预测性反应生物标志物发现方法
文献发布时间:2023-06-19 19:28:50
相关申请的引用
本申请要求2020年06月19日提交的美国临时专利申请号63/041,440;2020年09月17日提交的63/079,507;和2021年04月09日提交的63/172,904的申请日权益。上述相关申请的全部内容,包括在列表和附图中的任何序列,均通过引用并入本文。
背景技术
大部分针对疾病指征(如癌症、自身免疫性疾病或神经退行性疾病)的治疗干预措施仅对部分患者有效,而对其余患者最好是无效的(如果不是有害的话)。治疗可能无法从某种治疗中获益的患者不仅经济负担沉重(尤其是对于涉及专利创新单克隆抗体的某些昂贵治疗方案),而且可能对正在治疗的患者造成潜在危害,更不用说失去了用其他可能有效的替代疗法治疗这些患者的机会。
因此,需要为特定治疗方案确定合适的患者群体,以最大限度地提高成功治疗的机会。
发明内容
本发明的一个方面提供了一种用于鉴定使用T/B细胞靶向免疫调节疗法治疗疾病(例如,癌症)的预测性反应生物标志物(PRB)的方法反应生物标志物,所述方法包括下述步骤:(a)在所述疾病(例如,癌症)的治疗前样品中,鉴定具有与所述疾病(例如,癌症)的匹配治疗后样品中的克隆扩增T/B细胞的TCR(T细胞受体)/BCR(B细胞受体)克隆型相同的TCR/BCR克隆型的具有反应能力的T/B细胞,其中在所述匹配治疗后样品中所述克隆扩增T/B细胞在所述治疗后克隆扩增;和(b)生成在所述治疗前样品的所述具有反应能力的T/B细胞中上调和/或下调的基因的列表,以建立所述PRB;(c)任选地,使用逻辑回归系数对在所述PRB中的每个基因进行加权,所述逻辑回归系数通过将作为预测因子的所述的每个基因与作为结果的扩增状态和/或临床反应状态拟合而获得。
在某些实施方式中,步骤(a)包括:(1)通过在足以使T细胞靶向免疫调节化合物对所述疾病(例如,癌症)的未治疗样品的离体培养物的免疫调节作用显现的条件和时间段内将所述离体培养物内的T细胞群与所述化合物接触来生成所述匹配治疗后样品;(2)将从所述离体培养物分离、纯化或富集的单个T细胞包封到用于功能性质的单细胞分析的皮升液滴中,从而分别根据所述功能性质的存在或不存在将每个包封的单个T细胞分离到第一应答T细胞池和第二非应答T细胞池中;(3)针对在所述第一应答T细胞池和所述第二非应答T细胞池中的每个包封的单个T细胞确定TCR克隆型,从而将所述应答T细胞的TCR克隆型鉴定为在所述匹配治疗后样品中的克隆扩增T细胞的所述TCR克隆型;(4)将从所述治疗前样品中分离、纯化或富集的单个T细胞包封到皮升液滴中以鉴定具有与在所述匹配治疗后样品中所述克隆扩增T细胞的TCR克隆型相同的TCR克隆型的所述具有反应能力的T细胞。
在某些实施方式中,步骤(a)包括:(1)从适宜的供体获得所述疾病(例如,癌症)的所述治疗前样品和所述匹配治疗后样品,其中所述匹配治疗后样品已经在足以使所述化合物对所述治疗后样品内的T细胞群的免疫调节作用显现的条件和时间段内与所述化合物接触;(2)将从所述治疗后样品中分离、纯化或富集的单个T细胞包封到用于功能性质的单细胞分析的皮升液滴中,从而分别根据所述功能性质的存在或不存在将每个包封的单个T细胞分离到第一应答T细胞池和第二非应答T细胞池中;(3)针对在所述第一应答T细胞池和所述第二非应答T细胞池中的每个包封的单个T细胞确定TCR克隆型,从而将所述应答T细胞的TCR克隆型鉴定为在所述匹配治疗后样品中的克隆扩增T细胞的所述TCR克隆型;(4)将从所述治疗前样品中分离、纯化或富集的单个T细胞包封到皮升液滴中以鉴定具有与在所述匹配治疗后样品中所述克隆扩增T细胞的TCR克隆型相同的TCR克隆型的所述具有反应能力的T细胞。
在某些实施方式中,步骤(b)包括:(5)使用单细胞RNA测序(scRNA-seq)来鉴定在所述治疗前样品中的所述具有反应能力的T细胞中上调的基因的所述列表,其中在所述PRB中的每个基因具有>0.2的log2倍变化,并且在<40%的在所述匹配治疗后样品中不克隆扩增的具有TCR克隆型的T细胞中表达。
在某些实施方式中,所述scRNA-seq是使用专门对于所述化合物的作用机制设计的基因组套或者设计用于评估免疫调节的预定基因组套进行的。
在某些实施方式中,所述治疗前样品是血液样品。
在某些实施方式中,所述疾病是癌症,如实体瘤。
在某些实施方式中,所述离体培养物是从疾病组织新分离的。
在某些实施方式中,所述离体培养物是从储存的(例如,冻存的)疾病组织建立的。
在某些实施方式中,所述离体培养物是单细胞悬液。
在某些实施方式中,所述离体培养物是贴壁培养物。
在某些实施方式中,所述时间段是约12-24hr、24-36hr、36-48小时、长达3天、长达4天或长达5天。
在某些实施方式中,所述条件包括:所述化合物的一系列浓度之一,和/或具有或不具有与第二治疗剂的组合。
在某些实施方式中,所述第二治疗剂包括细胞因子(例如,IL-2、TNF)、TCR刺激(例如,通过CD3交联)和/或共刺激(例如,CTLA、B7、CD28等)。
在某些实施方式中,所述化合物是免疫调节抗体。
在某些实施方式中,步骤(2)通过微流控装置进行。
在某些实施方式中,所述单个T细胞是通过分离、纯化或富集特定T细胞亚群或群(如CD4
在某些实施方式中,所述单个T细胞是通过一般地对于T细胞进行分离、纯化或富集分离、纯化或富集的。
在某些实施方式中,所述功能性质包括:IFN-γ分泌和/或激活标志物的上调。
在某些实施方式中,所述基因组套包括:针对增殖的基因(如Ki67);针对干细胞样特性的基因(如TCF7);针对T细胞激活的基因(如CD25、颗粒酶B、穿孔蛋白、CD28、TNF、IL2、IFNG、4-1BB、CD38、CD69、OX-40、GITR、IL4、IL6);针对T细胞耗竭的基因(CD39、CTLA-4、EOMES、PD-1、TIGIT、2B4、LAG-3、T-bet、TIM3、TOX、CD160、IL-10);针对迁移的基因(如CD31、CD103、CXCR5、CXCR4、CCR7、CCR3、CCR4、CCR8、CCR5、CXCR3);及其组合。
在某些实施方式中,所述基因组套还包括CD4、CD8、FOXP3、CD62L、CD44、CD127、CD27、IL33R和/或PTPRC。
在某些实施方式中,在步骤(2)中,所述功能性质是用于根据所述功能性质的存在或不存在使用微流控装置物理分离所述应答T细胞和非应答T细胞。
在某些实施方式中,在步骤(2)中,所述功能性质是激活标志物或应答印记(其任一或两者可以通过例如单细胞RNA测序确定),并且其中所述应答T细胞和非应答T细胞不是物理分离的,而是基于所述激活标志物或应答印记的表达或缺乏表达而实质上区分的。
在某些实施方式中,所述方法还包括对所述治疗前样品和/或所述治疗后样品进行分析以表征所述样品的例如细胞类型组成(通过,例如,流式细胞术)和T细胞上的靶标表达。
本发明的另一个方面提供了一种用于选择使用T细胞靶向免疫调节化合物治疗疾病(例如,癌症)的患者的方法,所述方法包括:通过评估通过本发明的方法获得的所述疾病(例如,癌症)的预测性反应生物标志物(PRB)中基因的表达状态和/或表达水平获得所述患者的临床应答,以确定所述患者的所述临床反应评分是否超过了预先确定的阈值临床反应评分;
其中具有超过所述阈值评分的临床反应评分的患者鉴定为从所述治疗中获益并选择进行所述治疗,和/或
其中具有低于第二阈值评分的临床反应评分的患者鉴定为不会从所述治疗中获益并不选择进行所述治疗。
本发明的另一个方面提供了一种使用T细胞靶向免疫调节化合物治疗患者的疾病(例如,癌症)的方法,所述方法包括:(i)通过评估通过本发明的方法获得的所述疾病(例如,癌症)的预测性反应生物标志物(PRB)中基因的表达状态和/或表达水平获得所述患者的临床反应评分,以确定所述患者的所述临床反应评分是否超过了预先确定的阈值临床反应评分;其中具有超过所述阈值评分的临床反应评分的患者鉴定为从所述治疗中获益并选择进行所述治疗;和(ii)向在(i)中鉴定为从所述治疗中获益并选择进行所述治疗的所述患者施用治疗有效量的所述化合物。
应当理解的是,本发明的任何一个实施方式,包括仅在实例或权利要求中描述的那些,可以与本发明的任意一个或多个实施方式组合,除非明确否认或是不适当的。
附图说明
图1是显示本发明的一个实施方式的非限制性示意图。
图2是显示本发明的另一个实施方式的非限制性示意图。
图3用于说明T细胞对免疫调节剂(如OX-40激动剂)治疗的反应的预定基因组套中的示例性基因的列表。
图4是显示基于单细胞RNA测序(scRNA-seq)数据的比较在扩增的CD8+T细胞对比非扩增的CD8+T细胞的差异表达(显著增加或显著降低)基因的火山图。
图5A显示了在POPLAR试验的阿特珠单抗组中生存分析显示CD8 T细胞扩增标记高(Sig
图5B显示在IMvigor210试验的阿特珠单抗组中生存分析显示CD8 T细胞扩增标记高(Sig
图6显示了来自使用ICB治疗的患者的扩增和非扩增T细胞亚群的分类。基于其基因表达谱的相似性使用降维算法对来自BCC(a,c,e)的12,551个T细胞和来自SCC(b,d,f)的14,522个T细胞进行聚类和可视化。基于标志物基因表达(a,b)鉴定单细胞与CD8+、CD4+或调节性T细胞的关联,以及提示单细胞与患者标识符的关联(c,d)。基于scTCR-seq数据鉴定响应于抗PD-L1治疗扩增的T细胞,并用红色标记。所有其他T细胞显示为灰色(e,f)。
图7显示了差异表达的基因,其基于其治疗前基因表达谱来区分PD-1阻断后将扩增的T细胞。将样品分成BCC(a,b)和SCC(c,d),以及CD8+(a,c)和CD4+T细胞(b,d)。为选择最明确定义的差异表达基因,我们删除了具有高背景表达,倍数变化和统计学显著性两者通过阈值(灰色线)的基因。由此产生的差异表达基因的列表被命名并以颜色突出显示,其中蓝色表示扩增部分下调,红色部分表示扩增部分上调。
图8显示了在单细胞数据可视化上重叠的所鉴定的预测基因表达标记的评分值的图示。鉴定了两种不同标记,BCC患者样品中的CD8+T细胞标记,其是扩增的正预测因子(a),SCC患者样本中的CD4+T细胞标记,其是扩增的负预测因子(b)。标记评分值是相对的、无单位的,并由色标表示,评分值以红色表示,与扩增可能性大相关,蓝色表示较低扩增可能性。
图9显示了使用预测标记的临床试验数据的生存分析。针对每项临床试验中使用阿特朱单抗治疗的患者,显示了无进展生存期(PFS)的Kaplan-Meier曲线,通过其高于(Sig+)或低于(Sig-)相应临床试验中所有患者的中位数的其预测标记评分将患者分组。删除的观察结果以加号表示。显示了来自Cox比例风险模型分析的风险比(*pval<0.05,**pval<0.01,***pval<0.001)。在b中,基于在a中所示的两个标记将患者分成四组(低-低、低-高、高-低和高-高),首先显示CD8标记。显示了与低-低组相关的风险比和显著性。
图10显示了训练用于推导基因标记的逻辑回归模型的结果,该基因表达标记可以最佳地预测PD-1阻断后T细胞的扩增。该图显示了训练逻辑回归模型以预测在单细胞水平PD-1阻断后T细胞扩增的结果。基于差异基因表达的分析,对于每个T细胞亚群,已经推导了独立的标记。对于每个亚群,改变差异基因表达倍数变化的阈值,以研究更严格的截止值是否会影响预测性能。预测性能通过准确度(准确预测的比例,真阳性和真阴性,占病例总数)、灵敏度(真阳性率)和特异性(真阴性率)来衡量。
图11显示了在两个生物标志物阳性(BM+)和阴性(BM-)群中实验组和阳性对照组之间的风险比比较。两组患者的风险比和Cox比例风险模型的双侧统计检验均显示出显著性(*pval<0.05,**pval<0.01,***pval<0.001)。
图12显示了来自预治疗BCC和SCC样品的T细胞亚群,其中鉴定了患者的来源和扩增状态。基于其基因表达谱的相似性使用降维算法对细胞进行聚类和可视化。显示了单个细胞与患者标识符的关联(a-d)。已基于scTCR-seq数据鉴定出响应于抗PD-L1治疗而扩增的T细胞,并且以红色标记,所有其他T细胞以灰色显示(e-h)。
图13在流程图中示出了从单细胞数据推断预测基因表达标记的工作流程,该流程图示出了从scRNA-seq数据推断预测性基因表达标记工作流程中的步骤。
图14显示了将预测基因表达标记应用于来自临床试验的大量RNA-seq数据的流程图,该流程图示出了将预测性基因表达标记用于来自临床试验的大量RNA-seq数据的工作流程中的步骤。
图15显示了包含在从SCC患者样品中的CD4+T细胞鉴定的CD4非扩增标记中的FOXP3(a)和GZMA(b)的基因表达。每个基因的表达强度用色标表示。鉴定为Treg的簇由虚线圆圈表示。
图16显示了生物标志物阳性和阴性群之间的风险比的比较。两组患者的风险比和Cox比例风险模型的双侧统计检验均显示出显著性(*pval<0.05,**pval<0.01,***pval<0.001)。
具体实施方式
在一个方面中,本文所述的本发明提供了一种预测对治疗的临床反应的方法,如基于免疫疗法的抗癌T细胞,所述免疫疗法基于检查点阻断(例如,抗PD-L1或抗PD-1治疗),所述方法基于在免疫疗法后最终扩增的肿瘤中的T细胞克隆型与在免疫治疗之后最终不扩增的肿瘤中的T细胞克隆型之间的基因表达谱差异。
本发明的一个显著特征是,在未经处理的效应细胞(例T-/B-细胞)群中鉴定的基因标记或预测性反应生物标志物(PRB)的集合,其部分基于单细胞谱。
单一细胞分析的一个优点是,通过利用患者样品内的异质性,单细胞分辨率提高了统计学把握度,在患者样品中通常至少包含数百万甚至数千万个异质性细胞,包括疾病(例如,癌症)细胞、免疫细胞(T细胞、B细胞、NK细胞、巨噬细胞、中性粒细胞等)。疾病组织的复杂性和疾病的微环境(如肿瘤微环境或TME)在治疗过程中至少部分保留;同时,可以单独评估单个靶细胞的特异性应答,如作为免疫调节药物(例如,免疫检查点抑制剂)靶点的免疫细胞。
统计学把握度增加的直接获益是发现PRB所需的患者数量显著减少。
在某些实施方式中,适于本发明的方法(即,以鉴定PRB,并且基于PRB治疗患者)的样品可以是容易获得的体液,如血液样品,这进一步提高了可以进行本发明方法的容易性。此外,TME和PBMC隔室之间靶细胞(例如,免疫细胞)的运输使得可能的基于血液的生物标志物筛选能够增强临床成功。
本发明的另一个显著特征是从未治疗的样品中鉴定PRB,这有助于发现生物标志物,这些生物标志物否则基于通常依赖于与对照相比经治疗样品中的表达或活性差异的传统方法将无法鉴定。
就使用使用本发明方法鉴定的PRB而言,基于单细胞分析的结果,可以使用更容易获得的技术(如免疫组织化学(IHC)、批量qPCR或基因组套)在患者分层中实施鉴定的PRB。
本文所述的发明特别适合于鉴定免疫细胞(如T细胞或B细胞)中的PRB,但相同的方法可以容易地适用于治疗的其他靶细胞或效应细胞。
因此,在一个方面中,本发明提供了一种用于鉴定使用T/B细胞靶向免疫调节疗法治疗疾病或病症(例如,癌症)的预测性反应生物标志物(PRB)的方法反应生物标志物,所述方法包括下述步骤:(a)在疾病(例如,癌症、自身免疫性疾病或神经退行性疾病)的治疗前样品中,鉴定具有与所述疾病(例如,癌症)的匹配治疗后样品中的克隆扩增T/B细胞的TCR(T细胞受体)/BCR(B细胞受体)克隆型相同的TCR/BCR克隆型的具有反应能力的T/B细胞,其中在所述匹配治疗后样品中所述克隆扩增T/B细胞在所述治疗后克隆扩增;和(b)生成在所述治疗前样品的所述具有反应能力的T/B细胞中上调和/或下调的基因的列表,以建立所述PRB;(c)任选地,使用逻辑回归系数对在所述PRB中的每个基因进行加权,所述逻辑回归系数通过将作为预测因子的所述的每个基因与作为结果的扩增状态和/或临床反应状态拟合而获得。
如本文所用,“预测性反应生物标志物(PRB)”包括其表达或表达缺失可用于预测靶细胞(如免疫调节疗法中的T细胞或B细胞)或包含靶细胞的患者是否会对特定治疗产生应答的基因。PRB可以由一个基因组成,也可以由一组基因组成,这些基因可以统称为“基因标记”。
疾病或病症可以包括可通过免疫调节疗法治疗的任何疾病或病症,如小分子化合物或抗体(例如,抗PD-1Ab或抗PD-L1 Ab)。例如,疾病可以是癌症,如在实例中的那些癌症,包括BCC、SCC、NSCLC、RCC、转移性尿路上皮癌等。
如本文所用,“克隆型”指分别在B细胞和T细胞上的BCR或TCR链的特异性配对,因为每个成熟的B细胞和T细胞都在其表面表达一种可潜在地识别抗原的BCR和TCR。这样的B细胞和T细胞可以在适当的条件下并且在与抗原结合时进行克隆扩增,并且作为这种克隆扩增的后代的所有B细胞和T细胞共享相同的BCR/TCR配对,这将其与其他B/T细胞上的BCR/TCR区分开来。
“逻辑回归”是一种统计模型,其基本形式使用逻辑函数对二元因变量进行建模。在回归分析中,逻辑回归是估计逻辑模型的参数,其是二元回归的一种形式。数学上,二元逻辑模型有一个具有两个可能值的因变量,标记为“0”和“1”
在某些实施方式中,步骤(a)包括:(1)通过在足以使T细胞靶向免疫调节化合物对所述疾病(例如,癌症)的未治疗样品的离体培养物的免疫调节作用显现的条件和时间段内将所述离体培养物内的T细胞群与所述化合物接触来生成所述匹配治疗后样品;(2)将从所述离体培养物分离、纯化或富集的单个T细胞包封到用于功能性质的单细胞分析的皮升液滴中,从而分别根据所述功能性质的存在或不存在将每个包封的单个T细胞分离到第一应答T细胞池和第二非应答T细胞池中;(3)针对在所述第一应答T细胞池和所述第二非应答T细胞池中的每个包封的单个T细胞确定TCR克隆型,从而将所述应答T细胞的TCR克隆型鉴定为在所述匹配治疗后样品中的克隆扩增T细胞的所述TCR克隆型;(4)将从所述治疗前样品中分离、纯化或富集的单个T细胞包封到皮升液滴中以鉴定具有与在所述匹配治疗后样品中所述克隆扩增T细胞的TCR克隆型相同的TCR克隆型的所述具有反应能力的T细胞。
T细胞群可以包括所有T细胞或者T细胞亚群,如辅助性CD4
不同的靶细胞(例如,T细胞亚群)可针对相同的疾病或适应症产生不同的PRB。
在某些其他实施方式中,步骤(a)包括:(1)从适宜的供体获得所述疾病(例如,癌症)的所述治疗前样品和所述匹配治疗后样品,其中所述匹配治疗后样品已经在足以使所述化合物对所述治疗后样品内的T细胞群的免疫调节作用显现的条件和时间段内与所述化合物接触;(2)将从所述治疗后样品中分离、纯化或富集的单个T细胞包封到用于功能性质的单细胞分析的皮升液滴中,从而分别根据所述功能性质的存在或不存在将每个包封的单个T细胞分离到第一应答T细胞池和第二非应答T细胞池中;(3)针对在所述第一应答T细胞池和所述第二非应答T细胞池中的每个包封的单个T细胞确定TCR克隆型,从而将所述应答T细胞的TCR克隆型鉴定为在所述匹配治疗后样品中的克隆扩增T细胞的所述TCR克隆型;(4)将从所述治疗前样品中分离、纯化或富集的单个T细胞包封到皮升液滴中以鉴定具有与在所述匹配治疗后样品中所述克隆扩增T细胞的TCR克隆型相同的TCR克隆型的所述具有反应能力的T细胞。
许多微流控装置可用于产生皮升液滴,以包封细胞用于单细胞分析。参见,例如,Gérard等人,Nat Biotechnol 38:715-721,2020(通过引用并入)。
在某些实施方式中,步骤(b)包括:(5)使用单细胞RNA测序(scRNA-seq)来鉴定在所述治疗前样品中的所述具有反应能力的T细胞中上调的基因的所述列表,其中在所述PRB中的每个基因具有>0.2的log2倍变化,并且在<40%的在所述匹配治疗后样品中不克隆扩增的具有TCR克隆型的T细胞中表达。
单细胞转录组测序(或者简称scRNA-seq)提供了单个细胞的表达谱。与分析大量细胞中RNA表达的标准方法(如微阵列和批量RNA-seq分析)相比,scRNA-seq可以通过基因聚类分析来鉴定基因表达模式,上述方法可能会掩盖这些群体内单个细胞之间的关键差异。参见Eberwine等人,Nature Methods.11(1):25–27,2014(通过引用并入)。
在某些实施方式中,PRB是与积极治疗结果(例如,在PRB存在的情况下,预计治疗是有利的或成功的)相关的。例如,在下面的实施例章节中,表明与克隆扩增的活化CD8
在某些实施方式中,PRB是与消积治疗结果(例如,在PRB存在的情况下,预计治疗是不利的或不成功的)相关。例如,在下面的实施例章节中,表明与克隆扩增的活化CD4
在某些实施方式中,所述scRNA-seq是使用专门对于所述化合物的作用机制设计的基因组套或者设计用于评估免疫调节的预定基因组套进行的。
在某些实施方式中,所述基因组套包含代表T细胞激活、耗竭、迁移和/或增殖的基因。
在某些实施方式中,所述治疗前样品是血液样品。
在某些实施方式中,所述疾病是癌症,如实体瘤。
在某些实施方式中,所述离体培养物是从疾病组织新分离的。
在某些实施方式中,所述离体培养物是从储存的(例如,冻存的)疾病组织建立的。
在某些实施方式中,所述离体培养物是单细胞悬液。
在某些实施方式中,所述离体培养物是贴壁培养物。
在某些实施方式中,所述时间段是约12-24hr、24-36hr、36-48小时、长达3天、长达4天或长达5天。
在某些实施方式中,所述条件包括:所述化合物的一系列浓度之一,和/或具有或不具有与第二治疗剂的组合。
在某些实施方式中,所述第二治疗剂包括细胞因子(例如,IL-2、TNF)、TCR刺激(例如,通过CD3交联)和/或共刺激(例如,CTLA、B7、CD28等)。
在某些实施方式中,所述化合物是免疫调节抗体。
在某些实施方式中,步骤(2)通过微流控装置进行。
在某些实施方式中,所述单个T细胞是通过分离、纯化或富集特定T细胞亚群或群(如CD4
在某些实施方式中,所述单个T细胞是通过一般地对于T细胞进行分离、纯化或富集分离、纯化或富集的。
在某些实施方式中,所述功能性质包括:细胞因子(例如,IFN-γ)分泌和/或激活标志物的上调。在某些实施方式中,基因组套包括:针对增殖的基因(如Ki67);针对干细胞样特性的基因(如TCF7);针对T细胞激活的基因(如CD25、颗粒酶B、穿孔蛋白、CD28、TNF、IL2、IFNG、4-1BB、CD38、CD69、OX-40、GITR、IL4、IL6);针对T细胞耗竭的基因(CD39、CTLA-4、EOMES、PD-1、TIGIT、2B4、LAG-3、T-bet、TIM3、TOX、CD160、IL-10);针对迁移的基因(如CD31、CD103、CXCR5、CXCR4、CCR7、CCR3、CCR4、CCR8、CCR5、CXCR3);及其组合。
在某些实施方式中,基因组套还包括CD4、CD8、FOXP3、CD62L、CD44、CD127、CD27、IL33R和/或PTPRC。
在某些实施方式中,在步骤(2)中,所述功能性质是用于根据所述功能性质的存在或不存在使用微流控装置物理分离所述应答T细胞和非应答T细胞。
在某些实施方式中,在步骤(2)中,所述功能性质是激活标志物或应答印记(其任一或两者可以通过例如单细胞RNA测序确定),并且其中所述应答T细胞和非应答T细胞不是物理分离的,而是基于所述激活标志物或应答印记的表达或缺乏表达而实质上区分的。
在某些实施方式中,该方法还包括对治疗前样品和/或治疗后样品进行分析以表征样品的例如细胞类型组成(通过,例如,流式细胞术)和T细胞上的靶标表达。
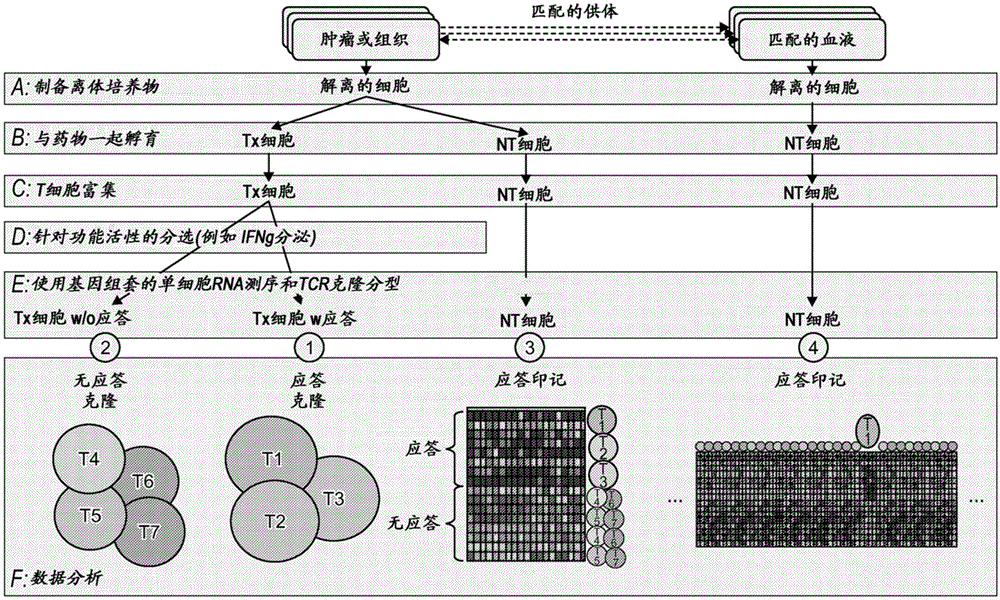
鉴于图1和图2中的示意图中描绘的非限制性示例性实施方式的以下描述,可以更好地说明本文描述的本发明。
图1描述了一个示例性实施例,其中获取疾病(如癌症)的新鲜、未经治疗的患者样本,以鉴定预测性反应生物标志物(PRB),其可能有助于对特定治疗的患者进行分层,以确保最大的治疗有效性。该实施例聚焦于T细胞作为潜在免疫调节药物(例如,抗PD-1或抗PD-L1抗体)的靶细胞,但相同的方法可以应用于其他细胞,如B细胞。为了简单起见,描述集中于T细胞,这不是限制性的。
具体地,图1说明了可用于使用体外细胞培养、单细胞筛选和匹配的TCR克隆分型和表型鉴定T细胞靶向免疫调节药物的预测性反应生物标志物(PRB)的示例性程序,所述预测性反应生物标志物如对靶T细胞(例如,OX40)表面上表达的免疫调节受体特异性的激动剂抗体。
在进入实验流程(A-F)之前,该过程首先从具有可触及实体瘤病变的癌症患者收集组织,如通过切除或活检。
在(A)制备离体培养物中:将新鲜组织解离成单细胞悬液,该悬液可任选地使用例如流式细胞术的细胞组成分析来进一步表征,以表征样品的细胞类型组成。在此阶段还可以评估T细胞上的靶标表达(例如,OX40受体表达)。样品的若干等分试样可以任选地冷冻和储存,以便将来重复实验。然后,剩余的解离细胞可以分布在一个或多个实验条件下。
(B)可以顺序或平行地测试多个实验条件。实验条件可以包括对照条件或未治疗条件(NT),例如,通过对任何基于抗体的治疗条件使用同种型匹配对照。实验条件还可以包括使用免疫调节药物(例如免疫调节抗体)的治疗条件(Tx)。这可以包括单独或与其他药物组合的不同浓度,或T细胞相关的补充条件,如细胞因子(例如,IL-2、TNF)、TCR刺激(例如,使用抗CD3交联)或共刺激(例如,CD28刺激)。无论用于特定治疗的特定条件如何,将离体细胞培养物与所选择的治疗条件孵育足够的时间,以便在评估单个细胞上的活性读取之前使治疗作用于靶细胞(T细胞)。例如,对于基因表达或早期激活标志物,典型的孵育时间可以在12-48小时范围内,但细胞可以培养长达3-5天,以显示典型的表型读取。
(C)T细胞富集:在免疫调节药物(例如,抗OX40激动剂抗体)通过其作用机制在靶细胞(T细胞)上诱导活性的足够时间过去之后,对于每个实验条件,将靶细胞从离体细胞培养物中的异质细胞群中富集,以在单细胞水平上进一步表征。这些细胞可以广泛地富集任何和所有T细胞,或者富集特定的T细胞亚群,如CD4
(D)对于每个治疗(Tx)条件,可以使用微流控装置将单个富集的靶细胞(T细胞)包封在皮升液滴中,并对每个细胞的一个或多个功能性质进行测定,如IFN-γ分泌或一个或多个激活标志物上调。根据这些功能性质的存在或不存在,可以将靶细胞(T细胞)物理或虚拟分选为针对特定治疗条件的第一应答池(1)(有应答的Tx细胞)和第二无应答池(2)(无应答的Tx)。同时,对于相同的靶细胞(例如,T细胞),可以类似地富集未治疗(NT)条件样品,并使用微流控装置(3)类似地包封在皮升液滴中。
(E)TCR克隆分型和/或单细胞RNA测序(例如,使用基因组套):Tx条件下的每个单个靶细胞(T细胞),包括物理或虚拟分选的部分(1)和(2),以及NT条件(3),现在可以进行TCR克隆分型,以鉴定其各自与克隆型的关联。同时,每个单个靶细胞(T细胞),特别是处于NT条件(3)的那些,可以通过单细胞RNA测序进行分析,任选地通过使用针对所研究药物的各自作用机制(如抗OX40激动剂抗体)专门设计的基因组套。作为免疫调节药物的OX40激动剂抗体的示例性基因组套显示在图3中。因此,NT条件(3)下的每个TCR克隆型与基因组套的表达谱(或如果不使用基因组套,则为全基因组表达谱)相关。
(F)数据分析。已知实体瘤含有与抗肿瘤抗原特异性相关的克隆扩增T细胞,这些扩增T细胞克隆型被认为是负责抗肿瘤活性的重要细胞。然而,经常发现这样的T细胞在肿瘤微环境中受到抑制,这可能是由于存在由肿瘤细胞表达的PD-L1,通过例如PD-1/PD-L1途径的检查点抑制。因此,可以使用免疫检查阻断剂(如抗PD-1抗体或抗PD-L1抗体)或其他免疫调节药物(如抗OX40激动剂抗体)来缓解这种抑制以重新刺激休眠的T细胞。
虽然不希望局限于任何特定的理论,但据信,为了鉴定PRB,应答部分(1)中扩增的T细胞克隆型(如图1中的T1-T3所示)是作为药物治疗结果的抗肿瘤活性的重要驱动因素。然而,另一方面,将存在将在无应答部分(2)中收集的无应答T细胞克隆型(如图1中的T4-T7所示)。通过比较部分(1)和(2)中的克隆型,可以鉴定/分配最终对药物治疗有应答的克隆型(如T1-T3中的那些)和最终对药物治疗无应答的克隆型(如T4-T7中的那些)。
重要的是,由于所有克隆型的所有T细胞随机分布在(A)中的Tx和NT实验条件中,重要的应答克隆将被扩增并多次出现在原始样品中,包括在NT条件下从未治疗过的样品中(3)。NT(3)中的应答者与最终鉴定为Tx(1)中应答者的应答者具有相同的克隆型,不同之处在于,NT(3)中的这些应答者之前没有暴露于治疗条件Tx。NT(3)中的其他克隆型可能是无应答者的克隆型。
因此,根据本发明的方法,可以鉴定NT条件(3)中的应答和无应答克隆型,即使NT条件下的T细胞中没有一个此前暴露于治疗条件Tx。
通过比较(3)中的基因表达谱,在具有与药物治疗应答相关的克隆型的细胞(由T1-T3示出)和具有与药物治疗无应答相关联的克隆型细胞(由T4-T7示出)之间,在T细胞克隆型被治疗条件Tx治疗之前,对其具有特异性应答的标志物可以被鉴定为药物治疗的PRB(预测性应答印记)。
在某些实施方式中,与在无应答(例如,T)细胞中相比,在应答(例如,T)细胞中PRB包含一个或多个表达显著增加或显著降低的基因。参见图4。
在某些实施方式中,PRB中的一个或多个(例如,基本上全部或全部)基因具有>0.2、>0.3、>0.4、>0.5、>0.6、>0.7、>0.8、>0.9、>1(2倍)或更大的log2倍数变化。
在某些实施方式中,PRB中的一个或多个(例如,基本上全部或全部)基因在具有TCR/BCR克隆型的T/B细胞中的表达<5%、10%、20%、30%、<40%、<50%、<60%,其在匹配治疗后样品中没有克隆扩增。
在某些实施方式中,PRB中的所有基因都具有大于0.2的log2倍数变化,并且在具有TCR/BCR克隆型的T/B细胞中表达,其在匹配治疗后样品中没有克隆扩增。
在某些实施方式中,基于基因的个体预测值对PRB中的基因进行个体加权。例如,一些差异表达的基因(例如,与无应答者相比,应答者显著增加或减少,基于应答者和无应答者中阈值log2倍值和表达普遍性%值)可能与期望的生物功能或结果(如T细胞扩增或患者对治疗有应答)强烈相关。这样的基因具有很高权重(例如,在0到1的范围内接近1,0是无权重,1是最大权重)。相反,一些基因可能与期望的生物学功能或结果具有相对较低的相关性,使得此类基因的表达增加仅与例如30%的有利生物学结果相关。这些基因虽然仍然是PRB的一部分,但可以被赋予较低权重(例如,在0-1的范围内为0.3)。根据这种权重分配方法,表达增加与期望的生物学功能或结果反相关的基因被赋予负权重(例如,-0.6)。与其中每个基因被赋予相等权重的相应PRB相比,这种PRB(其每个或大部分基因根据其各自的预测值进行加权)可能更准确地进行预测,而不管单个基因对于特定生物学结果的预测值如何。
将权重分配给PRB中的基因可以作为机器学习过程的一部分进行,如基于逻辑回归分类器的机器学习。在这种实施方式中,可以提供训练数据集以包括PRB中基因的身份、和/或其各自的表达水平(在应答者中的绝对值,或者与无应答者相比的相对值)以及相关的生物学结果(例如,是否克隆扩增具有/不具有这种增加/减少的基因表达的T/B细胞,患者是否对治疗有应答等)。鉴于能够提供这种训练数据集的大量(例如,数万,如果不是数十万的话)可用T/B细胞,PRB中单个基因的权重和PRB的预测能力可以是高度准确和统计学上强大的。
替代地或附加地,可以使用相同的方法来测定从来自相同供体的匹配血液样品获得的细胞。在得到的数据(4)中,可以再次鉴定最终对早期步骤的药物治疗有应答的克隆型(由T1示出)。基于血液中的相应基因表达标记(类似于上述NT条件(3)),与使用来自疾病组织(如肿瘤)的组织样品(如活检)的相应方法相比,可以基于血液样品鉴定PRB,这使得能够更实际地实施本发明的方法。
如上所述,部分(1)和(2)可通过使用具有分选能力的微流控装置基于预定功能性质物理分离。例如,功能性质可以是T细胞中细胞因子如IL-12或IFN-γ的表达,或B细胞抗体分泌,或细胞表面标志物的表达,所有这些都可以用可用于分选的荧光信号或染料标记。
或者,例如,如果这种表达的标志物不容易被检测或标记用于物理分选,则不需要物理分选。在这种情况下,可以基于应答者的某些基因或一组基因的表达,以及这种表达的缺乏或这种基因在无应答者中的表达水平差异,对应答者部分(1)和无应答者部分(2)进行虚拟分选。这可以通过在NT条件下使用相同的基因组套(3)来实现,或者使用不同的基因组套来实现,所述基因组套被设计为最佳地捕获基因表达中的部分(1)和(2)之间的差异。
在另一个实施方式中,代替使用此前未经治疗的新鲜疾病组织(例如,T细胞靶向免疫调节药物)在离体培养物中测试治疗,可以类似地使用经治疗(治疗后)样品和匹配的未经治疗(治疗前)样品,而不使用离体培养物。当储存的治疗过的样品和匹配的未治疗样品可用时,这尤其有用。图2是该示例性实施方式的图示,用于使用治疗前和治疗后患者样本、单细胞筛选以及匹配的TCR克隆分型和表型分型来鉴定T细胞靶向免疫调节药物(如抗OX40激动剂抗体)的预测性反应生物标志物(PRB)。
类似地,该方法从治疗前和治疗后收集的具有可触及病变(如实体瘤)的患者的匹配组织开始。同样,在样品进入实验工作流程(A-C)之前,可以使用任何常规手段(如切除或活检)获得样品。
(A)制备细胞:可以将治疗前和治疗后组织解离成单细胞悬液。这些样品的几个等分试样可以冷冻保存,以备将来重复实验。来自每个样品的细胞可以广泛富集T细胞或特定T细胞亚群(例如,CD4
(B)TCR克隆分型和/或单细胞RNA测序(例如,使用基因组套):可以对来自每个样本的细胞进行分析,以鉴定每个单个T细胞的TCR克隆型,以便鉴定其与克隆型的各自关联。此外,样品的细胞,特别是治疗前样品(NT(3)条件)的细胞,可以通过使用单细胞RNA测序,或者通过使用全转录组测序,或者使用针对所研究药物的各自作用机制专门设计的更专门的基因组套来进行分析。
(C)数据分析:如前所述,基于来自治疗后样品的数据,可以基于功能性质的存在或不存在来鉴定应答T细胞(1)或无应答T细胞(2),如其基因表达模式,例如,一种或多种激活标志物(例如,IL-2RA)、细胞因子(例如IFN-γ)的表达,和/或通过与治疗前样品的克隆型相比克隆扩增增加。
实体瘤含有与抗肿瘤抗原特异性相关的克隆扩增T细胞,这些扩增的T细胞克隆型被认为是负责抗肿瘤活性的重要细胞。因此,为了PRB鉴定方法的目的,认为应答者部分(1)中扩增的克隆型(由T1-T3所示)是药物治疗后抗肿瘤活性的重要驱动因素。另一方面,无应答者T细胞克隆型(由T4-T7所示)将在无应答者部分(2)中收集。
重要的是,由于所有克隆型的所有T细胞最初随机分布在(A)中的Tx(治疗后)和NT(匹配治疗前)实验条件中,并且重要的应答克隆被扩增并多次存在于原始样品中,因此可以从治疗前样品(3)中鉴定应答和无应答克隆型。通过比较(3)中与药物治疗应答者相关的克隆型(T1-T3所示)和与药物治疗无应答者相关(T4-T7所示)的克隆型之间的基因表达谱,在应答者接受治疗条件之前,可以鉴定对应答T细胞克隆型特异性的标志物(PRB)。因此,根据本发明的方法,所鉴定的标志物构成药物治疗的预测性反应生物标志物(PRB)。
替代地或附加地,可以使用相同方法(图2)分析从来自相同供体的匹配血液样品获得的细胞。在得到的数据(4)中,可以再次鉴定对早期步骤的药物治疗有应答的克隆型(由T1所示)。基于血液中相应的基因表达标记,可以确认所鉴定的应答印记(PRB)可以基于血液样本,与使用来自肿瘤的组织样品相比,这使得能够更实际地实施针对药物治疗的受试者PRB。
尽管以上描述使用免疫调节药物对T细胞的刺激作为说明性实例,但是可以容易地设想本发明的方法可以应用于其他免疫细胞,包括B细胞和先天免疫系统效应细胞,以及其他治疗条件,如不同药物与不同作用机制的组合,和/或不同给药方案和治疗计划。
因此,在更一般的意义上,本文描述的本发明提供了一种鉴定来自患者的治疗前样品中关于特定治疗的PRB的方法,以及一种使用如此鉴定的PRB来预测个体患者的治疗结果的方法,使得可以针对任何特定治疗对患者群体进行适当和有效的分层。
本发明的方法采用基于单细胞的方法,该方法从患者疾病部位的(例如,癌症患者的实体瘤组织)的材料/样品采集开始。任选地,还可以收集来自相同患者的匹配PBMC,以便于鉴定潜在的基于血液的生物标志物。接下来,可以处理组织以进行离体培养,并且平行或顺序地使用一种或多种实验条件/药物处理。在这个阶段,可以选择性地进行几个预测试,以便更好地表征样品。例如,可以使用流式细胞术以确定组织组成以表征组织内的驻留细胞类型(例如,肿瘤组织中的T细胞)。此外,可以定量靶细胞上的药物靶向表达,并且可以在该预测试步骤中优化实验条件。在用实验条件/药物处理组织样品的离体培养物之后,并且在经过足够的时间以诱导任何药物作用之后,收获细胞池。此时,常规/批量分析可用于表征治疗结果,如流式细胞术、qRT-PCR或ELISA。接下来纯化靶细胞(如T细胞或B细胞),并进入单细胞分析管线。在这一阶段,所有实验条件都在单细胞水平上进行了描述,既用于表征处理活性的读取,也用于表征对照条件下细胞的状态。例如,微流控系统可用于筛选处理的功能活性读取,如激活标志物或分泌的可溶性因子如细胞因子的表达。此外,基于测序的读取如TCR或BCR克隆分型和基因表达读取可用于表征细胞的活性和特征。最后,将来自不同实验条件和读取的获得的单细胞数据以及来自常规分析的数据进入计算分析,以基于与对照相比在处理条件中观察到的差异来鉴定预测性反应生物标志物(PRB)。
本发明的另一个方面提供了一种用于选择使用T细胞靶向免疫调节化合物治疗疾病(例如,癌症)的患者的方法,所述方法包括:通过评估通过本文所述的本发明的方法获得的所述疾病(例如,癌症)的预测性反应生物标志物(PRB)中基因的表达状态和/或表达水平获得所述患者的临床反应评分,以确定所述患者的所述临床反应评分是否超过了预先确定的阈值临床反应评分;其中具有超过所述阈值评分的临床反应评分的患者鉴定为从所述治疗中获益并选择进行所述治疗,和/或其中具有低于第二阈值评分的临床反应评分的患者鉴定为不会从所述治疗中获益并选择进行所述治疗。
本发明的又一个方面提供了一种使用T细胞靶向免疫调节化合物治疗患者的疾病(例如,癌症)的方法,所述方法包括:(i)通过评估通过本发明方法中任一项所述方法获得的所述疾病(例如,癌症)的预测性反应生物标志物(PRB)中基因的表达状态和/或表达水平获得所述患者的临床反应评分,以确定所述患者的所述临床反应评分是否超过了预先确定的阈值临床反应评分;其中具有超过所述阈值评分的临床反应评分的患者鉴定为从所述治疗中获益并选择进行所述治疗;和(ii)向在(i)中鉴定为从所述治疗中获益并选择进行所述治疗的所述患者施用治疗有效量的所述化合物。
实施例
实施例1
此处描述的实施例提供了本发明的一个非限制性实施方式的说明,并且绝非是限制性的。
在一项此前的研究中,对患有SCC(鳞状细胞癌)的4名癌症患者和患有BCC(基底细胞癌)的11名患者进行抗PD-1抗体疗法。
SCC患者治疗后平均约31天,BCC患者治疗后平均约54天,从患者中获得位点匹配的正常或未治疗样品,并对从这些样品中分离的超过26,000个T细胞(TIL,肿瘤浸润淋巴细胞)进行单细胞分析。具体而言,对来自每个样品的分离的T细胞进行单细胞RNA测序(scRNA-seq)和单细胞TCR测序(scTCR-seq),以鉴定治疗后扩增的TCR克隆型。结果表明,扩增的T细胞克隆主要由从肿瘤外部迁移的新克隆型组成。该研究没有检查肿瘤内的任何扩增T细胞克隆。
因此,出现了一个问题,即在通过抗PD-1抗体治疗进行检查点阻断之前,肿瘤中扩增的TCR克隆型的基因表达与未扩增的TCR克隆型不同。此外,这些基因表达差异在多大程度上可以用于预测抗PD-1或抗PD-L1治疗的临床反应。
本实施例表明,使用TCR克隆型作为匹配条件,可以在治疗前样品中鉴定治疗后扩增的T细胞。
在通常情况下,对于靶向T细胞的免疫调节药物(即,调节T细胞活性),来自BCC和SCC治疗前样品的T细胞被特定的T细胞标志物富集。在本实施例中,将T细胞进一步分为来自BCC/SCC治疗前样品的CD8
然后进行逻辑回归,以基因标记的基因作为预测因子,以T细胞扩增状态作为结果。将如此获得的系数作为每个基因的权重,用于未来预测。如果基因的上调与T细胞克隆扩增密切相关,则该基因具有比另一个与T细胞克隆扩增不密切相关的基因更高的预测价值或把握度。
接下来,从几项抗PD-L1(阿特朱单抗)临床试验中获得批量RNA-seq数据,即IMvigor210(治疗转移性尿路上皮癌,n=348)、IMmotion150(治疗肾细胞癌或RCC,针对仅阿特朱单抗n=86,针对阿特朱单抗+贝伐单抗(其是设计以阻断VEGF的抗血管生成药物)n=88)和POPLAR(治疗NSCLC,n=93)。对于这些研究,从治疗前的肿瘤样品收集批量RNA-seq数据,收集无进展生存期(PFS)数据作为治疗终点。
有了这些数据,PRB(即,T细胞扩增的标记)可以与实际临床反应或PFS数据相关联。
具体而言,将scRNA-seq数据中每个基因标记的权重应用于临床试验的批量RNA-seq中的相应基因,以计算每个患者的评分,作为PRB/基因标记中基因的加权总和。根据患者的中位评分,将评分高于中位数的一半患者称为Sig
结果表明,单个CD8 T细胞扩增标记(PRB)预测了不同试验的阳性临床反应。具体而言,在具有348名患者的IMvigor210试验中,将CD8
*Wu等人,Nature 579:274-278(2020).
在具有86名患者的IMmotion 150试验(仅阿特朱单抗)中也获得了类似的结果,HR比率为0.87,但p值(p=0.58)不支持统计学显著性。在使用阿特朱单抗+贝伐单抗的相同试验(n=88名患者)中,HR是0.68(p=0.12)。
另一方面,以下数据显示CD4 T细胞扩增标记预测阴性临床反应,其支持本发明的另一个实施方式,其中CD4
特别是,在IMvigor210试验中,应用从CD4
在PRB中使用GZMA基因的POPLAR试验中,HR为1.44(p=0.11)。
在仅使用阿特朱单抗的IMmotion150试验中,PRB(GZMA)导致Sig
总之,上述数据表明,抗PD1治疗后CD8或CD4 T细胞中扩增的上调基因可以分别预测阳性或阴性临床反应。来自一种癌症的基因标记或PRB(例如,BCC和SCC数据集)适用于其他肿瘤类型(RCC、NSCLC、转移性尿路上皮癌)。因此,结合克隆型和表型分析可用于鉴定与患者结果相关的预测性应答印记。本文描述的方法为发现预测性反应生物标志物提供了无偏倚和系统的方法。
参考文献
1.clinicaltrials.gov/ct2/show/NCT02108652,“A Study of Atezolizumab inParticipants With Locally Advanced or Metastatic Urothelial Bladder Cancer(Cohort 2).”
2.Balar等人,Atezolizumab as first-line treatment in cisplatin-ineligible patients with locally advanced and metastatic urothelialcarcinoma:a single-arm,multicentre,phase 2 trial.The Lancet,389(10064):67-76,2017.
3.clinicaltrials.gov/ct2/show/NCT01903993,“A Randomized Phase 2Studyof Atezolizumab(an Engineered Anti-PDL1 Antibody)Compared With Docetaxel inParticipants With Locally Advanced or Metastatic Non-Small Cell Lung CancerWho Have Failed Platinum Therapy–‘POPLAR’.”
4.Weinstock等人,U.S.Food and Drug Administration Approval Summary:Atezolizumab for Metastatic Non–Small Cell Lung Cancer.Clin Cancer Res.23(16):OF1-OF6,2017.
实施例2
使用克隆型条形码的单细胞免疫分析鉴定预测免疫检查点阻断应答的生物标志物标记。
迫切需要鉴定患者治疗应答的预测性生物标志物,以提高现有和新型实验疗法的成功概率。单细胞分析为肿瘤微环境中的药物应答提供了新的生物学见解,但其发现生物标志物的潜力尚未被用于治疗目的。本实施例描述了一种从单细胞数据中发现预测性反应生物标志物的新方法,该方法使用每个T细胞固有的T细胞受体序列来匹配治疗前和治疗后肿瘤样品之间的克隆型。结果,从小患者队列中鉴定出用于免疫检查点阻断的预测性基因表达标记,并使用三项更大的临床研究的数据验证了其预测性能。
本文提出的结果表明,将克隆分型与基因组分析相结合是一种使用单细胞技术进行生物标志物鉴定的新方法。这种方法提高了成功的概率,减少了临床试验的规模,因此显著影响了新型免疫调节疗法的未来临床发展。
为了研究哪些T细胞通过克隆扩增对ICB产生应答,我们使用了来自14名皮肤癌症患者的数据,这些患者分别接受了抗PD-1治疗前后的位点匹配肿瘤活检。通过单细胞RNA测序(scRNA-seq)针对基因表达对每次活检进行分析,并通过单细胞TCR测序(scTCR-seq)进行克隆型分析。从基底细胞癌(BCC)和鳞状细胞癌(SCC)分离样品进行进一步分析。然后,使用聚类和降维算法来基于基因表达谱鉴定具有共同特征的细胞群体,并找到由T细胞亚群和患者特异性标识符分隔的聚类。参见图6。通过鉴定治疗前和治疗后样品之间匹配的T细胞克隆型来量化ICB引发的克隆扩增,以计算治疗诱导的克隆型大小的倍数增加。参见图6。在BCC数据中,ICB后,所有CD8+T细胞有2.02%,所有常规CD4+T细胞有0.63%和所有FOXP3+CD4+Treg有0.99%扩增,其平均倍数增加分别为3.82、2.75和4.54。虽然CD8+T细胞在BCC中主要有应答,但在SCC中有更广泛的T细胞应答,在ICB后,所有CD8+T淋巴细胞中有5.14%,所有CD4+T细胞中有5.04%,所有Treg中有2.70%扩增,其平均倍数增加分别为4.88、5.26和4.06。在两个数据集中,通过克隆扩增对ICB作出应答的Treg克隆型的绝对数量都是有限的。因此,将Treg和常规CD4+T细胞组合用于后续分析。为了提高分辨率,对每个主要T细胞亚群重复克隆型扩增分析,在BCC中,大多数应答的CD8+T细胞来自一名患者。参见图12。然而,该患者仅有6.74%的单个CD8+T细胞被鉴定为应答。在SCC数据中,应答细胞在患者中更分散。总之,在BCC和SCC队列中,基于scTCR-seq克隆型分析,CD8+和CD4+T细胞克隆型均在ICB应答中扩增。
接下来,研究了使用预治疗基因表达谱来区分对克隆扩增的ICB有应答的T细胞和无应答的T细胞的可能性。使用CB方法,将治疗前样本中的TCR克隆型分为ICB后有应答的部分和无应答的部分。参见图12。然后使用scRNA-seq基因表达谱鉴定在一组应答细胞中差异表达的基因。为了鉴定差异表达的基因,计算每个基因的两个部分之间的基因表达倍数变化,并使用几个过滤步骤来确保所鉴定的基因表达清晰(不受单细胞数据稀疏性或高基线表达的影响)、有意义(具有可察觉的对数倍数变化)以及统计学上稳健的信号(基于多次测试校正的秩和检验的显著性)。参见图13。在BCC患者样品中,与ICB后未扩增的克隆型相比,发现在CD8+T细胞中8个基因(IFNG、ADGRE2、TNF、FGL2、RHOB、ADRB2、NFKBIZ、STAT1)上调和9个基因(TRAT1、MRPL18、PEBP1、CMTM7、CCR7、CYSTM1、AHSA1、CMSS1)下调。参见图7。而相比之下,在CD4+T细胞中没有差异表达的基因。参见图7。这可能是由于该亚群中扩增细胞的频率较低(0.7%)。在SCC样品中,在CD8+T细胞中未发现差异表达的基因,但在CD4+T细胞内鉴定出5个下调的(LAIR2、ACP5、IL1R2、GZMA、FOXP3)和2个上调的(G0S2、BAG3)基因,这些基因与ICB后未扩增的克隆型相关。见图7。总之,在单细胞水平上鉴定出两组不同的差异表达基因,一组与BCC中的CD8+T细胞相关,另一组与SCC中的CD4+T细胞相关联,这两组基因与对ICB应答的克隆扩增相关。
考虑到CD4+和CD8+T细胞中差异表达的基因集,找到哪一个单细胞基因表达标记最能预测对ICB的应答,这一点很重要,稍后可以应用于大量基因表达数据,以预测临床试验中患者对ICB的应答。批量RNA-seq数据不仅代表肿瘤浸润的T细胞,还代表TME中所有细胞的混合物,包括免疫细胞、基质细胞和恶性细胞。为了获得T细胞特异性预测标记,通过计算其与T细胞谱系标志物的相关性,将差异表达基因的列表限制为表达更限于T细胞亚群的基因。为了获得在单细胞水平上最能预测ICB后T细胞扩增的基因表达标记,训练了剩余差异表达基因的逻辑回归模型。参见图13。此外,还研究了更严格的差异表达截止值是否会影响逻辑回归模型的预测性能,这会导致标记中的基因数量减少。参见图10。对于CD8+T细胞扩增标记,使用只有一个基因IFNG的最简约模型作为扩增的正预测因子具有最高的准确度和特异性。图8,参见图12。对于CD4+T细胞标记,发现具有两个基因GZMA和FOXP3的模型具有最佳的准确度和特异性;这些基因的CD4+T细胞转录都是扩增的负预测因子(即,在治疗前,这些转录物在有应答的克隆型中下调,而在ICB后没有应答的克隆型中下调)。图8,参见图12。
总之,在单细胞水平上鉴定了预测对ICB应答的基因表达标记:一个标记的表达是CD8+T细胞扩增的正预测因子(CD8扩增标记),相反,另一个标记的表达是CD4+T细胞扩增的负预测因子(CD4非扩增标记)。
为了研究来源于单细胞数据的两个基因表达标记(图8)是否能够预测ICB治疗结果,将CD8扩增标记和CD4非扩增标记应用于使用抗PD-L1检查点抑制剂(阿特朱单抗)的三个II期临床试验的数据,其中治疗前患者样品上的批量RNA-seq是可用的:在局部晚期或转移性非小细胞肺癌中进行的POPLAR研究16(NCT01903993),在局部晚期或转移性尿路上皮性膀胱癌中进行的IMvigor210研究21(NCT02951767,NCT02108652)以及在晚期肾细胞癌中进行的IMmotion150研究22(NCT01984242)。对于每项研究,生成每名患者的标记评分,使用中位截止值来区分标记高和标记低的患者,并使用生存分析研究该标记是否能够预测治疗结果。参见图14。对于CD8扩增标记,标记高的患者倾向于具有更长的无进展生存期(PFS)。参见图9。对于CD4非扩增标记,标记高的患者也倾向于具有更长PFS。参见图9。这令人吃惊,因为高标记意味着肿瘤中的CD4+T细胞在ICB后扩增的概率较低。FOXP3和GZMA的表达(这两个基因是导致这一标记的基因)存在于在不同的常规CD4+T细胞簇中,然而,Treg簇中也存在一些重叠。参见图15。因此,不能排除源自Treg亚群内的该非扩增标记的潜在预测重要性。虽然IMvigor210和POPLAR中的两个标记显著(p值<0.05)富集了有应答的患者,但只有IMmotion150试验中具有CD4非扩增标记的患者在联用组(阿特朱单抗+贝伐单抗)中达到显著水平。
接下来,研究了在联用中应用这两种标记是否能进一步改善治疗结果预测。将患者分为四组,首先显示CD8标记:两个标记的评估高于中位数(Pos-Pos),一个标记的评分高于中位数(Pos-Neg,Neg-Pos),或者两个标记的评分低于中位数(Neg-Neg)。Pos-Pos组合标记可以进一步改进IMvigor210以及IMmotion150的组合组的预测。参见图9。对于OPLAR,与低-低组相比,两个标记中至少一个阳性的所有组都具有显著改善的结果。参见图9。基于这些观察结果,推导出了考虑两个标记的组合标记。当与其他公开标记以及包括CD274(PD-L1)、PDCD1(PD-1)、CD8A、CD4和FOXP3在内的目标基因表达标志物比较时,与其他标记或目标标志物相比,组合标记始终实现了相似或更高的风险比。参见图16。值得注意的是,对于IMmotion150阿特朱单抗单药治疗组,没有一个标记能够提供显著降低的风险比(HR)。总之,当应用于治疗前批量RNA序列数据时,来自少数患者的治疗前scRNA序列数据的基因表达标记可以预测ICB治疗患者的更大队列的结果。此外,根据肿瘤类型,组合标记可以导致进一步改善的预测。
为了进一步证明来自单细胞数据的预测标记如何影响新实验药物的临床开发,使用我们的组合标记对患者分层进行分析,以确定其是否可以提高临床试验中获得显著改善的PFS的概率。为此,使用了POPLAR和IMmotion150研究的患者数据亚群,其中治疗前批量RNA-seq数据可用于实验和标准护理阳性对照组。使用组合标记将每个试验中接受ICB治疗的患者分为生物标志物阳性(BM+)和生物标志物阴性(BM-)组。对于每个试验和生物标志物组,在阳性对照组和实验组之间进行生存分析,并将结果与未分层患者群体的结果进行比较。参见图11。在未分层的情况下,在三个分析中的任一个中,实验组未显著改善HR。根据组合标记对患者进行分层后,与未分层相比,BM+组的总体HR降低。IMmotion150试验组合组达到了统计学显著性,表明在该患者群体中,阿特朱单抗和贝伐单抗的组合比多重靶向受体酪氨酸激酶(RTK)抑制剂舒尼替尼更有益。相反,与未分层相比,在BM-组中发现了HR增加。在POPLAR的BM-组中达到了显著性,这表明阿特朱单抗在该患者组中的获益不如化疗多西他赛。相应地,与未分层相比,POPLAR的BM+组的HR明显下降的趋势;然而,其没有正式达到显著性(p=0.06)。我们还将我们的组合标记与此前公开的标记和其他目标生物标志物进行了比较。参见图11。该标记始终表现出与其他相关标志物更好或相似的性能。值得注意的是,对于IMmotion150阿特朱单抗单药治疗组,没有一个标记能够提供显著降低的HR。总之,有证据表明,与未分层患者群体中的患者相比,来自少数患者的单细胞数据的基因表达标记可以前瞻性地鉴定出获益于实验治疗的患者群体。
使用接受ICB的患者治疗前后的单细胞数据,本实施例显示了一种新的方法来鉴定预测基因表达标记,该标记可作为分层患者的生物标志物应用,以改善临床试验的结果。本文描述的克隆型条形码(CB)方法使用每个T细胞固有的TCR序列来匹配治疗前和治疗后肿瘤样品之间的克隆型,以鉴定响应于治疗而扩增的T细胞。CB方法优先考虑TME中检测到的扩增和再出现T细胞克隆型的基因表达模式,以预测治疗结果。CB方法也可用于鉴定针对自身免疫性疾病中致病性B细胞的治疗的预测生物标志物。
单细胞分析使CB方法能够利用单细胞水平上的异质性来获得预测基因表达标记。与需要更多患者样品以比较不同患者组的传统方法相比,将来自相同患者的治疗前和治疗后样品的T细胞组连接起来的能力为集中分析和鉴定最关键的预测参数提供了一种巧妙的方式。因此,这种方法允许人们从较小的患者队列(如从早期临床研究中获得的那些)中进行无偏倚的预测标记搜索。使用CB方法发现标记异常简单,易于解释和实际临床实施(即,免疫组化或原位杂交),但如果不是更复杂标记的一部分,则可能会丢失。尽管本研究中使用的单细胞数据在治疗前和治疗后匹配的T细胞克隆型的数量上仍然有限,但具有更大数量的异型细胞的scRNA-seq数据集的生成将产生更精细的标记,从而更清楚地将T细胞生物学与患者对免疫疗法的应答联系起来。此外,更优化和标准化的单细胞数据生成和分析将进一步富集数据集,并提供更有意义的分析。
从机理上讲,IFNG被鉴定为接受ICB治疗的BCC患者CD8+T细胞克隆扩增的最佳预测因子,表明存在积极识别同源肿瘤抗原的功能性肿瘤反应细胞,但其扩增被PD-1/PD-L1途径抑制。因此,令人吃惊的是,尽管治疗前CD8+T细胞克隆扩增的比例更大(BCC和SCC中CD8+T克隆分别为2.02%和5.14%),但SCC患者中的CD8+T淋巴细胞标记未被鉴定。SCC中的T细胞克隆应答比BCC更广泛,在SCC中常规CD4+T细胞和Treg扩增的比例更大,这可能表明这些癌症适应症之间存在不同的ICB作用机制。尽管BCC中CD8+T细胞的扩增可能是ICB的直接后果,但其可能是SCC的间接后果,可能是由于CD4+T细胞的帮助、调节性T细胞抑制的降低,或来由于我们CB方法中未捕获的免疫细胞亚群(如NK细胞)的抗肿瘤活性。
CD4非扩增标记的解释可能更复杂。FOXP3和GZMA不一定对应于相同的细胞亚群,GZMA本身仍然具有Treg集簇之外的预测把握度。虽然转录因子FOXP3是Treg的特征,但FOXP3转录也在活化后的常规T细胞中表达,事实上,在SCC scRNA seq数据中的常规CD4 T细胞簇中发现了FOXP3。GZMA编码细胞溶解蛋白颗粒酶A并与效应T细胞相关,其此前已被鉴定为ICB治疗的正预测因子或预测生物标志物,尽管这通常归因于CD8+T细胞。然而,由于Treg集簇内FOXP3和GZMA之间存在一些重叠,因而该Treg亚群可能具有潜在的预测重要性。据报道,活化Treg产生的颗粒酶A使这些细胞更容易受到自身造成的凋亡的影响。
此外,已经证明,使用CB方法鉴定的预测基因表达标记可以在更大的临床试验中得到验证。CD8扩增标记和CD4非扩增标记都可以预测治疗的有利结果,并且这两种标记的组合可以导致进一步改善的结果。有趣的是,这些预测标记是根据皮肤癌症患者的数据得出的,但在其他三种实体癌症类型的较大队列中,其仍然显示出预测性。然而,最佳的标记组合取决于接受治疗的患者群体。
最后,证明了预测特征如何应用于患者分层,以提高临床试验成功的概率。尽管这些标记始终表现得更好或类似于其他相关标记,但单独的PD-1表达本身也是一个强有力的预测生物标志物。基于对ICB机理的理解这并不意外。重要的是,CB方法的优势在于,其可以以无偏倚的方式鉴定表现类似良好的生物标志物标记。这可能与鉴定新实验性疗法的生物标志物最相关,因为对其机制可能不太了解或更复杂。因此,提出的CB方法是单细胞数据用于生物标志物鉴定的一种有前景的新用途,这将影响新型免疫调节疗法的未来临床发展。
方法
基于scTCR-seq数据的克隆扩增鉴定
从Yost等人获得来自单个T细胞的TCR克隆型标记。基于其匹配的TCR CDR3序列来鉴定同一患者治疗前和治疗后T细胞共有的克隆型。治疗后克隆型大小(共享相同TCR CDR3序列的T细胞数量)大于治疗前克隆型大小的克隆型被认为是对ICB的应答而扩增。对于在SCC数据集中的患者su013,治疗前样品中有4488个T细胞被分析,但治疗后样品中只有69个T细胞;T细胞计数的这种不平衡部分解释了为什么su013患者在治疗后没有克隆型被认为经历了扩增。
scRNA-seq数据的聚类和降维
所有BCC细胞类型的scRNA-seq数据的聚类如之前Seurat(3.1.1版)所述。基于该第一轮聚类和典型T细胞标志物(CD8A、CD4、FOXP3)的表达,我们鉴定了T细胞簇。从完整的数据集中提取治疗前样品中特异性的T细胞,然后重新聚类。在这轮聚类中,使用Seurat中默认的可变基因选择标准。将这些可变基因用于主成分分析(PCA)。将前20个主成分用于基于共享最近邻的聚类,k=20,分辨率=3。将相同的主成分用于最小距离=0.1和相邻数量=20的UMAP投影的可视化。SCC scRNA-seq数据集已对T细胞具有特异性。仅从该数据集提取治疗前T细胞后,使用Seurat进行聚类。这些治疗前的T细胞以与BCC治疗前T细胞相同的方式聚集。在将治疗前T细胞聚类后,基于CD8A、CD4或FOXP3的表达,分别将每个簇注释为CD8+T细胞、常规CD4+T细胞或Treg。
差异基因表达分析
仅使用治疗前scRNA-seq数据,进行差异基因表达分析以确定扩增的T细胞群与未扩增的T细胞群中显著上调或下调的基因。采取了几个过滤步骤,从测试中去除不太可能与生物学相关的基因。测试仅限于在被比较的两个群体中至少10%的细胞中检测到的基因。将由HGNC定义的非编码基因去测试中去除。将人T细胞受体α可变(TRAV)基因、人T细胞受体β可变(TRBV)基因和人白细胞抗原(HLA)基因也从测试中去除。对于通过筛选的基因,进行Wilcoxon秩和检验,以检测扩增T细胞群与未扩增的T细胞人之间的差异基因表达。将Benjamini Hochberg校正用于调整生成的p值,以控制错误发现率。将差异表达基因定义为log2倍数变化>0.6且调整后p值<0.01的基因。此外,对于被认为在扩增的T细胞群中显著上调的基因,其需要在不到30%的非扩增的T细胞群中表达。同样,对于在扩增的T细胞群中被认为是显著下调的基因,其需要在不到30%的扩增的T细胞群中表达。
基于差异表达基因产生预测标记
差异基因表达分析产生了两个差异表达基因的列表:一个来自BCC CD8+T细胞数据集,另一个来自SCC CD4+T细胞数据集。对于每个列表,将倍数变化>1.5的基因用作逻辑回归分类器中的预测因子,该分类器预测了每个细胞的扩增状态。分类器为每个基因计算了一个系数,该系数反映了每个基因的基因表达水平对每个细胞扩增状态(扩增与非扩增)的预测程度。正系数表明表达该基因的细胞更有可能扩增。负系数表明表达该基因的细胞更有可能不扩增。然后以1.75的倍数变化阈值重复该过程,然后再以2.0的倍数变化阈值重复该过程;随着倍数变化阈值的增加,逻辑回归分类器中使用的基因数量减少。记录每个逻辑回归分类器在每个倍数变化阈值下的系数、准确度、灵敏度和特异性。对于BCC CD8+T细胞数据集,我们比较了逻辑回归分类器的性能,并选择了准确度、灵敏度和特异性没有显著下降的最简约模型。在本例中表现最好的分类器包含一个基因IFNG,其具有正系数。将其定义为CD8-扩增标记。使用SCC CD4+T细胞数据集的相同分类器选择标准,我们将CD4非扩增标记定义为包含两个基因GZMA和FOXP3,每个基因都具有负系数。对于在扩增标记中的每个基因,将其逻辑回归系数的绝对值作为其权重。
通过将基于单细胞的基因表达标记应用于批量RNA-seq数据预测患者对ICB的应答
将来自三项ICB临床试验的批量肿瘤RNA序列数据用于评估CD8扩增标记、CD4非扩增标记以及这两种标记组合在预测患者应答中的有效性。在POPLAR(NCT01903993)中,使用阿特朱单抗(n=93)或多西他赛(n=100)治疗患有局部晚期或转移性非小细胞肺癌的患者。在IMvigor210(NCT02951767,NCT02108652)中,使用阿特朱单抗治疗患有局部完全或转移性尿路上皮性膀胱癌的患者(n=354)。在IMmotion150(NCT01984242)中,单独使用阿特朱单抗(n=86)或使用阿特朱单抗和贝伐单抗两者(n=88)治疗患有晚期肾细胞癌的患者。对于每项临床试验中的患者,计算标记(CD8+T细胞扩增标记或CD4+T细胞扩增标记)中基因的加权和作为标记评分。确定每项临床试验的中位标记评分,将评分高于该中位数的患者置于生物标志物阳性组,而将同一试验中的其余患者置于生物标志物阴性组。如Wu等人此前所述,对每个临床试验组中生物标志物阳性组和生物标志物阴性组进行生存分析。将Cox比例风险模型拟合到每个临床试验的每个组的生存数据中;报告了Wald统计所得的风险比和p值。这些生存分析的结果以Kaplan-Meier图表示。为了在临床试验中研究CD8+和CD4+T细胞扩增标记之间的相互作用,我们将每个组中的患者分配至四组之一:两个标记的评分均高于中位数(阳性-阳性),一个标记的评分高于中位数(阳性-阴性,阴性-阳性),或者两个标记的评分均低于中位数(阴性-阴性)。如上所述进行生存分析和Kaplan-Meier作图,将阴性-阴性组作为其他三组中每一个的基线阳性对照。作为参考,将CD274(PD-L1)、PDCD1(PD-1)、CD8A、CD4和FOXP3分别作为其自身的未加权生物标志物处理。如上所述,对每个临床试验的每个组中的生物标志物阳性组和生物标志物阴性组进行生存分析。
通过预测标记预测试验结果(实验组对比阳性对照组)
为了比较IMmotion150中实验组(阿特朱单抗,阿特朱单抗+贝伐单抗)和阳性对照组(舒尼替尼)之间这些T细胞扩增特征的有效性,将CD8+和CD4+T细胞扩增标记合并为单个标记,将每个基因的各自权重转移到合并的标记。首先,为了建立基线,对每个实验组和阳性对照组进行生存分析,以检测生存率的任何分层前差异。接下来,将来自IMmotion150的患者基于合并的标记分层为生物标志物阳性组和生物标志物阴性组。对于每个生物标志物分层组,如上所述对阿特朱单抗治疗的患者与多西他赛治疗的患者进行生存分析。对于POPLAR,将相同过程用于生存分析,以检测实验组和阳性对照组之间这些T细胞扩增标记的有效性。唯一的区别是,没有合并CD8+和CD4+T细胞扩增标记,而是将其分开;任何一种标记(阳性-阳性、阳性-阴性、阴性-阳性)评分高于中位数的患者被认为是生物标志物阳性组,其余患者被认为是生物标志物阴性组。这一分层策略与之前的观察结果一致,即POPLAR中任一扩增标记高于中位数的患者,其生存模式彼此之间比阴性-阴性组的患者更相似。为了比较IMmotion150和POPLAR中实验组和阳性对照组之间的其他标记的有效性,将Wu等人的19基因双扩增标记作为未加权的单一标记应用,而将CD274(PD-L1)、PDCD1(PD-1)、CD8A、CD4和FOXP3各自作为其自身的未加权标记处理。
参考文献
1.Topalian,S.L.等人Safety,Activity,and Immune Correlates of Anti–PD-1Antibody in Cancer.New Engl J Medicine 366,2443–2454(2012).
2.Rizvi,N.A.等人Mutational landscape determines sensitivity to PD-1blockade in non–small cell lung cancer.Science 348,124–128(2015).
3.Rooney,M.S.,Shukla,S.A.,Wu,C.J.,Getz,G.&Hacohen,N.Molecular andGenetic Properties of Tumors Associated with Local Immune CytolyticActivity.Cell 160,48–61(2015).
4.Miao,D.等人Genomic correlates of response to immune checkpointblockade in microsatellite-stable solid tumors.Nat Genet 50,1271–1281(2018).
5.Kümpers,C.等人Immune Cell Infiltration of the Primary Tumor,Not PD-L1 Status,Is Associated With Improved Response to Checkpoint Inhibition inMetastatic Melanoma.Frontiers Medicine 6,27(2019).
6.Kansy,B.A.等人PD-1status in CD8+T cells associates with survivaland anti-PD-1 therapeutic outcomes in head and neck cancer.Cancer Res 77,canres.3167.2016(2017).
7.Gros,A.等人PD-1identifies the patient-specific CD8+tumor-reactiverepertoire infiltrating human tumors.J Clin Invest 124,2246–2259(2014).
8.Kumagai,S.等人The PD-1expression balance between effector andregulatory T cells predicts the clinical efficacy of PD-1blockadetherapies.Nat Immunol 1–13(2020)doi:10.1038/s41590-020-0769-3.
9.Yi,M.等人Biomarkers for predicting efficacy of PD-1/PD-L1inhibitors.Mol Cancer 17,129(2018).
10.Sharma,P.,Hu-Lieskovan,S.,Wargo,J.A.&Ribas,A.Primary,Adaptive,andAcquired Resistance to Cancer Immunotherapy.Cell 168,707–723(2017).
11.Higgs,B.W.等人Interferon Gamma Messenger RNA Signature in TumorBiopsies Predicts Outcomes in Patients with Non-Small-Cell Lung Carcinoma orUrothelial Cancer Treated with Durvalumab.Clin Cancer Res 24,clincanres.3451.2017(2018).
12.Postel-Vinay,S.等人Challenges of phase 1clinical trials evaluatingimmune checkpoint-targeted antibodies.Ann Oncol 27,214–224(2016).
13.Wong,K.M.,Capasso,A.&Eckhardt,S.G.The changing landscape of phaseI trials in oncology.Nat Rev Clin Oncol 13,106–117(2016).
14.Chabner,B.A.Approval After Phase I:Ceritinib Runs the Three-MinuteMile.Oncol 19,577–578(2014).
15.Huang,A.C.等人T-cell invigoration to tumour burden ratioassociated with anti-PD-1 response.Nature 545,60–65(2017).
16.Fehrenbacher,L.等人Atezolizumab versus docetaxel for patients withpreviously treated non-small-cell lung cancer(POPLAR):a multicentre,open-label,phase 2randomised controlled trial.Lancet 387,1837–1846(2016).
17.Fairfax,B.P.等人Peripheral CD8+T cell characteristics associatedwith durable responses to immune checkpoint blockade in patients withmetastatic melanoma.Nat Med 26,193–199(2020).
18.Wu,T.D.等人Peripheral T cell expansion predicts tumourinfiltration and clinical response.Nature(2020)doi:10.1038/s41586-020-2056-8.
19.Sade-Feldman,M.等人Defining T Cell States Associated with Responseto Checkpoint Immunotherapy in Melanoma.Cell 175,998-1013.e20(2018).
20.Yost,K.E.等人Clonal replacement of tumor-specific T cellsfollowing PD-1blockade.Nat Med 25,1251–1259(2019).
21.Mariathasan,S.等人TGFβattenuates tumour response to PD-L1 blockadeby contributing to exclusion of T cells.Nature 554,544–548(2018).
22.McDermott,D.F.等人Clinical activity and molecular correlates ofresponse to atezolizumab alone or in combination with bevacizumab versussunitinib in renal cell carcinoma.Nat Med 24,749–757(2018).
23.Baumeister,S.H.,Freeman,G.J.,Dranoff,G.&Sharpe,A.H.CoinhibitoryPathways in Immunotherapy for Cancer.Annu Rev Immunol 34,1–35(2014).
24.Kreiter,S.等人Mutant MHC class II epitopes drive therapeuticimmune responses to cancer.Nature 520,692–696(2015).
25.Alspach,E.等人MHC-II neoantigens shape tumour immunity andresponse to immunotherapy.Nature 574,696–701(2019).
26.Francisco,L.M.等人PD-L1 regulates the development,maintenance,andfunction of induced regulatory T cells.J Exp Medicine 206,3015–3029(2009).
27.Zappasodi,R.等人Non-conventional Inhibitory CD4+Foxp3–PD-1hi TCells as a Biomarker of Immune Checkpoint Blockade Activity.Cancer Cell 34,691(2018).
28.Pesce,S.等人PD/1-PD-Ls Checkpoint:Insight on the Potential Role ofNK Cells.Front Immunol 10,1242(2019).
29.Subrahmanyam,P.B.等人Distinct predictive biomarker candidates forresponse to anti-CTLA-4and anti-PD-1immunotherapy in melanoma patients.JImmunother Cancer 6,18(2018).
30.Morgan,M.E.等人Expression of FOXP3 mRNA is not confined to CD4+CD25+T regulatory cells in humans.Hum Immunol 66,13–20(2005).
31.Roufas,C.等人The Expression and Prognostic Impact of ImmuneCytolytic Activity-Related Markers in Human Malignancies:A ComprehensiveMeta-analysis.Frontiers Oncol 8,27(2018).
32.Inoue,H.等人Intratumoral expression levels of PD-L1,GZMA,and HLA-Aalong with oligoclonal T cell expansion associate with response to nivolumabin metastatic melanoma.Oncoimmunology 5,00–00(2016).
33.Karreci,E.S.等人Human regulatory T cells undergo self-inflicteddamage via granzyme pathways upon activation.Jci Insight 2,e91599(2017).
- 一种适用于早期发现、早期预测或早期诊断重度慢阻肺的生物标志物及其应用和筛选方法
- 发现用于前列腺癌诊断和治疗之生物标志物和药物靶标的方法及其确立的生物标志物测定