一种宏基因组多重比对序列重分配的方法及应用
文献发布时间:2023-06-19 19:28:50
技术领域
本申请属于生物信息学技术领域,具体涉及一种宏基因组多重比对序列重分配的方法及应用。
技术背景
宏基因组测序(metagenomics next generation sequencing,mNGS)是一种不依赖于培养,通过无偏采样,快速准确检测病原体的新型诊断技术。在检测不明原因,难培养及共感染病原体中,mNGS具有明显优势,是未来十年病原检测领域重要工具。
物种鉴定和丰度估计是mNGS生物信息学分析关键环节,其准确性直接影响病原检测的灵敏度和特异度。由于mNGS病原比对数据库为宏参考基因组,比对结果中包含大量多重比对序列,因此,对多重比对序列进行有效重分配既是mNGS生信分析的重难点,也是病原检测效能提升的优化点。
目前,常用物种鉴定和丰度估计方法包括:Blast,Kraken2+Bracken,MetaPhIAn2等。其中Blast方法基于局部比对,准确度高,但运行效率低,无多重比对序列重分配功能;Kraken2基于Kmer和LCA映射,运行效率高,但在物种水平的分辨率低,大量多重比对序列被标识到物种水平之上;Bracken序列丰度重估计模块存在一定缺陷,如:对低丰度或无唯一比对序列的物种会出现漏检,自下而上(株到种水平)序列重分配不依赖于概率分配模型;MetaPhIAn2仅依赖于Marker基因序列,特异度高,但灵敏度低。
因此,充分挖掘宏基因组参考序列数据库和mNGS唯一比对序列信息,对多重比对序列进行重分配,可有效提高物种鉴定和丰度估计准确性。
鉴于此,提出本申请。
发明内容
为解决上述技术问题,本申请通过生物信息学分析研究,建立一套宏基因组多重比对序列重分配方法,该方法可显著提高物种鉴定和丰度估计的准确性,有效提升病原检测效能。
具体的,本申请提出如下技术方案:
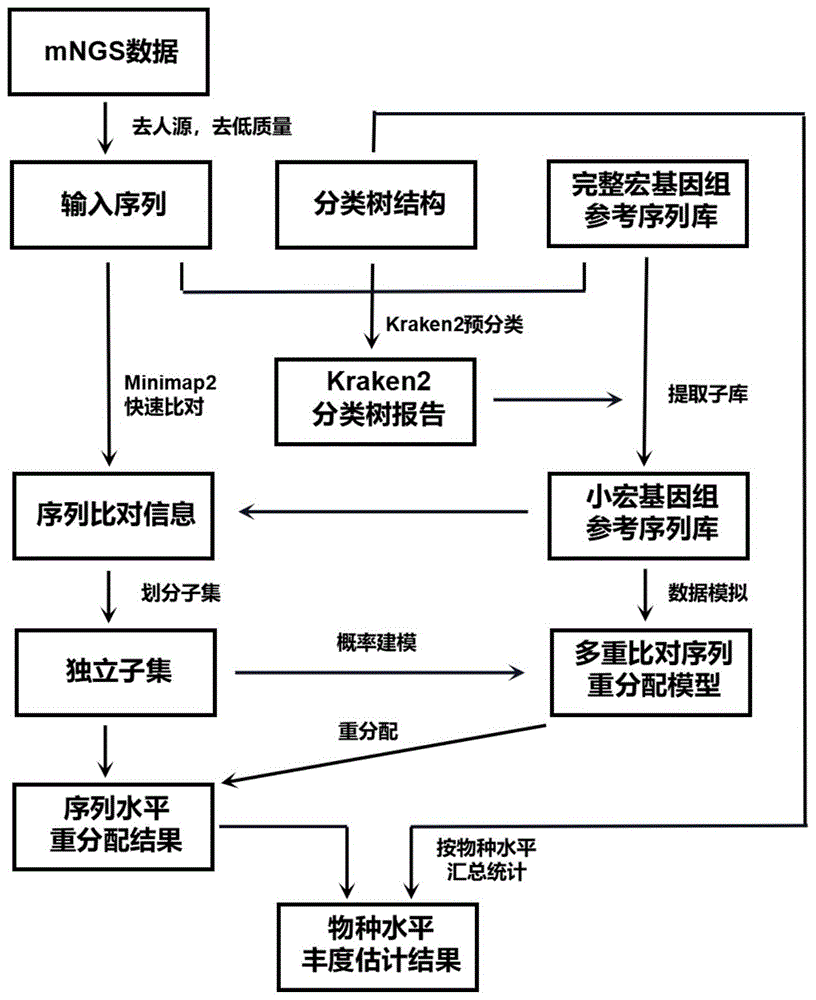
本申请首先提供一种宏基因组多重比对序列重分配的方法,包括如下步骤:
1)快速分类:基于参考序列数据库,对宏基因组测序数据进行快速分类,得到分类树报告;
2)提取小库:基于分类树报告,从完整宏基因组参考序列数据库中提取目标参考序列,得到小宏基因组参考序列数据库;
3)快速比对:基于小宏基因组参考序列数据库,对宏基因组测序数据进行快速比对,得到序列比对信息;
4)划分子集:基于序列比对信息,划分最小分类单元和比对序列均封闭的独立子集;
5)构建模型:基于小宏基因组参考序列数据库和独立子集,模拟计算独立子集内每个最小分类单元唯一比对率,构建多重比对序列重分配概率模型;
6)序列重分配:基于多重比对序列重分配概率模型和序列比对信息,计算每条多重比对序列后验分配概率,将后验分配概率作为随机函数参数为该条序列重分配最小分类单元标识;
优选的,该方法还包括如下步骤:
步骤7)丰度估计:基于唯一比对序列和多重比对序列重分配结果评估物种丰度。
进一步的,所述1)具体为:基于完整宏基因组参考序列数据库,采用Kraken2对宏基因组测序数据进行快速分类,得到分类树报告;
优选的,所述参考序列数据库为完整宏基因组参考序列数据库;所述完整宏基因组参考序列数据库为去冗余去低质量的nt数据库+专业病毒库+专业寄生虫库。
更优选的,所述宏基因组测序数据为去除人源序列,以及去出低质量序列后的测序数据;
进一步的,所述Kraken2参数优选设置为confidence=0.5;
更优选的,所述Kraken2分类树报告信息包括:各分类层级水平分类树信息(分类名称,分类编号),分类到该层级水平及以下的总序列数,分类到该层级水平的序列数以及分类到该层级水平以下的序列数。
进一步的,所述2)中,所述提取是按照宏基因组参考序列纳入标准进行提取;优选的,所述参考序列纳入标准包括:
a、分类树报告中列出的所有物种在完整宏基因组参考序列数据库中收录的参考序列;
b、上述列出的所有物种所包含的所有物种亚型,血清型和株在完整宏基因组参考序列数据库中收录的参考序列;
c、分类树报告中列出的未分配序列数>1000的科所包含的所有物种,物种亚型,血清型和株在完整宏基因组参考序列数据库中收录的参考序列;
d、分类树报告中列出的未分配序列数>100的属所包含的所有物种,物种亚型,血清型和株在完整宏基因组参考序列数据库中收录的参考序列。
进一步的,所述3)中的比对为采用minimap2对宏基因组测序数据进行快速比对;
优选的,所述序列比对信息包括:
a、序列比对的结果,包括:比对序列名称、参考序列名称、最小分类单元名称、比对位置、比对质量、详细比对信息;
b、最小分类单元,包括:物种、物种亚型、血清型和株。
进一步的,所述4)中的独立子集包括以下任一或多个特点:
a、独立子集与独立子集间最小分类单元无交集;
b、独立子集与独立子集间序列无交集;
c、所有独立子集的并集为序列比对信息。
所述步骤4)中,划分子集的规则为最小分类单元和比对序列均封闭,子集的划分依赖于序列比对信息,与分类树结构无关。
进一步的,所述5)中所述模拟计算包括以下步骤:
a、提取独立子集内每个最小分类单元的参考序列,生成独立子集参考序列数据库;
b、基于独立子集参考序列数据库模拟生成测序序列;
c、通过minimap2将模拟序列比对到独立子集内最小分类单元的参考序列上,得到独立子集内模拟序列的序列比对结果;
d、基于模拟序列的比对结果,计算独立子集内每个最小分类单元的唯一比对率。
进一步的,所述5)中构建包括以下任一或多个特点:
a、在每个独立子集内建模;
b、基于唯一比对序列和唯一比对率估计独立子集中每个最小分类单元的序列丰度;
c、基于唯一比对率和每个最小分类单元的丰度估计每个最小分类单元的多重比对序列丰度;
d、每个最小分类单元的多重比对序列丰度作为先验重分配概率;
e、基于每条多重比对序列的比对结果生成每个最小分类单元的观察值概率;
f、基于先验重分配概率和观察值概率,构建多重比对序列重分配概率模型。
进一步的,所述7)中的估计包括如下步骤:
a、先估计每个最小分类单元的序列丰度,即唯一比对序列数与多重比对重分配序列数之和;
b、基于分类树构建最小分类单元的层次关系,每个层次关系的根节点为物种;在物种水平统计序列丰度,作为物种序列丰度输出结果。
本申请还提供一种宏基因组多重比对序列重分配的系统,包括如下组件:
组件1)快速分类组件:用于基于完整宏基因组参考序列数据库,采用Kraken2对宏基因组测序数据进行快速分类,得到分类树报告;
组件2)提取小库组件:用于基于分类树报告,从完整宏基因组参考序列数据库中提取目标参考序列,得到小宏基因组参考序列数据库;
组件3)快速比对组件:用于基于小宏基因组参考序列数据库,对宏基因组测序数据进行快速比对,得到序列比对信息;
组件4)划分子集组件:用于基于序列比对信息,划分最小分类单元和比对序列均封闭的独立子集;
组件5)构建模型组件:用于基于小宏基因组参考序列数据库和独立子集,模拟计算独立子集内每个最小分类单元唯一比对率,构建多重比对序列重分配概率模型;
组件6)序列重分配组件:用于基于多重比对序列重分配概率模型和序列比对信息,计算每条多重比对序列后验分配概率,将后验分配概率作为随机函数参数为该条序列重分配最小分类单元标识;
优选的,该方法还包括如下步骤:
组件7)丰度估计组件:用于基于唯一比对序列和多重比对序列重分配结果估计物种丰度。
进一步的,所述具体组件中的详细限定参考上述方法。
个申请还提供一种计算机存储介质,所述计算机存储介质存储有计算机程序,所述计算机程序包括程序指令,所述程序指令当被处理器执行时,实现上述任一项所述方法。
个申请还提供一种电子设备,所述电子设备包括存储器和处理器,所述存储器和处理器相连,所述存储器用于存储计算机程序,所述处理器用于调用所述计算机程序,实现上述任一项所述方法。
本申请有益技术效果:
1)本申请充分整合Kraken2快速预分类和minimap2快速比对优势,在小宏基因组参考序列数据库范围内获取每条序列详细比对信息;
2)本申请基于每条序列详细比对信息划分独立子集,在独立子集范围内进行多重比对序列重分配,有效减少分类树结构错误对序列重分配影响;
3)本申请基于小宏基因组参考序列数据库进行数据模拟和概率建模,拟合计算唯一比对率,有效减少病原比对数据库不平衡对序列重分配影响;
4)本申请充分挖掘分类树结构,病原比对数据库及序列比对信息,基于概率模型对每条多重比对序列进行重分配,有效降低假阴性和假阳性,进而提高病原检测准确性。
附图说明
图1、一种宏基因组多重比对序列重分配方法及应用示意图;
图2实施例1中大肠杆菌和福氏志贺菌模拟数据集的不同丰度估计方法结果比较;
图3实施例2中鲍曼不动杆菌和威氏不动杆菌模拟数据集不同丰度估计方法结果比较。
具体实施方式
下面将结合实施例对本申请的实施方案进行详细描述,但是本领域技术人员将会理解,下列实施例仅用于说明本申请,而不应视为限制本申请的范围。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市场购买获得的常规产品。
部分术语定义,除非在下文中另有定义,本申请具体实施方式中所用的所有技术术语和科学术语的含义意图与本领域技术人员通常所理解的相同。虽然相信以下术语对于本领域技术人员很好理解,但仍然阐述以下定义以更好地解释本申请。
本申请中的术语“大约”表示本领域技术人员能够理解的仍可保证论及特征的技术效果的准确度区间。该术语通常表示偏离指示数值的±10%,优选±5%。
如本申请中所使用,术语“包括”、“包含”、“具有”、“含有”或“涉及”为包含性的(inclusive)或开放式的,且不排除其它未列举的元素或方法步骤。术语“由…组成”被认为是术语“包含”的优选实施方案。如果在下文中某一组被定义为包含至少一定数目的实施方案,这也应被理解为揭示了一个优选地仅由这些实施方案组成的组。
此外,说明书和权利要求书中的术语第一、第二、第三、(a)、(b)、(c)以及诸如此类,是用于区分相似的元素,不是描述顺序或时间次序必须的。应理解,如此应用的术语在适当的环境下可互换,并且本申请描述的实施方案能以不同于本申请描述或举例说明的其它顺序实施。
下面结合具体实施例来阐述本申请。
实验例:本申请方法体系建立
本申请通过生信分析优化,初步建立一套适用于宏基因组多重比对序列重分配方法,该方法具体包括了如下步骤:
步骤1)快速分类:基于完整宏基因组参考序列数据库,采用Kraken2对对宏基因组测序数据进行快速分类,得到分类树报告;
其中,所述参考序列数据库为完整宏基因组参考序列数据库;完整宏基因组参考序列数据库优选为去冗余去低质量的nt数据库+专业病毒库+专业寄生虫库。宏基因组测序数据优选为去人源,去低质量后的测序数据;所述Kraken2参数优选设置为confidence=0.5;所述Kraken2分类树报告信息包括:各分类层级水平分类树信息(分类名称,分类编号),分类到该层级水平及以下的总序列数,分类到该层级水平的序列数以及分类到该层级水平以下的序列数。
步骤2)提取小库:基于Kraken2分类树报告,按照宏基因组参考序列纳入标准,从完整宏基因组参考序列数据库中提取目标参考序列,得到小宏基因组参考序列数据库;
其中,参考序列纳入标准包括:
a、分类树报告中列出的所有物种在完整宏基因组参考序列数据库中收录的参考序列;
b、上述列出的所有物种所包含的所有物种亚型,血清型和株在完整宏基因组参考序列数据库中收录的参考序列;
c、分类树报告中列出的未分配序列数>1000的科所包含的所有物种,物种亚型,血清型和株在完整宏基因组参考序列数据库中收录的参考序列;
d、分类树报告中列出的未分配序列数>100的属所包含的所有物种,物种亚型,血清型和株在完整宏基因组参考序列数据库中收录的参考序列。
其中,小宏基因组参考序列数据库优选的为完整宏基因组参考序列数据库的子集。
步骤2)还可以包括基于测序数据建库类型及参考序列来源信息对小库进行二次提取。
步骤3)快速比对:基于小宏基因组参考序列数据库,采用minimap2对宏基因组测序数据进行快速比对,得到序列比对信息;
序列比对的结果包括:比对序列名称,参考序列名称,最小分类单元名称,比对位置,比对质量,详细比对信息(M:匹配,S:软比对,I:插入;D:缺失);最小分类单元包括:物种,物种亚型,血清型和株。另外,步骤3)可基于序列比对信息进一步对序列比对结果进行对比质量过滤。
步骤4)划分子集:基于序列比对信息,划分最小分类单元和比对序列均封闭的独立子集;划分子集的输入为上述快速比对的序列比对信息,输出为最小分类单元和比对序列均封闭的独立子集。
其中,最小分类单元和比对序列均封闭的独立子集特点包括:
1)独立子集与独立子集间最小分类单元无交集;
2)独立子集与独立子集间序列无交集;
3)所有独立子集的并集为序列比对信息。
优选的,划分子集的规则为最小分类单元和比对序列均封闭,子集的划分依赖于序列比对信息,与分类树结构无关。
步骤5)构建模型:基于小宏基因组参考序列数据库和独立子集,模拟计算独立子集内每个最小分类单元唯一比对率,构建多重比对序列重分配概率模型;
模拟计算独立子集内每个最小分类单元唯一比对率包括:
1)提取独立子集内每个最小分类单元的参考序列,生成独立子集参考序列数据库;
2)基于独立子集参考序列数据库模拟生成测序序列;
3)通过minimap2将模拟序列比对到独立子集内所有最小分类单元的参考序列上,得到独立子集内模拟序列的序列比对结果;
4)基于模拟序列的比对结果,计算独立子集内每个最小分类单元的唯一比对率。
多重比对序列重分配概率模型构建包括:
1)在每个独立子集内建模;基于唯一比对序列和唯一比对率估计独立子集中每个最小分类单元的序列丰度;
2)基于唯一比对率和每个最小分类单元的丰度估计每个最小分类单元的多重比对序列丰度;
3)每个最小分类单元的多重比对序列丰度作为先验重分配概率;基于每条多重比对序列的比对结果生成每个最小分类单元的观察值概率;
4)基于先验重分配概率和观察值概率,构建多重比对序列重分配概率模型。
另外,步骤5)中,基于独立子集参考序列数据库模拟生成测序序列可参照子集范围内物种及序列信息设置模拟数据通量和读长。
步骤6)序列重分配:基于多重比对序列重分配概率模型和序列比对信息,计算每条多重比对序列后验分配概率,将后验分配概率作为随机函数参数为该条序列重分配最小分类单元标识;
多重比对序列重分配包括:
1)对每条多重比对序列进行重分配;
2)基于多重比对序列重分配概率模型,生成每条多重比对序列的后验重分配概率;
3)后验重分配概率作为随机分配函数的参数为每条多重比对序列重分配最小分类单元标识。
步骤7)丰度估计:基于唯一比对序列和多重比对序列重分配结果估计物种丰度。
基于唯一比对序列和多重比对序列重分配结果估计物种丰度的步骤包括:
1)估计每个最小分类单元的序列丰度,即唯一比对序列数与多重比对重分配序列数之和;
2)基于分类树构建最小分类单元的层次关系,每个层次关系的根节点为物种;
3)在物种水平统计序列丰度,作为物种序列丰度输出结果。
实施例1:大肠杆菌和福氏志贺菌的丰度估计
1)大肠杆菌和志贺杆菌测序数据模拟
从NCBI官网下载大肠杆菌参考基因组(GCF_000005845.2_ASM584v2_genomic.fna)和福氏志贺菌参考基因组(GCF_000006925.2_ASM692v2_genomic.fna)。采用art_illumina模拟器按不同序列梯度生成模拟数据集,序列读长为75bp,测序模式为单端,测序平台为HS2500。
2)分别采用Kraken2,Kraken2+Bracken,本申请多重比对序列重分配方法对上述模拟数据集进行物种检测和丰度估计。
3)丰度估计结果统计。
如统计结果和图2所示,与大肠杆菌模拟序列理论值相比,Kraken2在不同序列梯度下,丰度估计均出现显著低估(丰度估计值分别为理论值0.04倍,0.05倍,0.039倍和0.036倍)。
与大肠杆菌模拟序列理论值相比,Kraken2+Bracken在低丰度(100条模拟序列)时,出现漏检;在中低丰度(500条模拟序列)时,出现显著高估(丰度估计值为理论值1.874倍);在中高丰度(2500条模拟序列)和高丰度(12500条模拟序列)时,出现一定低估(丰度估计值分别为理论值0.797倍和0.626倍)。
与大肠杆菌模拟序列理论值相比,本申请新方法在低丰度,中低丰度,中高丰度和高丰度时,丰度估计值与理论值均高度接近(丰度估计值分别为理论值1.03倍,1.03倍,0.975倍和.967倍)。
与福氏志贺菌模拟序列理论值相比,Kraken2在不同序列梯度下,丰度估计均出现严重低估(丰度估计值分别为理论值0.03倍,0.016倍,0.012倍和0.016倍)。
与福氏志贺菌模拟序列理论值相比,Kraken2+Bracken在低丰度(100条模拟序列)和中低丰度(500条模拟序列)时,出现漏检;在中高丰度和高丰度时出现轻度高估(丰度估计值分别为理论值1.14倍和1.29倍)。
与福氏志贺菌模拟序列理论值相比,本申请方法在低丰度和中低丰度时出现低估(丰度估计值分别为理论值0.27倍和0.268倍);在中高丰度和高丰度时,丰度估计值与理论值均高度接近(丰度估计值分别为理论0.972倍和0.967倍)。
与Kraken2和Kraken2+Bracken相比,本申请方法在各序列丰度梯度下,对大肠杆菌和福氏志贺菌的丰度估计值与理论值均更接近。
实施例1中,大肠杆菌和志贺杆菌基因组相似度高达98%,但在分类树上被错误的分配到同科不同属上。
Kraken2预分类是基于Kmer和LCA映射,由于上述大肠杆菌和专辑杆菌的分类树分类信息错误,大量大肠杆菌和志贺杆菌序列会被标识为科水平的LCA,导致种水平的序列丰度被严重低估。
另外,当大肠杆菌和志贺杆菌输入序列丰度较低时,Kraken2和或Bracken会出现漏检或丰度低估,导致假阴性。
由实施例1可知,本申请提供的多重比对序列重分配方法通过提取小库重比对和多重比对序列重分配,有效规避或减轻了分类树错误可能导致的漏检,丰度低估和假阴性。
实施例2:鲍曼不动杆菌和威氏不动杆菌的丰度估计
从NCBI官网下载鲍曼不动杆菌参考基因组(GCF_008632635.1_ASM863263v1_genomic.fna)和威氏不动杆菌参考基因组(GCF_000368585.1_Acin_vene_CIP_110063_V1_genomic.fna)。采用art_illumina模拟器按不同序列梯度生成模拟数据集,序列读长为75bp,测序模式为单端,测序平台为HS2500。
2)分别采用Kraken2,Kraken2+Bracken,本申请多重比对序列重分配方法对上述模拟数据集进行物种检测和丰度估计。
3)丰度估计结果统计。
如统计结果和图3所示,与鲍曼不动杆菌模拟序列理论值相比,Kraken2在不同序列丰度梯度下,丰度估计均出现显著低估(丰度估计值分别为理论值0.09倍,0.136倍,0.124倍和0.122倍)。
与鲍曼不动杆菌模拟序列理论值相比,Kraken2+Bracken在低丰度(100条模拟序列)时出现漏检,在中低丰度,中高丰度和高丰度时,丰度估计值与理论值接近(丰度估计值分别为理论值1.032倍,1.039倍,1.019倍)。
与鲍曼不动杆菌模拟序列理论值相比,本申请新方法在不同序列梯度下,丰度估计与理论值接近(丰度估计值分别为理论值0.95倍,1倍,0.819倍,0.975倍)。
与威氏不动杆菌模拟序列理论值相比,Kraken2在不同序列丰度梯度下,丰度估计均出现显著低佑(丰度估计值分别为理论值0.44倍,0.42倍,0.439倍和0.423倍)。
与威氏不动杆菌模拟序列理论值相比,Kraken2+Bracken在低丰度时出现显著高估(丰度估计值为理论值1.44倍),在中低丰度,中高丰度,高丰度时,丰度估计值均出现显著低估(丰度估计值分别为理论值0.494倍,0.52倍和0.503倍)。
与威氏不动杆菌模拟序列理论值相比,本申请新方法在不同序列丰度梯度下,丰度估计值均出现低估(丰度估计值分别为理论0.630倍,0.732倍,0.717倍和0.695倍)。
与Kraken2和Kraken2+Bracken相比,本申请新方法在低丰度鲍曼不动杆菌序列丰度估计上具有明显优势。
与Kraken2+Bracken相比,本申请新方法在威氏不动杆菌序列丰度估计上与理论值更接近。
实施例2中,鲍曼不动杆菌和威氏不动杆菌为同属异种,在分类树上分类信息正确,但在宏基因组参考序列数据库中,鲍曼不动杆菌在种水平和株水平收录的参考序列数显著高于威氏不动杆菌。
由于鲍曼不动杆菌和威氏不动菌在参考序列收录数量上高度不平衡,Kraken2方法会将大量来自鲍曼不动杆菌的序列映射到属水平或株水平,导致映射到种水平的序列数显著低于威氏不动杆菌。
minimap2也会将大量来自鲍曼不动杆菌的序列标识为多重比对序列。
Bracken进行丰度估计包括两个部分:从属到种的估计采用贝叶斯概率模型(乘法运算),从株到种的估计采用简单的向上递归(加法运算)。
由于物种和株水平均有对应参考序列,物种和株参考序列收录数量和结构不平衡会引起Bracken概率模型乘法运算和向上递归加法运算的比例失衡,导致一定的估计偏倚。
本申请新方法将物种和株均视为同一级别最小分类单元,各自有独立参考序列,多重比对序列的重分配在独立封闭子集内进行,有效规避或降低分类树上下层级结构和参考序列数据和比例失调带来的重分配偏倚。
另一方面,与鲍曼不动杆菌相比,威氏不动杆菌收录的参考序列相当少或不全,Bracken在重分配过程中,唯一比对率计算基于完整宏基因组参考序列库,可能导致唯一比对率的估算及丰度估计偏倚。
本申请新方法唯一比对率的计算基于独立子集和小宏基因组参考序列库,减少了其它无关物种参考序列影响,唯一比对率的模拟计算和序列丰度估计会更接近理论值或真实值。
由实施例2可知,本申请提供的多重比对序列重分配方法通过提取小库重比对和多重比对序列重分配,有效规避或减轻了宏基因组参考序列数据库收录的参考序列不平衡可能导致的漏检或丰度低估。
最后应说明的是:以上各实施例仅用以说明本申请的技术方案,而非对其限制;尽管参照前述各实施例对本申请进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本申请各实施例技术方案的范围。