高品质聚丙烯腈及其可控合成方法与应用
文献发布时间:2023-06-19 19:27:02
技术领域
本发明涉及聚丙烯腈碳纤维技术领域,尤其涉及一种高品质聚丙烯腈及其可控合成方法与应用。
背景技术
碳纤维是指含碳量在90%以上的高强度、高模量纤维,因其具有高强度、高模量、低密度、耐高温等一系列优异性能,在航空航天、国防军事、能源、土木建筑、体育等领域得到了广泛的应用。用于生产碳纤维的前驱体主要包括聚丙烯腈碳纤维、沥青基碳纤维以及黏胶基碳纤维三大类。其中由聚丙烯腈加工成的碳纤维因工艺相对简单、性能最优,成为碳纤维生产的主流。在聚丙烯腈基碳纤维的制备工艺流程中,聚丙烯腈原丝的质量是决定高品质碳纤维制备的关键因素,生产高品质聚丙烯腈原丝需要同时具备高品质聚丙烯腈纺丝原液的生产技术及先进的纺丝工艺。其中,高品质聚丙烯腈纺丝原液的合成是关键。
当前工业上合成聚丙烯腈的方法主要包括均相聚合的一步法和水相悬浮聚合的两步法两大类。均相聚合指单体在特定溶剂中发生聚合,直接得到均一的丙烯腈聚合物溶液,所得到的聚合物无需纯化、分离,经脱单脱泡后可直接用于纺丝,该法工艺流程简单、生产成本较低,但存在所得聚合产物分子量低、反应热不易扩散等不足。两步法指先通过在水相悬浮液中进行聚合,再将聚合物溶解在合适的溶剂中并进行纺丝。该法得到的聚合物一般具有较高的相对分子质量,可以很好的满足高端领域的要求,但该法步骤繁琐、成本高,同时产品中少量残留物的存在会影响后续炭化处理时所得碳纤维的最终性能。要合成高品质碳纤维,所使用聚丙烯腈纺丝原液需满足分子量高、分子量分布窄、无金属离子等杂质残留等要求。
相比于传统的自由基聚合,可控活性自由基聚合能够很好的实现对聚合物分子量、分子量分布、结构等的有效调控,有望用于高品质碳纤维纺丝原液的制备。在众多的可控活性聚合方法中,可逆加成-断裂链转移(RAFT)聚合因具有适用单体范围广、分子设计能力强、具有高的末端链活性等特性,得到了广泛的关注。目前,现有技术在使用RAFT聚合合成聚丙烯腈时,多采用热引发的方式。比如,现有技术CN 110078860 A中公开了一种具有低多分散指数(PDI)的丙烯腈(PAN)聚合物的制备方法,其在RAFT聚合中属于热引发,在使用时须额外添加引发剂,通过引发剂分解产生引发自由基引发反应进行,但该法的可控性较差,同时需要加热等条件带来额外的能量消耗。
基于光的清洁、环保、低能耗等特点,使用光作为外部刺激调控聚合反应进行的光控RAFT法近年来得到了快速发展。当前,光控RAFT聚合体系主要由单体、光催化剂、链转移剂组成,催化剂经光照后与链转移剂作用从而引发聚合反应的进行。但在这一体系中,催化剂的残留会带来聚合物的污染,影响最终产品的性能。且目前光控RAFT聚合体系所得产物分子量分布较宽,可控性较差,同时体系不具备耐氧性,不利于规模化放大。
现有技术中张剑雄在《2-氰基-2-丙基十二烷基三硫代碳酸酯(TTP)调控的(甲基)丙烯酸酯的光解RAFT聚合研究》中发现了TTP在光解RAFT聚合中的应用,但其应用于甲基丙烯酸酯,所得聚合物分子量及单体转换率均不高,且其分子量及分子量分布的可控性较差。
而对于聚丙烯腈而言,其单体极性强,同时其聚合物不溶于其单体溶液,对聚合体系提出了更高的要求,同时在分子量及分子量分布的控制上也存在更大的难度。
发明内容
本发明提供一种高品质聚丙烯腈及其可控合成方法与应用,用以解决现有技术中光控RAFT聚合体系所得产物分子量分布较宽,可控性较差,同时体系不具备耐氧性,不利于规模化放大的缺陷,实现高品质聚丙烯腈的可控合成。
本发明提供一种聚丙烯腈的合成方法,包括:将丙烯腈单体、链转移剂与氧清除剂等在溶剂中进行混合得到混合溶液,而后在光源下发生聚合反应;其中,所述链转移剂为2-氰基-2-丙基十二烷基三硫代碳酸酯、2-(十二烷基三硫代碳酸酯基)-2-甲基丙酸和4-氰基-4-[(十二烷基硫烷基硫羰基)硫烷基]戊酸中的一种或多种;丙烯腈单体与链转移剂的摩尔比为200~10000:1。
现有技术中对聚丙烯腈合成中分子量的控制通常通过调整反应时间、单体浓度、引发剂浓度等来实现;而本发明经过大量研究后偶然发现,在上述体系下,使用光源作为外部刺激引发聚合反应进行,能够仅通过调控丙烯腈单体与链转移剂的摩尔比来控制聚合物分子量,而且可控性大大提高;较现有技术而言,本发明体系下单体转换率显著提高,且制备得到的丙烯腈聚合物分子量可控性有效增强,同时能够保证分子量分布更窄。
作为优选,丙烯腈单体与链转移剂的摩尔比为500~6000:1。
作为优选,以混合溶液的总质量为基准,丙烯腈单体浓度为5~40wt%;更优选地,浓度为15~26wt%。
本发明发现,当丙烯腈浓度在上述范围内时,所得的聚丙烯腈的分子量可控性更强,且分子量分布更窄。
作为优选,所述氧清除剂为葡萄糖氧化酶、吡喃糖氧化酶、三乙醇胺和三甲胺中的一种或多种。
更优选地,以混合溶液总质量为基准,所述氧清除剂用量为0.05~2wt%;更优选地为0.1~0.5wt%。
作为优选,所述光源为紫外或可见光。
更优选地,所述光源的波长为420~460nm。
作为优选,聚合反应时间为0.5~48h,更优选地为6~24h。
在本发明中,由于所述聚合反应为光引发,因此对反应温度没有特殊限制。作为优选,所述聚合反应的温度为0~80℃,更优选地为20~45℃。
在本发明中,上述反应体系在不同的溶剂体系中均具有普适性,本领域技术人员能够根据实际需要选择不同的有机溶剂使用。
作为优选,所述有机溶剂包括二甲基亚砜、二甲基乙酰胺、碳酸乙烯酯、N,N-二甲基甲酰胺中的一种或多种。
作为优选,所述合成方法还包括:在所述聚合反应结束后,将反应产物进行沉淀处理;其中,所使用的沉淀剂为水与小分子醇类的混合物。
在所述聚合反应结束后,其中,所使用的沉淀剂为水与小分子醇类的混合物。
更优选地,所述小分子醇类包括甲醇、乙醇或异丙醇。
作为优选,所述水醇体积比为10~0.1:1,更优选地为2~0.5:1。
在具体实施中,将反应产物进行沉淀处理后,本领域技术人员能够根据实际情况进行后续过滤、干燥等常规后处理操作。
本发明进一步提供一种聚丙烯腈,其由上述的合成方法制得。
作为优选,所述聚丙烯腈分子量为10,000~420,000g/mol,分子量分布为1.1~1.5。
本发明还提供一种聚丙烯腈原丝的制备方法,其包括:由聚丙烯腈纺丝原液制备得到聚丙烯腈原丝;在聚合得到所述聚丙烯腈纺丝原液时,使用上述的聚丙烯腈的合成方法,且在所述混合溶液中还包括共聚单体。
作为优选,所述共聚单体为丙烯酸丁酯、衣康酸、丙烯酸、甲基丙烯酸甲酯、丙烯酸甲酯、乙酸乙烯酯、甲基丙烯酸、甲基丙烯酸正丁酯、甲基丙烯酸异丁酯和丙烯酰胺中的一种或多种。
更优选地,以丙烯腈单体与共聚单体的总量为基准,所述共聚单体的用量为0.5~20mol%,优选地为2~5mol%。
本发明进一步提供上述制备方法制得的聚丙烯腈原丝。
在具体实施过程中,本领域技术人员能够根据实际情况将上述聚丙烯腈原丝通过碳化、煅烧等常规操作制备得到聚丙烯腈碳纤维。
在本发明中,所述聚丙烯腈与聚丙烯腈原丝均能通过使用本发明所述反应体系以及限定用量下的链引发剂与丙烯腈单体,采用外部光源作为刺激引发RAFT聚合反应的进行,从而实现一种绿色环保、且可控性强的高品质碳纤维纺丝原液丙烯腈聚合物的合成方法。
基于上述技术方案,本发明的有益效果在于:
本发明提供了一种高品质聚丙烯腈及其可控合成方法与应用,通过使用光控RAFT聚合技术,重新构建反应体系,使得丙烯腈聚合物分子量具有高度可控性,且分子量分布窄,同时无需额外添加引发剂或光催化剂,产物纯净,为合成高品质碳纤维奠定了基础。其次,上述体系具有极高的单体转化率(>95%),可简化脱单等后处理工艺流程,极大的节约成本,从而能满足工业化生产的需要。此外,上述体系无需加热亦无需搅拌,使用低能耗的光作为外部刺激调控反应,极大的节约了能耗。同时,本发明所构建的体系对于不同的溶剂体系具有普适性,且对反应温度没有特殊要求,不需要额外进行加热等。
附图说明
为了更清楚地说明本发明或现有技术中的技术方案,下面将对实施例所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
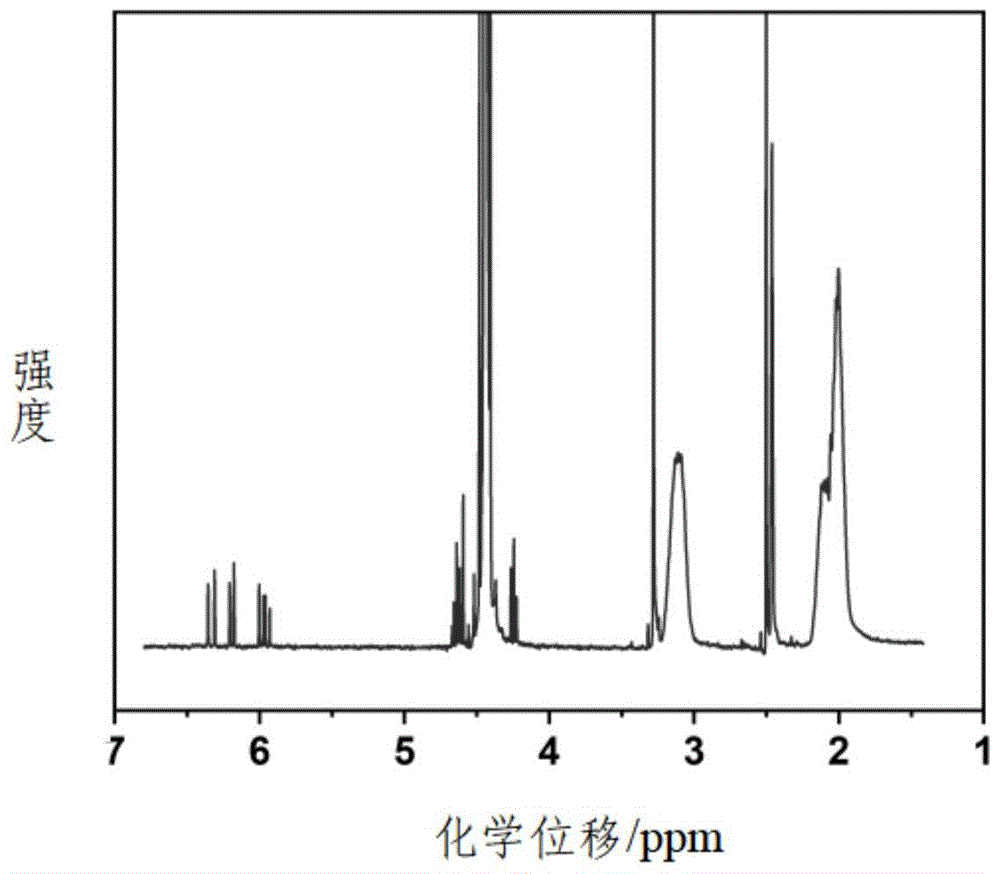
图1是本发明实施例1提供的测定单体转换率的核磁谱图;
图2是本发明实施例2提供的不同单体与链转移剂比例条件下所合成丙烯腈聚合物的GPC曲线图;
图3是本发明实施例3提供的所合成聚合物的GPC曲线图。
具体实施方式
为使本发明的目的、技术方案和优点更加清楚,下面将结合本发明中的附图,对本发明中的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
若未特别说明,实施例中所用的各原料均为市售常规原料,所用的技术手段为本领域技术人员所熟知的常规手段。
本发明实施例中采用凝胶渗透色谱测定丙烯腈聚合物的分子量和分子量分布;采用核磁测定不同条件下的单体转换率。
实施例1
本实施例提供一种聚丙烯腈的可控合成方法,具体包括如下步骤:
将丙烯腈单体用氧化铝粉末过滤,量取1.5mL丙烯腈单体,7.5μL 2-氰基-2-丙基十二烷基三硫代碳酸酯,4.5mg葡萄糖氧化酶和3.0mL二甲基亚砜加入10mL石英反应管中,在4mW cm
用二甲基亚砜稀释所得产物,用甲醇与水的混合溶液为稀释剂沉淀样品,抽滤后干燥收集最终产物。采用核磁测定单体转化率,如图1所示,所得单体转换率为96%,表明该体系具有很高的单体转换率,展现出很强的工业应用价值。同时,所得聚合物分子量为59,500g/mol;分子量分布为1.23。
实施例2
本实施例提供一种聚丙烯腈的可控合成方法,具体包括如下步骤:
将丙烯腈单体用氧化铝粉末过滤,量取1.5mL丙烯腈单体,3.0mL二甲基亚砜,分别量取2μL、4μL、8μL、16μL的2-氰基-2-丙基十二烷基三硫代碳酸酯以及4mg葡萄糖氧化酶依次加入10mL石英反应管中,将反应管放入光反应器中,在4mW cm
不同单体与链转移剂比例条件下所得聚合物分子量及分子量分布、单体转换率如表1所示,相应摩尔比条件下所得丙烯腈聚合物的GPC曲线如图2所示。由表1及图2可知,该方法具有极高的单体转换率,所得聚合物分子量分布窄且符合预先设定的分子量;当反应时间一定时,丙烯腈单体与链转移剂的摩尔比越大,聚合物分子量越大,表明该方法可设计性强,能够通过调控丙烯腈单体与链转移剂的摩尔比来对聚丙烯腈的分子量进行设计。
表1丙烯腈单体M与链转移剂不同摩尔比所得聚合物的分子量及分布
实施例3
本实施例提供一种聚丙烯腈纺丝原液的制备方法,具体包括如下步骤:
将丙烯腈单体、甲基丙烯酸甲酯单体用氧化铝粉末过滤,量取1.5mL丙烯腈单体,0.1mL甲基丙烯酸甲酯,4.5μL 2-氰基-2-丙基十二烷基三硫代碳酸酯,6mg葡萄糖氧化酶和3mL二甲基亚砜加入10mL石英反应管中,用橡胶塞密封后将反应管放入光反应器中,在4mWcm
实施例4
本实施例提供一种聚丙烯腈纺丝原液的制备方法,具体包括如下步骤:
将丙烯腈单体、丙烯酸丁酯用氧化铝粉末过滤,量取1.35mL丙烯腈单体,0.05mL丙烯酸丁酯,称量14.8mg 2-(十二烷基三硫代碳酸酯基)-2-甲基丙酸,6μL三乙醇胺和3.0mL二甲基亚砜加入10mL石英反应管中,将反应管放入光反应器中,在4mW cm
实施例5
本实施例提供一种聚丙烯腈纺丝原液的制备方法,具体包括如下步骤:
将丙烯腈单体用氧化铝粉末过滤,量取1.5mL丙烯腈单体,称量100mg衣康酸,12μL三乙醇胺4.2mg 2-(十二烷基三硫代碳酸酯基)-2-甲基丙酸和3.0mL二甲基亚砜加入10mL石英反应管中,将反应管放入光反应器中,在5mW cm
实施例6
本实施例提供一种聚丙烯腈纺丝原液的制备方法,具体包括如下步骤:
将丙烯腈单体、丙烯酸甲酯单体用氧化铝粉末过滤,量取1.4mL丙烯腈单体,20μL丙烯酸甲酯,称量60mg衣康酸,10mg葡萄糖氧化酶,4.2mg 2-(十二烷基三硫代碳酸酯基)-2-甲基丙酸和3.0mL二甲基亚砜加入10mL石英反应管中,将反应管放入光反应器中,在4mWcm
实施例7
本实施例提供一种聚丙烯腈纺丝原液的制备方法,具体包括如下步骤:
将丙烯腈单体用氧化铝粉末过滤,量取1.35mL丙烯腈单体,50mg衣康酸,10mg葡萄糖氧化酶,2μL 2-氰基-2-丙基十二烷基三硫代碳酸酯,和3.0mL二甲基亚砜加入10mL石英反应管中,将反应管放入光反应器中,在4mW cm
实施例8
本实施例提供一种聚丙烯腈纺丝原液的制备方法,具体包括如下步骤:
将丙烯腈单体、甲基丙烯酸甲酯单体用氧化铝粉末过滤,量取1.35mL丙烯腈单体,0.05mL甲基丙烯酸甲酯,称量8mg 2-(十二烷基三硫代碳酸酯基)-2-甲基丙酸,10mg葡萄糖氧化酶和3.0mL二甲基亚砜加入10mL石英反应管中,将反应管放入光反应器中,在2mW cm
在具体实施过程中,本领域技术人员能够根据需要,使用本领域常规方法将实施例3~8提供的聚丙烯腈纺丝原液制备成聚丙烯腈原丝,在此不再赘述。
最后应说明的是:以上实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的精神和范围。