhang,J.&Li,Y.Therapeutic uses of FGFs.Semin.Cell Dev.Biol.53,144-154(2016).
38.Presta,M.et al.Fibroblast growth factor/fibroblast growth factorreceptor system in angiogenesis.Cytokine Growth Facto rRev.16,159-178(2005).
39.Carmeliet,P.Fibroblast growth factor-1 stimulates branching andsurvival of myocardial arteries:A goal for therapeutic angiogenesis?Circ.Res.87,176-178(2000).
40.Schumacher,B.,Pecher,P.,von Specht,B.U.&Stegmann,T.Induction ofneoangiogenesis in ischemic myocardium by human growth factors:first clinicalresults of a new treatment of coronary heart disease.Circulation 97,645-650(1998).
41.Mori,S.et al.A Dominant-Negative FGF1 Mutant(the R50E Mutant)Suppresses Tumorigenesis and Angiogenesis.PLoS One 8,(2013).
42.Comerota,A.J.et al.Naked plasmid DNA encoding fibroblast growthfactor type 1 for the treatment of end-stage unreconstructible lowerextremity ischemia:Preliminary results of a phase I trial.J.asc.Surg.35,930-936(2002).
43.Nikol,S.et al.Therapeutic angiogenesis with intramuscular NV1FGFimproves amputation-free survival in patients with critical limbischemia.Mol.Ther.16,972-978(2008).
44.Belch,J.et al.Effect of fibroblast growth factor NV1FGF onamputation and death:A randomised placebo-controlled trial of gene therapy incritical limb ischaemia.Lancet 377,1929-1937(2011).
45.Rask-Andersen,M.,Zhang,J.,Fabbro,D.&Schio,H.B.Advances in kinasetargeting:current clinical use and clinical trials.Trends Pharmacol.Sci 35,604-620(2014).
46.Saksela,O.,Moscatelli,D.,Sommer,A.&Rifkin,D.B.Endothelial cell-derived heparan sulfate binds basic fibroblast growth factor and protects itfrom proteolytic degradation.J.Cell Biol.107,743-51(1988).
47.Whitelock,J.M.,Murdoch,A.D.,Iozzo,R.V.&Underwood,P.A.Thedegradation of human endothelial cell-derived perlecan and release of boundbasic fibroblast growth factor by stromelysin,collagenase,plasmin,andheparanases.J.Biol.Chem.271,10079-10086(1996).
48.Park,S.et al.Limitations of yeast surface display in engineeringproteins of high thermostability.Protein Eng.Des.Sel.19,211-217(2006).
49.Kowalski,J.M.,Parekh,R.N.&Wittrup,K.D.Secretion Efficiency inSaccharomyces cere V isiae of Bovine Pancreatic Trypsin Inhibitor MutantsLacking Disulfide Bonds Is Correlated with ThermodynamicStability.Biochemistry 37,1264-1273(1998).
50.Lee,J.,Dubey,V.K.,Longo,L.M.&Blaber,M.A Logical OR Redundancywithin the Asx-Pro-Asx-Gly Type I??-Turn Motif.J.Mol.Biol.377,1251-1264(2008).
51.Dubey,V.K.,Lee,J.,Somasundaram,T.,Blaber,S.&Blaber,M.Spackling theCrack:Stabilizing Human Fibroblast Growth Factor-1 by Targeting the N and Cterminus β-Strand Interactions.J.Mol.Biol.371,256-268(2007).
52.Chao,G.et al.Isolating and engineering human antibodies usingyeast surface display.Nat.Protoc.1,755-768(2006).
53.Deventer,J.A.Van&Wittrup,K.D.Yeast surface display for antibodyisolation:Library construction,library screening,and affinitymauraion.Methods Mol.Biol.1131,151-181(2014).
54.Lavinder,J.J.,Hari,S.B.,Sullivan,B.J.&Magliery,T.J.High-throughputthermal scanning:a general,rapid dye-binding thermal shift screen for proteinengineering.J Am Chem Soc.131,3794-3795(2009).
55.Ericsson,U.B.,Hallberg,B.M.,Detitta,G.T.,Dekker,N.&Nordlund,P.Thermofluor-based high-throughput stability optimization of proteins forstructural studies.Anal.Biochem.357,289-298(2006).
56.Yun,Y.et al.Fibroblast Growth Factors:Bioloby,Function,andApplication for Tissue Regeneration.J.Tissue Eng.(2010).doi:10.4061/2010/218142
57.Sharrocks,A.D.Cell Cycle:Sustained ERK Signalling Represses theInhibitors.16,540-542
58.Hervio,L.S.et al.Negative selectivity and the evolution ofprotease cascades:the specificity of plasmin for peptide and proteinsubstrates.Chem.Biol.7,443-453(2000).
59.Breitling,J.&Aebi,M.N-Linked Protein Glycosylation intheEndoplasmic Reticulum.Cold Spring Harb.Perspect.Biol.(2013).doi:10.1101/cshperspect.a013359
60.Yamaji,S.et al.A Novel Fibroblast Growth Factor-1(FGF1)Mutant thatActs as an FGF Antagonist.PLoS One 5,(2010).
61.Weber,J.D.,Raben,D.M.,Phillips,P.J.&Baldassare,J.J.Sustainedactivation of extracellular-signal-regulated kinase 1(ERK1)is required forthe continued expression of cyclin D1 in G 1 phase.Biochem.J.68,61-68(1997).
62.Ho,S.N.,Hunt,H.D.,Horton,R.M.,Pullen,J.K.&Peasea,L.R.Site-directedmutagenesis by overlap extension using the polymerase chain reaction.Gene 77,51-59(1989).
- 用于治疗的工程化成纤维细胞生长因子变体与工程化肝细胞生长因子变体的组合
- 作为受体拮抗剂的工程化纤维细胞生长因子变体