一种可发酵合成Terrequinone A的重组大肠杆菌及其制备方法以及应用
文献发布时间:2023-06-19 19:30:30
技术领域
本发明属于生物技术领域,具体涉及一种可发酵合成Terrequinone A的重组大肠杆菌及其制备方法以及应用。
背景技术
双吲哚醌类化合物是一类具有抗逆转录病毒、抗糖尿病或细胞毒性生物活性的真菌天然产物。双吲哚醌自可皆霉素(Cochliodinol)被发现以来,其家族成员不断壮大,而在这其中,从沙漠植物根际真菌土曲霉(Aspergillus terreus)中首次分离出来的Terrequinone A,对四种人类癌细胞(NCIH460、MCF-7、SF-268和MIA Pa Ca-2)具有中等细胞毒性,是一种潜在的抗癌药物(He et al.,Journal of Natural Products,2004,67(12):1985-1991),也是唯一一种有生物合成信息的双吲哚醌类化合物。2004年,Bok等人利用转录调控因子LaeA对构巢曲霉(Aspergillus nidulans)的次生代谢组进行了挖掘,识别了Terrequinone A的合成基因簇tdiABCDE(Bok et al.,Eukaryotic Cell,2004,3(2):527-535)。
Terrequinone A作为一种天然产物,其结构复杂、官能团较多,采用化学合成法所需步骤多、产率低以及环境不友好,随着Terrequinone A在生物体内进行合成和修饰的特殊途径被阐明,利用生物催化法合成Terrequinone A成为一种有效的尝试,Balibar等人分别过表达和研究了tdiABCDE基因簇中的5个基因,通过酶的体外分步反应成功合成了Terrequinone A(Carl J Balibar等人,Terrequinone A biosynthesis through L-tryptophan oxidation,dimerization and bisprenylation,Nature Chemical Biology,2007,3(9):584-592)。然而,体外合成需要外源性地添加昂贵的辅因子和辅因子再生酶系;另外,游离酶催化体系中一种酶催化的产物往往是下一个酶的底物,其底物产物的传递通常受到空间的限制,降低了产物的合成效率。因此,需要寻求更加高效的方法来生产Terrequinone A。大肠杆菌作为一种优良的生物反应器,通过对其进行合成途径的改造或引入新的代谢途径,可用于生物合成高价值天然产物,它能完美地规避上述两个问题。
发明内容
本发明的目的在于提供一种可发酵合成Terrequinone A的重组大肠杆菌及其制备方法以及应用,从而解决现有技术中Terrequinone A的体外合成方法存在的生产成本较高、合成效率低下的问题。
为了解决上述问题,本发明提供如下技术方案:
根据本发明的第一方面,提供一种可发酵合成Terrequinone A的重组大肠杆菌的制备方法,包括以下步骤:S1:对tdiA基因、tdiB基因、tdiC基因、tdiD基因、tidE基因和sfp基因按大肠杆菌表达模式优化,分别获得核苷酸序列如SEQ ID NO.1~6所示的tdiAS基因、tdiBS基因、tdiCS基因、tdiDS基因、tidES基因和sfpS基因,将所述六个基因分别与T7启动子和终止子连接,构建基因表达盒T7 tdiAS、T7 tdiBS、T7 tdiCS、T7 tdiDS、T7 tidES和T7sfpS;S2:将步骤S1获得的六个基因表达盒按T7 tdiDS、T7 tdiAS、T7sfpS、T7 tdiBS、T7tdiCS和T7 tidES的顺序串联,连接入大肠杆菌表达载体,构建重组质粒pC04;S3:将步骤S2获得的重组质粒pC04转化入大肠杆菌BL21-AI中,获得一种可发酵合成Terrequinone A的重组大肠杆菌。
优选地,步骤S2中,所述大肠杆菌表达载体为pCAMBIA1301。
优选地,步骤S2中,在T7 tdiDS的5’-端连接EcoRⅠ内切酶位点,在T7 tidES的3’-端连接HindⅢ内切酶位点,获得EcoRⅠ-T7 tdiDS-T7tdiAS-T7 sfpS-T7 tdiBS-T7 tdiCS-T7 tidES-HindⅢ。
根据本发明的第二方面,提供一种用于生产Terrequinone A的重组质粒pC04,所述重组质粒pC04由基因表达盒按T7 tdiDS、T7 tdiAS、T7sfpS、T7 tdiBS、T7 tdiCS和T7tidES的顺序串联并连接入大肠杆菌表达载体上构建而成,其中,所述基因表达盒T7tdiAS、T7 tdiBS、T7 tdiCS、T7 tdiDS、T7 tidES和T7 sfpS由核苷酸序列如SEQ ID NO.1~6所示的tdiAS基因、tdiBS基因、tdiCS基因、tdiDS基因、tidES基因和sfpS基因分别与T7启动子和终止子连接而成。
根据本发明的第三方面,提供一种根据上述制备方法获得的可发酵合成Terrequinone A的重组大肠杆菌。
根据本发明的第四方面,提供一种利用重组大肠杆菌发酵合成Terrequinone A的方法,将所述重组大肠杆菌接种于含50μg/ml卡那霉素的M9液体培养基中进行发酵培养,加入L-阿拉伯糖诱导,以L-色氨酸为底物,即可发酵合成Terrequinone A。
优选地,所使用的M9液体培养基每升含有:15g甘油(glycerol),6g Na
优选地,加入的所述L-阿拉伯糖的浓度为0.2%,在25℃温度下诱导培养14~18h。
优选地,在诱导培养后,加入浓度为0.5g/L的L-色氨酸,在30℃培养22~26h,即可。
本发明中,根据大肠杆菌的密码子偏好性对tdiA基因、tdiB基因、tdiC基因、tdiD基因、tidE基因和sfp基因作为整体进行优化,优化原则包括:(一)按照大肠杆菌密码子偏爱性优化基因,优化基因密码子,提高基因翻译效率;(二)消除茎环结构、转录终止信号、同一基因或相邻基因200bp以内的逆向重复序列,并使基因内部的GC/AT均衡,提高RNA的稳定性;(三)使基因编码蛋白符合N端原则,以提高翻译蛋白的稳定性;(四)优化mRNA二级结构自由能,以提高基因表达效率;(五)在满足上述四个原则的基础上,选择消除四个基因内部的EcoRI和HindIII内切酶识别位点,便于构建表达盒;优化后获得tdiAS、tdiBS、tdiCS、tdiDS、tidES和sfpS基因。
与现有技术相比,本发明具有如下有益效果:
本发明利用合成生物学技术,对tdiA基因、tdiB基因、tdiC基因、tdiD基因、tidE基因和sfp基因按大肠杆菌表达模式优化,优化后分别与大肠杆菌T7启动子和终止子连接,构建基因表达盒,再与大肠杆菌表达载体连接获得多基因重组质粒,构建基因工程菌,编码非核糖体肽合成酶的tdiAS基因、编码甲代丙烯基-L-色氨酸合成酶的tdiBS基因、编码氧化还原酶的tdiCS基因、编码氨基转移酶的tdiDS基因、编码伴侣蛋白的tidES基因和编码磷酸泛酰巯基乙胺基转移酶的sfpS基因均能活性表达,以L-色氨酸为底物,可生产具有抗癌细胞生物活性的Terrequinone A,在生物制药领域具有潜在的应用价值。
He等人对土曲霉(Aspergillus terreus)经过28天的培养后,从5.4L的发酵液中分离得到6.0mg(即1.1mg/L)Terrequinone A(He et al.,Journal of Natural Products,2004,67(12):1985-1991)。与天然菌株Aspergillus terreus相比,本发明的重组大肠杆菌在经过48h的培养后,发酵液中Terrequinone A的含量可达到1.8mg/L,大大缩短了Terrequinone A的合成周期,并提高了Terrequinone A在发酵液中的含量。
附图说明
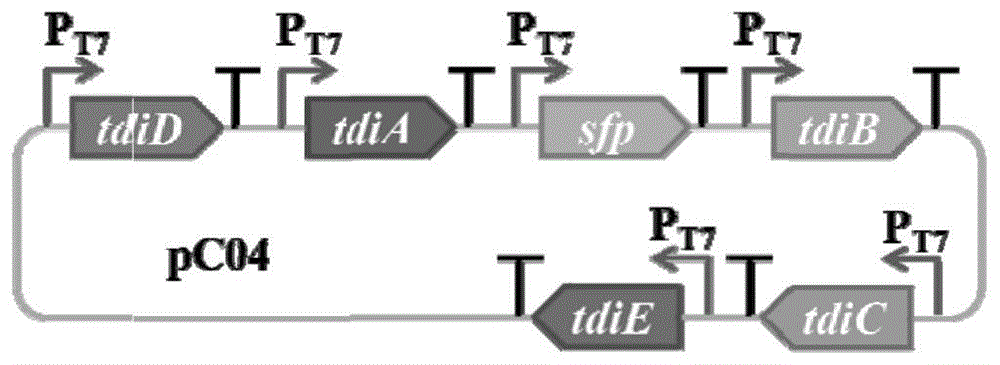
图1为本发明实施例3中重组质粒pC04的结构示意图;
图2为本发明实施例3中大肠杆菌BL-4转入的Terrequinone A合成途径;
图3为本发明实施例4中大肠杆菌BL-4外源基因的PCR检测结果,M为Marker;
图4为本发明实施例5中Terrequinone A的质谱测定鉴定图(正离子m/z=491.2);
图5为本发明实施例5中菌培养液经氯仿萃取后的HPLC图谱。
具体实施方式
下面结合说明书附图和具体实施例对本发明作进一步说明。
实施例中所使用的试验方法如无特殊说明,均为常规分子生物学方法;所使用的材料、试剂等,如无特殊说明,为可从商业途径得到的试剂和材料。
实施例1六段基因的优化
基于tdiA(Genbank:EF550581.1)、tdiB(Genbank:EF550582.1)、tdiC(Genbank:EF550583.1)、tdiD(Genbank:EF550584.1)、tidE基因(Genbank:(Genbank:EF550585.1)和sfp基因(GenBank:X65610.1,Bacillus subtilis)的编码序列,按照以下原则对上述六个基因作为整体进行优化:
(一)按照大肠杆菌密码子偏爱性优化基因,优化基因密码子,提高基因翻译效率;(二)消除茎环结构、转录终止信号、同一基因或相邻基因200bp以内的逆向重复序列,并使基因内部的GC/AT均衡,提高RNA的稳定性;(三)使基因编码蛋白符合N端原则,以提高翻译蛋白的稳定性;(四)优化mRNA二级结构自由能,以提高基因表达效率;(五)在满足上述四个原则的基础上,选择消除八个基因内部的EcoRI和HindⅢ内切酶识别位点,便于构建重组质粒。
优化后,获得tdiAS、tdiBS、tdiCS、tdiDS、tidES和sfpS基因,所述tdiAS基因的核苷酸序列如SEQ ID NO.1所示;所述tdiBS基因的核苷酸序列如SEQ ID NO.2所示;所述tdiCS基因的核苷酸序列如SEQ ID NO.3所示;所述tidDS基因的核苷酸序列如SEQ ID NO.4所示;所述tdiES基因的核苷酸序列如SEQ ID NO.5所示;所述sfpS基因的核苷酸序列如SEQID NO.6所示。
实施例2基因表达盒的构建
在每个优化的基因序列前端连接大肠杆菌T7启动子序列,在每个优化的基因序列末端连接大肠杆菌T7终止子序列,构建基因表达盒T7tdiAS、T7 tdiBS、T7 tdiCS、T7tdiDS、T7 tidES和T7 sfpS,由南京金斯瑞公司化学合成。
T7启动子序列(SEQ ID NO.7)为:
5’-TAATACGACTCACTATAGG-3’。
T7终止子序列(SEQ ID NO.8)为:
5’-TAGCATAACCCCTTGGGGCCTCTAAACGGGTCTTGAGGGGTTTTTTG-3’。
实施例3构建重组质粒并转化
利用“聚丙烯酰胺凝胶电泳(PAGE)介导的重叠延伸PCR”技术(彭日荷等人,Adirect and efficient PAGE-mediated overlap extension PCR method for genemultiple-site mutagenesis,Appl Microbiol Biotechnol.2006,73(1):234-40),获得的六个基因表达盒按T7 tdiDS、T7 tdiAS、T7sfpS、T7 tdiBS、T7 tdiCS和T7 tidES的顺序串联,并在T7 tdiDS的5’-端连接EcoRⅠ内切酶位点,在T7 sfpS的3’-端连接HindⅢ内切酶位点,获得EcoRⅠ-T7 tdiDS-T7 tdiAS-T7 sfpS-T7 tdiBS-T7 tidCS-T7 tdiES-HindⅢ。
为获得EcoRⅠ-T7 tdiDS-T7 tdiAS-T7 sfpS-T7 tdiBS-T7 tidCS-T7tdiES-HindⅢ,设计的引物序列(SEQ ID NO.9~SEQ ID NO.20)如下:
T7 tdiDS:
P1:5’-GAATCCTAATACGACTCACTATAGGATGGGTTCTATTG-3’;
P2:5’-CTTAGATGGTGCCATCCTATAGTGAGTCGTATTACAAAAAACCCCTC-3’;
T7 tdiAS:
P3:5’-TTGAGGGGTTTTTTGTAATACGACTCACTATAGGATGGCACCATCTAAG-3’;
P4:5’-ACCGTAGATTTTCATCCTATAGTGAGTCGTATTACAAAAAACCCCTC-3’;
T7 sfpS:
P5:5’-TTGAGGGGTTTTTTGTAATACGACTCACTATAGGATGAAAATCTACGGT-3’;
P6:5’-GTATTCAGTAGCCATCCTATAGTGAGTCGTATTACAAAAAACCCCTC-3’;
T7 tdiBS:
P7:5’-TTGAGGGGTTTTTTGTAATACGACTCACTATAGGATGGCTACTGAATAC-3’;
P8:5’-AAGAGCTGCGTGCATCCTATAGTGAGTCGTATTACAAAAAACCCCTC-3’;
T7 tdiCS:
P9:5’-TTGAGGGGTTTTTTGTAATACGACTCACTATAGGATGCACGCAGCTCTT-3’;
P10:5’-ATGGTCTGTTAACCATCCTATAGTGAGTCGTATTACAAAAAACCCCTC-3’;
T7 tdiES:
P11:5’-TTGAGGGGTTTTTTGTAATACGACTCACTATAGGATGGTTAACAGACCAT-3’;
P12:5’-AAGCTTCAAAAAACCCCTCAAGACCCGTTTAGAGG-3’。
第一步PCR:分别以T7 tdiDS、T7 tdiAS、T7 sfpS、T7 tdiBS、T7 tidCS和T7 tdiES基因片段为模板,添加对应的引物进行PCR扩增。PCR扩增程序:94℃30s,68℃60s,共10个循环;
将第一步PCR获得的6个基因片段进行凝胶电泳,胶回收;
第二步PCR:以胶回收获得的6个基因片段的混合物作为模板,以P1和P12为引物进行PCR扩增。PCR扩增程序:94℃预变性1min;94℃30s,58℃30s,72℃30s,共25个循环;最后72℃再延伸10min。
将上述串联的六个基因片段用EcoRⅠ和HindⅢ双酶切并连接到经同样酶切的载体pCAMBIA1301上,获得含有六个基因的重组质粒pC04(图1)。
采用热激法将pC04转入大肠杆菌BL21-AI中,涂布在含50μg/ml卡那霉素抗性的固体LB平板上,37℃过夜培养后挑取阳性克隆,即重组大肠杆菌BL-4,其转入的TerrequinoneA合成途径如图2所示。
实施例4重组大肠杆菌的鉴定
通过PCR鉴定外源基因在大肠杆菌中的成功转入,挑取单菌落接种于50ml含50μg/ml卡那霉素的LB液体培养基中,37℃培养至菌液OD
根据各基因的特异性片段设计的PCR检测所用引物序列(SEQ ID NO.21~SEQ IDNO.32)如下:
tdiA-F:5’-TCCGTCAAGTGCATGGATGTC-3’;
tdiA-R:5’-CAGACCACGCTCACGCAGGAC-3’;
tdiB-F:5’-GCACTGAAGAAGCTGGGTAAC-3’;
tdiB-R:5’-ACGGAAACCGAAGTCACCAGC-3’;
tdiC-F:5’-ATCTCTCGTAAGCCAATCTGC-3’;
tdiC-R:5’-GACGATGACGACACGGGAACC-3’;
tdiD-F:5’-ATGTTCGTCTGGCTGGAACTC-3’;
tdiD-R:5’-GCAACGATCGACCAGACCAGC-3’;
tdiE-F:5’-AAGACCTTGGGTTTGTGGAAC-3’;
tdiE-R:5’-GACGTCGGAACCAGGTGCAGC-3’;
sfp-F:5’-TCTCACTCTGGACGTTGGGTG-3’;
sfp-R:5’-TGCAGATGCGATGAGACGTTG-3’。
所用扩增程序:94℃预变性3min;94℃30s,54℃30s,72℃30s,共30个循环;最后72℃再延伸10min。
图3结果显示,本发明的重组大肠杆菌BL-4扩增出上述六个基因tdiAS、tdiBS、tdiCS、tdiDS、tidES和sfpS,表明重组质粒pC04转入了大肠杆菌中,所获得的重组大肠杆菌BL-4中包含该六个外源基因。
实施例5利用重组大肠杆菌BL-4发酵合成Terrequinone A
挑取实施例3中重组大肠杆菌BL-4的单菌落,接种于50ml含50μg/ml卡那霉素的优化后的M9液体培养基(15g甘油(glycerol),6g Na
取200μl发酵液,加入两倍体积甲醇,用超声波破碎细胞(功率为400W,超声4s,间歇8s,重复99轮),4℃10000rpm离心1min,取上清,用等体积氯仿萃取,减压蒸馏后用甲醇重溶,用于Terrequinone A检测。
通过LC-MS检测,将样品质谱图(图4)与文献报道的Terrequinone A质谱图(Balibar et al.,Nature Chemical Biology,2007,3(9):584-592)作比较,鉴定为Terrequinone A。具体LC-MS检测条件:TSQ Quantum-Accela型液质联用仪;岛津Shim-packGIST C18色谱柱(150mm×2.1mm,3μm);流动相为A为含0.1%的甲酸水溶液,流动相B为含0.1%的甲酸乙腈,采用梯度洗脱:0-3min:20%B;3min-13min:20%B-90%B。流速0.2μL/min。柱温为35℃。进样量为1μL。离子源采用ESI+模式,电喷雾电压为3500V,鞘气流速为13mL/min,离子传输管温度为275℃,扫描模式为全扫描模式,扫描分辨率选择为0.4Da,采集的质荷比范围为m/z=300-500Da,扫描时间为0.5s。
通过HPLC检测Terrequinone A含量,具体HPLC检测条件:安捷伦1100高效液相色谱系统;C18柱(
以上所述的,仅为本发明的较佳实施例,并非用以限定本发明的范围,本发明的上述实施例还可以做出各种变化。凡是依据本发明申请的权利要求书及说明书内容所作的简单、等效变化与修饰,皆落入本发明专利的权利要求保护范围。本发明未详尽描述的均为常规技术内容。
- 一种重组大肠杆菌及合成四氢嘧啶的应用
- 广藿香醇合成酶突变体及重组大肠杆菌发酵制备广藿香醇的方法
- 一种大肠杆菌重组核酸、重组大肠杆菌及培养方法和生物合成L-苏氨酸的方法