一种采用高效液相色谱法测定他达拉非中甲胺的方法
文献发布时间:2023-06-19 19:27:02
技术领域
本发明属于药物分析检测技术领域,具体涉及一种采用高效液相色谱法测定他达拉非中甲胺的方法。
背景技术
他达拉非是环磷酸鸟苷(cGMP)特异性磷酸二酯酶5(PDE5)的选择性、可逆性抑制剂,其起效迅速,且不受高脂饮食和酒精摄入影响,是FDA和CFDA批准的唯一长效PDE5抑制剂,具有勃起功能障碍(ED)适应症外,还获批用于治疗肺动脉高压和良性前列腺增生。今年来,药品质量安全问题备受关注,因此,对他达拉非原料药的质量控制有了更高的要求。
他达拉非的合成工艺中需要加入过量甲胺增加收率,而甲胺属于低毒类碱性物质,对皮肤黏膜具有刺激性和腐蚀性、可影响中枢神经系统的兴奋性、引起哮喘等过敏反应,因此需要严格控制他达拉非中甲胺的残留量。中国专利CN108169395B公开了他达拉非油管物质的分析检测方法,其利用结合紫外检测器的高效液相色谱法进行杂质检测,可以高灵敏度的将他达拉非与杂质分离。薛峰峰等人采用气相色谱法测定他达拉非原料药中甲胺与三乙胺的残留量,其进样口温度为200℃,检测器温度为260℃,流速为3.46mL/min,但该条件接近该气相色谱柱的耐受条件,导致色谱柱在使用过程中柱效明显下降,使得该方法重现性较差。同时由于甲胺为易挥发气体,且没有紫外吸收,因此考虑采用现将甲胺衍生化后,再用HPLC对衍生化后的目标峰进行检测。
发明内容
本发明的目的是提供一种测定他达拉非中甲胺含量的方法,该方法灵敏度高,重复性好,结果准确,能够用于他达拉非生产中甲胺的质量控制。
本发明的技术方案如下:
一种测定他达拉非中甲胺含量的方法:先将甲胺衍生化后再进行高效液相色谱检测。
进一步地,衍生化方法为在他达拉非中加入对甲苯磺酰氯和甲胺,振摇后再加入KOH溶液,摇匀后稀释定容即可。
更进一步地,衍生化的溶液中他达拉非的浓度为1-2mg/mL,对甲苯磺酰氯的浓度为0.02-0.04mg/mL,甲胺的浓度为20-40mg/mL,KOH的浓度为0.005-0.01mg/mL,其中甲胺的质量分数为25%-30%。
进一步地,衍生化后的溶液制备时,先将各种试剂制备成储备溶液,然后再用溶剂稀释定容;所述溶剂为乙腈-水(50:50)。
进一步地,所述高效液相色谱的色谱条件为:色谱柱为C18,流动相A为0.1%磷酸,流动相B为乙腈,检测波长为210-254nm,柱温为30℃±5℃,流速1.0mL/min±0.1mL/min。
更进一步地,检测波长为226nm,柱温为30℃,流速1.0mL/min,进样量为10μL。
进一步地,色谱柱为Waters XbridgeC18色谱柱,以十八烷基硅烷键合硅胶为填充剂固定相。
进一步地,检测过程中,采用流动相进行梯度洗脱,具体洗脱程序如表1所示:
表1
本发明还提供一种将所述方法在他达拉非原料药生产质量控制中的应用。
本发明具有以下有益效果:
本发明提供的高效液相色谱法测定他达拉非中甲胺的方法,将他达拉非原料药中甲胺杂质衍生化为N-甲基对甲苯磺酰胺,并将其与邻近杂质的分离度达到2.34,确定大于1.5,成功将甲胺与其他杂质分离,并准确的检测出甲胺的含量。检测限LOD为0.00301μg/mL,约相当于样品浓度的0.0006%,定量限为0.01002μg/mL,约相当于样品浓度的0.002%。同时通过线性实验证明该方法在定量限至限度浓度的150%(0.0100μg/mL-0.7514μg/mL)的范围内线性良好。
该检测方法的准确度高,多次实验的目标峰的平均回收率为100.1%,RSD为2.1%,小于3.0%;同具有良好的重复性,能够满足实际样品检测的需要,适用于他达拉非原料药中甲胺杂质测定,测定结果可靠,提高了产品质量标准,控制了甲胺杂质超标风险,有利于保证合成药物产品安全有效。
附图说明
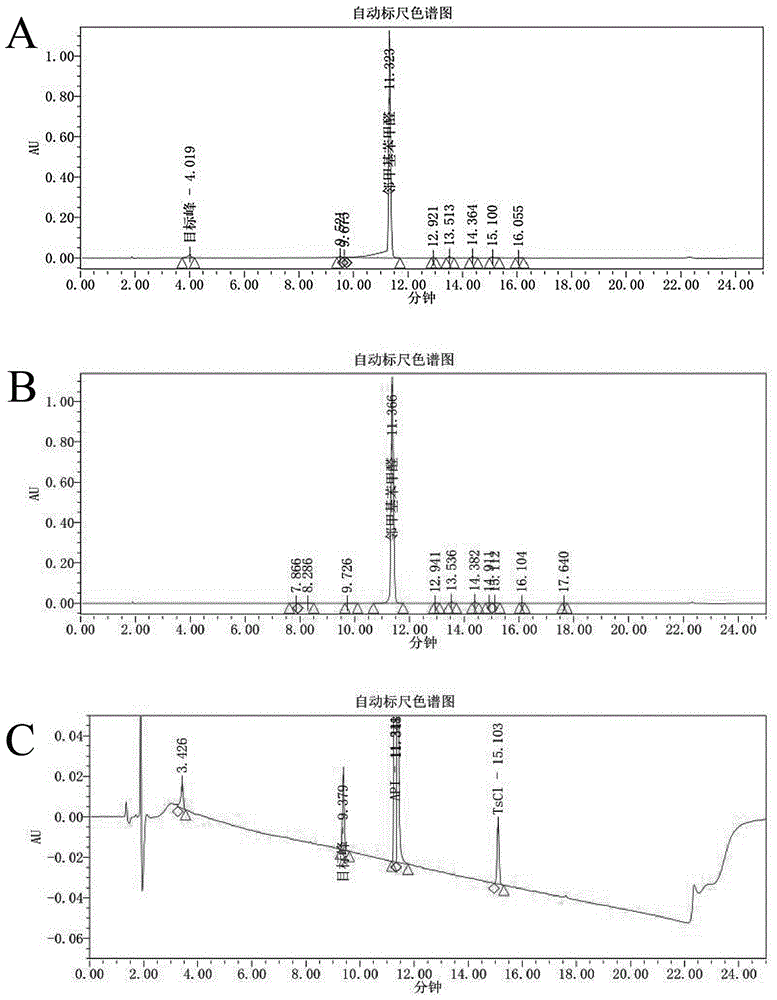
图1为实施例1的测试图谱,图中A为以邻甲基苯甲醛为衍生化试剂,0.1%三氟乙酸为流动相A的色谱图,B为以邻甲基苯甲醛为衍生化试剂,纯水为流动性A的色谱图,C为以对甲苯磺酰氯为衍生化试剂,0.1%三氟乙酸为流动相A的色谱图。
图2为实施例3中确定检测波长的测试图谱。
图3为实施例3中不同流动相种类的检测图谱,图中A为三氟乙酸,B为磷酸。
图4为实施例3中采用3-1~3-6梯度洗脱的检测图谱。
图5为实施例4中N-甲基对甲苯磺酰胺线性关系图。
具体实施方式
下面将结合实施例对本发明的实施方案进行详细描述,但是本领域技术人员将会理解,下列实施例仅用于说明本发明,而不应视为限定本发明的范围。
下述实施例中使用的液相色谱仪为Waters e2695,UV和PDA检测器;电子分析天平为梅特勒-托利多XSE 205;他达拉非样品、对照品(SM01、SM02、TL01、TL02、IP-A、IP-C、IP-D、IP-I、异构体A、杂质E、杂质H、杂质G);甲胺水溶液(AR级);邻甲基苯甲醛(AR级)、对甲苯磺酰氯(AR级)、乙腈(HPLC级)、氢氧化钾(AR级)、磷酸(AR级)。
下述实施例中常用的各溶液的配方如下:
甲胺储备液(浓度为0.05mg/mL,其中甲胺水溶液浓度按25%计算):准确称取甲胺水溶液2.0292g,置于100mL量瓶中,加纯化水定容摇匀;精密量取该溶液1.0mL置于100mL量瓶中,加纯化水定容,摇匀即得。
对甲苯磺酰氯(TsCl)储备液的配制(浓度为0.5mg/mL):精密称定TsCl 50mg,置于100mL量瓶中,加乙腈稀释至刻度,摇匀得到对甲苯磺酰氯储备液。
KOH储备液的配制(浓度为0.1mg/mL):精密称定KOH25mg,置于100mL量瓶中,加纯化水稀释至刻度,摇匀得到KOH储备液。
供试样品溶液的配制:取他达拉非样品25mg,置于50mL量瓶中,精密量取对甲苯磺酰氯储备液1.0mL,盖好瓶盖后,振摇1-2min,加入氢氧化钾储备液1.0mL,摇匀,放置10min后用乙腈-水(50:50)稀释至刻度,摇匀得到供试样品溶液。
对照溶液的配制:精密量取对甲苯磺酰氯储备液1.0mL,置于50mL量瓶中,再迅速精密量取甲胺储备液1.0mL,盖好瓶盖后,振摇约1min,加入氢氧化钾储备液1.0mL,摇匀,放置10min后用乙腈-水(50:50)稀释至刻度,摇匀得到对照溶液。
准确度80%溶液:精密称定他达拉非样品25mg,置于50mL量瓶中,精密量取对甲苯磺酰氯储备液1.0mL,再迅速精密量取甲胺储备液0.8mL,盖好瓶盖后振摇lmin,加入KOH储备液1.0mL摇匀,放置10min后用乙腈-水(50:50)稀释至刻度,摇匀即得。
准确度100%溶液:精密称定他达拉非样品25mg,置于50mL量瓶中,精密量取对甲苯磺酰氯储备液1.0mL,再迅速精密量取甲胺储备液1.0mL,盖好瓶盖后振摇1min,加入KOH储备液1.0mL摇匀,放置10min后用乙腈-水(50:50)稀释至刻度,摇匀即得。
准确度120%溶液:精密称定他达拉非样品25mg,置于50mL量瓶中,精密量取对甲苯磺酰氯储备液1.0mL,再迅速精密量取甲胺储备液1.2mL,盖好瓶盖后振摇1min,加入KOH储备液1.0mL摇匀,放置10min后用乙腈-水(50:50)稀释至刻度,摇匀即得。
下述实施例中色谱柱中采用十八烷基硅烷键合硅胶(Waters Xbridge C18,4.6*150mm,5μm)为固定相。
实施例1衍生化试剂的选择
分别采用邻甲基苯甲醛和对甲苯磺酰氯作为衍生化试剂,检测峰面积从而定量检测甲胺,确定最适衍生化试剂,方法如下:
(1)邻甲基苯甲醛衍生化试剂的配制:在顶空瓶中加入2mL邻甲基苯甲醛(浓度为0.5mg/ml)和1mL甲胺水溶液(0.05mg/ml)、1mL KOH储备液,压盖,室温反应1h后用50%乙腈水溶液稀释,得到衍生化试剂;
(2)对甲苯磺酰氯衍生化试剂的配制:准确称取他达拉非(API)50mg置于50mL量瓶中,量取对甲苯磺酰氯储备液2mL,再准确量取甲胺储备液1mL,盖好瓶盖后振摇30s,再加入KOH储备液1mL,摇匀后用乙腈-水(50:50)稀释至刻度,摇匀后作为衍生化试剂;
(3)分别取两种衍生化试剂10μL注入色谱仪中,用C18为填充剂,以0.1%三氟乙酸或纯水为流动相A,乙腈为流动相B,按表2进行梯度洗脱,检测波长为278nm。
表2梯度洗脱
以三氯乙酸为流动相时(图1A),加入甲胺后生成的目标峰与邻甲基苯甲醛的峰前移,故需要进行方法优化。
采用纯水代替0.1%三氯乙酸(图1B)时,发现4.0min目标峰小时,这可能是由于该反应是可逆反应,生成的目标产物不稳定易分解,故邻甲基苯甲醛不适宜作为衍生化试剂。
对甲苯磺酰氯作为衍生化试剂(图1C)时,在三氟乙酸体系下反应正常进行,故可以作为衍生化试剂,但目标峰的响应较低,保留时间为9.379min,峰面积为170465,因此采用对甲苯磺酰氯作为衍生化试剂时,其衍生化方法和色谱条件均有待优化。
实施例2衍生化反应条件的优化
采用对甲苯磺酰氯作为衍生化试剂,用C18为填充剂,以0.1%三氟乙酸为流动相A,乙腈为流动相B,按照表1的梯度进行洗脱,检测波长为226nm。
一、KOH用量的确定
由于对甲苯磺酰氯和甲胺反应会生成HCl,抑制反应的正向进行,因此需要加入KOH作为缚酸剂。因此在加入过量对甲苯磺酰氯的前提下,需要对KOH用量进行考察确定,KOH的用量与色谱结果见表3。
甲胺+TsCl+API:称取API 50mg,置于50mL量瓶中,量取对甲磺氯储备液2mL,再准确量取甲胺储备液1mL,盖好瓶盖后,摇匀,用乙腈-水(50:50)稀释至刻度,摇匀即得;
甲胺+TsCl+KOH+API:称取API 50mg,置于50mL量瓶中,量取对甲磺氯储备液2mL,再准确量取甲胺储备液1mL,盖好瓶盖后,振摇3s,再加入KOH储备液1mL,摇匀,用乙腈-水(50:50)稀释至刻度,摇匀即得;
甲胺+TsCl+KOH:量取对甲磺氯储备液2mL,再准确量取甲胺储备液1mL,盖好瓶盖后,振摇3s,再加入不同量的KOH储备液,摇匀,用乙腈-水(50:50)稀释至刻度,摇匀即得。
表3KOH用量与色谱峰面积
结果表明,随着甲胺:TsCl:KOH的摩尔比逐渐上升,目标峰面积先逐渐增大再逐渐降低,其中当摩尔比约为1:3:5时,目标峰的面积最大,因此选择甲胺:TsCl:KOH的摩尔比约为1:3:5进行衍生化。
二、衍生化反应时间的确定
由于对甲苯磺酰氯和甲胺反应的时间可能对反应进行程度存在影响,因此当反应溶液中加入KOH溶液后放置不同的时间后分别进行色谱,探究衍生化时间对回收率的影响,结果见表3;在对甲苯磺酰氯衍生化试剂中加入KOH溶液后立即稀释定容,然后连续进样6针进行色谱检测,确定峰面积RSD,结果见表4,其中甲胺:TsCl:KOH的摩尔比为1:3:5。
表4
表5
实验结果表明(表4),加入KOH溶液后立即稀释时,由于先后操作时间差异,导致对照溶液中峰面积偏小,所得回收率在107.6%-114.4%之间,均偏大,说明在该操作时间内反应仍在进行。但将立即稀释后的溶液倒入进样小瓶,连续进样6针,峰面积RSD为0.36%(表5),说明稀释后反应基本稳定,不再进行。因此应重点考察稀释前的放置时间。
当放置10min后进行稀释,回收率在101.8%-104.2%之间,说明放置10min后,反应基本趋于平衡(表4),因此,选择放置10min作为反应时间。
综上所述,衍生化的反应条件为将甲胺和TsCl和按照摩尔比为1:3的比例混合均匀,然后再加入5倍摩尔比的KOH溶液,混匀后制备得到衍生化试剂,然后放置10min后再进行稀释,最后将稀释得到的溶液进行高效液相色谱。
实施例3液相色谱条件的确定
采用对甲苯磺酰氯作为衍生化试剂,用C18为填充剂,以乙腈为流动相B,进行洗脱,确定检测波长、流动相的种类、洗脱梯度以及样品的浓度和进样量。
一、波长的确定
采用0.1%三氟乙酸为流动相A,并按照表1中的梯度进行洗脱,得到LCMS结果,确定9.35min为N-甲基对甲苯磺酰胺的目标峰,MS+1(分子量)为186.16。根据PDA检测器中目标峰的扫描图,确定目标峰的检测波长为226nm(图2)。
二、流动相的确定
分别采用0.1%三氟乙酸和0.1%磷酸作为流动相A,按照表2中的梯度进行洗脱,检测基线偏移情况。
结果如图3所示,三氟乙酸在低波长下有末端吸收,本方法选择226nm波长时,基线偏移较大,而采用0.1%磷酸作为流动相A时,基线偏移量较小,因此在后续实验中采用磷酸代替三氟乙酸作为流动相A,从而提高方法的灵敏度。
三、洗脱梯度的确定
由于HPLC方法中,226nm波长下,他达拉非API及其杂质会出峰,可能会干扰甲胺衍生物的检测,因此在考察方法专属性时,结合杂质混合衍生化溶液中杂质峰与目标峰的分离情况来选择合适的洗脱梯度。
SM01储备液:取SM01对照品约2mg,置于10mL EP管中,加稀释剂4mL使之溶解,摇匀即得。SM02、TL01、TL02、IP-A、IP-C、IP-D、IP-I、杂质E、杂质H、杂质G、异构体A储备液同法配制。
SM01定位溶液:取SM01储备液1mL,置于10mL量瓶中,加稀释剂定容,摇匀即得。SM02、TL01、TL02、IP-A、IP-C、IP-D、IP-I、杂质E、杂质H、杂质G、异构体A定位溶液同法配制。
杂质混合衍生化溶液配制:精密称取他达拉非50mg,置50mL量瓶中,量取对甲苯磺酰氯储备液1mL,再分别量取杂质储备液(SM01、SM02、TL01、TL02、IP-A、IP-C、IP-D、IP-I、杂质E、杂质H、杂质G)各0.5mL,盖好瓶盖后,振摇约1min,再加入KOH储备液1mL,摇匀,放置10min后用乙腈-水(50:50)稀释至刻度,摇匀即得。
其他检测条件均不变,采用同一批次样品,分为6组,分别为3-1、3-2、3-3、3-4、3-5和3-6;在不同梯度条件下,对杂质混合衍生化溶液进行色谱检测,具体洗脱梯度如下表6-11:
表6 3-1对应梯度条件
表7 3-2对应梯度条件
表8 3-3对应梯度条件
表9 3-4对应梯度条件
表10 3-5对应梯度条件
表11 3-6对应梯度条件
结果表明(图4),SM02、IP-D以及目标峰的位置相邻,受到洗脱梯度的影响而稍微改变三者之间的位置;其中在3-1的梯度洗脱下,SM02和IP-D重合,保留时间为4.590min,会干扰目标峰的检测(图4A);在3-2的梯度洗脱下,SM02和IP-D重合,保留时间为4.581min,仍会干扰目标峰的检测,没有改善(图4B);在3-3的梯度洗脱下,SM02和IP-D分开,保留时间依次为4.868min、5.166min,此时,目标峰介于两者之间仍会干扰目标峰的检测(图4C);在3-4的梯度洗脱下,IP-D和SM02分开,IP-D出峰在SM02之前,保留时间依次为3.620min、3.955min,此时目标峰仍介于两者之间会受到干扰(图4D);在3-5的梯度洗脱下,SM02和IP-D分开,保留时间依次为4.515min、4.800min,此时,目标峰介于两者之间仍会干扰目标峰的检测(图4E);在3-6的梯度洗脱下,出峰顺序依次为SM02、目标峰、IP-D,可以发现,三种物质已完全分离、说明该洗脱梯度能满足实验要求(图4F)。
四、样品浓度和进样量的确定
用乙腈-水(30:70)配制他达拉非样品浓度为1mg/mL溶液时,样品能完全溶解,但放置1h后,有固体析出,故进入色谱仪后,样品溶液可能会析出。因此样品浓度确定调整为0.5mg/mL,甲胺限度为0.10%,浓度为0.5μg/mL。按照该浓度配制对照溶液,分别进样10μL和20μL,检测高效液相色谱法的灵敏度,结果如表12:
表12不同进样量下目标峰的峰面积和对照溶液中目标峰的S/N
结果显示(表12),降低样品浓度且进样量为10μL时,目标峰S/N为5293,此时浓度相当于样品浓度的0.05%,灵敏度符合要求,故确定样品浓度为0.5mg/mL,进样量为10μL。
实施例5
(1)色谱条件:色谱柱以十八烷基硅烷键合硅胶为填充剂(Waters Xbridge C18,4.6mm×250mm,5μm或效能相当的色谱柱)为固定相;流动相A:0.1%磷酸,流动相B:乙腈;流速:1.0mL/min;柱温:30℃;检测波长:226nm;进样体积:10μL;洗脱梯度如表10所示。
(2)溶液制备:
空白溶液/稀释剂:乙腈-水(50:50)
衍生化空白溶液:精密量取对甲苯磺酰氯储备液1.0mL,置于50mL量瓶中,盖好瓶盖后,振摇1min,加入KOH储备液1.0mL,摇匀放置10min后用乙腈-水(50:50)稀释至刻度,摇匀即得。
供试品溶液:称取他达拉非样品约25mg,置于50mL量瓶中,精密量取对甲苯磺酰氯储备液1.0mL,盖好瓶盖后,振摇1min,加入KOH储备液1.0mL,摇匀放置10min后用乙腈-水(50:50)稀释至刻度,摇匀即得。
(3)专属性实验
将空白溶液、衍生化空白溶液、实施例3(三)中的SM01定位溶液和杂质混合衍生化溶液分别进行色谱,统计出峰的保留时间以及峰与峰之间的分离度,结果见表13:
表13专属性实验结果
结果表明:空白溶液、衍生化空白溶液对目标峰的检测无干扰;杂质混合衍生化溶液中,目标峰与相邻峰之间的分离为2.34,大于1.5,具有专属性。
(4)系统适应性
取连续6针对照溶液进入色谱仪,检测目标峰面积与RSD,结果见表14:
表14系统适应性结果
结果显示,连续6针对照溶液中,目标峰的峰面积RSD为0.30%,小于1.0%,说明该方法系统适用性良好。
(5)定量限检测限实验
定量限溶液:精密量取对甲苯磺酰氯储备液1.0mL,置于50mL量瓶中,再迅速精密量取甲胺储备液适量,盖好瓶盖后振摇1min,加入KOH储备液1.0mL,摇匀放置10min后用乙腈-水(50:50)稀释至刻度,摇匀,使目标峰信噪比大于10,即得定量限溶液;平行配制6份溶液。
检测限溶液:精密量取对甲苯磺酰氯储备液1.0mL,置于50mL量瓶中,再迅速精密量取甲胺储备液适量,盖好瓶盖后振摇1min,加入KOH储备液1.0mL,摇匀放置10min后用乙腈-水(50:50)稀释至刻度,摇匀使目标峰信噪比大于3,即得检测限溶液。
将定量限和检测限溶液分别进行色谱,检测目标峰的相关参数并计算定量限和检测限的浓度。
表15定量限检测限结果
结果显示:6份检测限溶液中目标峰的S/N平均值为6.03,检测限为3.01ng/mL,相当于样品浓度的0.0006%。
定量限溶液中目标峰的S/N平均值为37.60,峰面积的RSD为0.61%,定量限为10.02ng/mL,相当于样品浓度的0.002%。
(6)线性实验
线性范围考察设计为定量限浓度至限度浓度的150%范围。设计6个浓度点,分别为定量限,10%,50%,80%,100%,150%对照品溶液浓度。
线性浓度10%:精密量取对甲苯磺酰氯储备液1.0mL,置于50mL量瓶中,再迅速精密量取甲胺储备液0.1mL,盖好瓶盖后振摇1min,加入KOH储备液1.0mL摇匀,放置10min后用乙腈-水(50:50)稀释至刻度,摇匀即得。
线性浓度50%:精密量取对甲苯磺酰氯储备液1.0mL,置于50mL量瓶中,再迅速精密量取甲胺储备液0.5mL,盖好瓶盖后振摇1min,加入KOH储备液1.0mL摇匀,放置10min后用乙腈-水(50:50)稀释至刻度,摇匀即得。
线性浓度80%:精密量取对甲苯磺酰氯储备液1.0mL,置于50mL量瓶中,再迅速精密量取甲胺储备液0.8mL,盖好瓶盖后振摇lmin,加入KOH储备液1.0mL摇匀,放置10min后用乙腈-水(50:50)稀释至刻度,摇匀即得。
线性浓度100%:精密量取对甲苯磺酰氯储备液1.0mL,置于50mL量瓶中,再迅速精密量取甲胺储备液1.0mL,盖好瓶盖后,振摇1min,加入KOH储备液1.0mL摇匀,放置10min后用乙腈-水(50:50)稀释至刻度,摇匀即得。
线性浓度150%:精密量取对甲苯磺酰氯储备液1.0mL,置于50mL量瓶中,再迅速精密量取甲胺储备液1.5mL,盖好瓶盖后振摇1min,加入KOH储备液1.0mL摇匀,放置10min后用乙腈-水(50:50)稀释至刻度,摇匀即得。
将上述6个浓度点的对照品溶液分别进行色谱,并进行2次平行实验,统计峰面积,结果见表16:
表16线性实验结果
结果显示(表16和图5),线性方程为y=231669x+2923.7,相关系数R为0.9962,Y轴截距占100%限度响应值浓度比值小于10%,表明在定量限至限定浓度的150%(0.0100μg/mL-0.7514μg/mL)范围内线性良好。
(7)准确度实验
空白样品:精密称定他达拉非样品25mg,置于50mL量瓶中,精密量取对甲苯磺酰氯储备液1.0mL,盖好瓶盖后振摇1min,加入KOH储备液1.0mL摇匀,放置10min后用乙腈-水(50:50)稀释至刻度,摇匀即得。
取准确度80%、100%以及120%的溶液分别进行色谱,每种准确度溶液进行3次重复实验,记录峰面积并计算回收率和RSD,结果见表17:
表17准确度实验结果
结果表明,3种准确度溶液共9份样品的目标峰的回收率均在95.92%-102.17%之间,平均回收率为100.1%,RSD为2.1%,小于3.0%,准确度高。
(8)重复性实验
取(7)中的对照溶液和准确度100%溶液,平均分成6份,然后分别进入液相色谱仪中进行色谱,记录色谱图中的峰面积,计算含量检测实验的重复性,结果见表18:
表18重复性实验结果
6份重复性样品中目标峰的平均含量为0.10%,RSD为1.8%,小于3.0%,说明该方法重复性良好。
(9)溶液稳定性实验
取(7)中的对照溶液和准确度100%溶液,室温放置一段时间后分别进入液相色谱仪中,检测溶液中目标峰的含量,考察溶液稳定性,结果见表19:
表19溶液稳定性实验结果
对照溶液和准确度100%溶液在室温下放置26h,准确度100%溶液中,目标峰含量RSD为0.45%,小于3.0%,说明溶液在室温下放置26h,稳定性良好。
(10)耐用性实验
分别取衍生化空白溶液、杂质混合衍生化溶液、对照溶液和准确度100%溶液进行不同色谱条件耐用性考察。色谱条件为:柱温:温度±2C;流速:1mL/min±0.1mL/min;流动相:比例±2%。
表20耐用性实验结果
结果表明,衍生化空白溶液对目标峰的检测无干扰。各色谱条件下,杂质混合衍生化溶液中目标峰与相邻杂质之间的分离度均大于1.5。各色谱条件下,准确度100%溶液中目标峰含量RSD为0.09%。
(10)衍生化方法耐用性实验
分别取对照溶液和准确度100%溶液考察衍生化方法的耐用性。衍生化条件变化如下:
1)迅速加甲胺后振摇时间:0.5min,1min,1.5min;
2)加入KOH后放置时间:5min,10min,15min。
表21衍生化方法耐用性实验结果
准确度100%溶液中,迅速加甲胺后振摇1±0.5min后目标峰的回收率在96.46%-99.39%之间,说明振摇时间对回收率影响较小,对结果准确度影响不大。加入KOH后放置10±5min后目标峰的回收率在95.85%-99.39%之间。说明放置时间对回收率影响较小,对结果准确度影响不大。说明该衍生化方法耐用性良好。
- 一种高效液相色谱法分离测定度他雄胺软胶囊中度他雄胺及有关物质的方法
- 一种高效液相色谱法测定甲磺草胺原药中甲磺草胺的方法
- 一种高效液相色谱法测定甲磺草胺原药中甲磺草胺的方法