一种氨基酸共聚物及其应用
文献发布时间:2023-06-19 19:30:30
技术领域
本申请涉及核酸递送材料技术领域,特别是涉及一种氨基酸共聚物及其应用。
背景技术
核酸药物是生物医药发展的前沿领域,包括反义核酸(ASO)、小干扰RNA(siRNA)、微小RNA(miRNA)、信使RNA(mRNA)等;核酸药物是基因治疗的一种形式,也是继小分子药物、蛋白药物、抗体药物之后的新一代制药技术。核酸药物能够直接作用于致病靶基因或者靶mRNA,在基因水平上发挥治疗疾病的作用;核酸药物从转录后水平进行基因沉默或激活治疗,相比传统蛋白水平发挥作用的药物具有高特异性、高效性、长效性等明显优势。核酸治疗技术创新和临床试验正在积极开展,多项核酸治疗项目相继在美国、欧盟、中国等国家获得批准上市。核酸治疗在各类重大疾病的治疗中具有十分广阔的应用前景,例如恶性肿瘤、感染性疾病、心血管疾病、自身免疫性疾病、代谢性疾病等。
核酸治疗的核心是核酸药物。常见的核酸药物主要有质粒DNA(plasmid DNA,pDNA)、信使RNA(message RNA,mRNA)、小干扰RNA(small interfering RNA,siRNA)等。其中,DNA可以携带具有特定治疗作用的核苷酸序列,在宿主的靶细胞内转录mRNA从而翻译成具有明确生物学功能的蛋白质,通过补充体内缺失蛋白或修正异常蛋白等方式治疗疾病。siRNA则作用于靶细胞的mRNA,通过特异性剪切靶标基因的mRNA以下调其靶标蛋白的表达,从而修复或杀死病变细胞,达到疾病治疗的目的。这些核酸分子作用位点明确,能够被用于多种疾病的治疗。基于mRNA的治疗,则是利用体外合成或者化学修饰后的mRNA分子进入细胞质中,利用细胞质内的自有核苷酸进行转录表达,生成机体所需要的蛋白质。mRNA药物开发的主要技术门槛在于稳定性和递送技术,相关技术难点还有待进一步的被攻克,以实现mRNA药物作为蛋白质补充或替代疗法以治疗相关疾病。尤其是随着癌症基因测序和抗原表位发现等技术的发展,mRNA药物也可用于针对肿瘤患者的个性化治疗,并具有巨大的应用潜力。
然而,核酸药物一般不稳定,在体内应用时,易被血液循环中的核酸酶降解;另外,核酸分子自身携带负电荷,与同样带负电荷的细胞膜存在静电排斥作用,进一步增加了其进入细胞的难度;核酸药物存在的这些障碍很大程度上限制了其在临床应用上的转化。因此,如何克服核酸分子本身物理化学性质带来的挑战,将其安全高效地递送至目标作用部位是亟待解决的问题之一。
核酸药物的体内递送主要面临3大个难关:1)核酸的分子量和负电荷使其不能自由通过生物膜;2)RNA容易被血浆和组织中RNase酶降解,被肝脏和肾脏快速清除和被免疫系统识别;3)进入细胞后被“困”在内体中最终被溶酶体降解而无法发挥功能。药物递送系统是克服核酸类药物发展面临的技术障碍的关键,目前,解决递送问题主要有两种策略:一是对核酸分子进行化学修饰改造,提高其稳定性并逃避免疫系统的识别;另外一种策略就是利用药物递送系统,比如说脂质纳米颗粒(LNP),GalNAc(N-乙酰化的半乳糖胺)偶联技术,脂质多聚复合物和聚合物纳米颗粒等。
近年来,多种合成材料已被陆续开发出来以用于核酸药物的体内递送。其中树形分子因其精确可控的结构、优异的单分散性和多价协同效应,被广泛应用于核酸药物递送领域,如商业化转染试剂SuperFectTM的主要成分就是聚酰胺-胺类(PAMAM)树形分子。PAMAM树形分子具有独特的结构优势:1)PAMAM表面丰富的正电荷,使得其能通过静电相互作用有效负载核酸药物分子,保护其不被核酸酶降解,并且有利于核酸药物制剂的细胞摄取;2)PAMAM表面大量可修饰的反应位点,可以引入不同性质的功能基团,且利用树形分子特有的多价协同放大效应可将功能性作用放大,提高递送体系的效率;3)PAMAM因内部丰富的叔胺基团具有较好的质子缓冲能力,有助于PAMAM与核酸复合物通过“质子海绵效应”从酸性细胞器中逃逸,有效释放负载的核酸药物;4)PAMAM结构中大量的酰胺结构,使其具有仿生“类蛋白”性能和良好的生物安全性。由于这些独特的结构性能,PAMAM树形分子在核酸药物输送中展现出独特的优势和巨大的潜力。
然而,传统的球形PAMAM树形分子很难适应不同的核酸分子的递送需求;并且,传统的PAMAM树形分子在体内难以降解,具有潜在的机体毒性;此外核酸与树形分子形成的组装体的释放特异性和递送效率仍然需要进一步的提高和改善。
同时,基于mRNA的核酸药物分子结构较大,带有负电荷,穿透细胞膜的难度较高;并且,在进入细胞后还需要从内体中逃逸至细胞质中,再加上外源的核酸分子具有免疫原性,会激活人体免疫系统的反应。因此,如何让核酸药物在体内循环过程中更加稳定且有效避免被机体免疫系统的识别和清除存在诸多的挑战。
目前现有的递送技术专利壁垒较高,且存在一定的缺陷。如LNP技术需要多组分进行配比组装,稳定性容易收到影响,并且过敏反应严重,大多只能靶向肝肾和网状内皮系统。GalNAc技术只能靶向肝脏实质性细胞,并且需要对核酸序列进行全化学修饰,合成难度大。其他递送技术,如PNP(多肽纳米粒子)靶向性不强;外泌体递送技术目前还处于早期探索阶段;PEI(聚乙烯亚胺)递送存在供应商稀缺,还有很大的毒性,因此严重阻碍了核酸药物的临床转化需求,急需开发一种新型的核酸递送技术。
发明内容
本申请的目的是提供一种改进的氨基酸共聚物及其应用。
为了实现上述目的,本申请采用了以下技术方案:
本申请的第一方面公开了一种氨基酸共聚物,该氨基酸共聚物为树形分子结构,且具有式(I)所示通式;
式(I):(PEG)
其中,PEG为聚乙二醇,PEG的分子量为2-10kDa,n表示PEG分子的聚合度;Lys为赖氨酸,构成氨基酸共聚物的末端树枝状结构,作为基团连接的键或连接子,x是0-100的整数,x等于树形赖氨酸连接子的代数;Arg为精氨酸,构成树形分子结构的末端,作为树形分子的树突,a是0-100的整数,a表示末端树突精氨酸的个数;并且,x和a不同时为0。
需要说明的是,本申请的氨基酸共聚物采用PEG作为聚合物基元,用于核酸纳米药物时,一方面,PEG可控制纳米颗粒的粒径并充当空间屏障起稳定作用;另一方面,PEG可以防止储存过程中纳米颗粒发生聚集;此外,PEG还能延长纳米颗粒在体内的循环时间。并且,本申请的氨基酸共聚物中,赖氨酸和精氨酸,都是人体必需的氨基酸,降解后的氨基酸产物基本无毒,并可为人体所用,生物相容性高;在实际的生物医学应用中,与LNP和其他多聚物载体相比,本申请的氨基酸共聚物具有更高的生物安全性。本申请的氨基酸共聚物用于核酸纳米药物时,在水溶液中,赖氨酸的氨基或者精氨酸的胍基与核酸分子中的磷酸基通过离子键相互作用而结合,加上分子间的疏水键等其他相互作用,自组装形成具有一定尺寸大小的纳米颗粒。并且,自组装的核酸纳米药物颗粒通过内吞作用进入细胞,在内涵体的酸性条件下,促使碱性氨基酸发生质子化,进而导致内涵体的破坏,释放核酸分子。采用本申请的氨基酸共聚物进行核酸的封装递送,可避免LNP复杂体系下多种脂质辅料的使用,制剂工艺更加简单,解决了复杂体系在体内的多途径代谢的问题。此外,与代数相同的传统树枝状大分子PAMAM和PPI(聚丙烯亚胺)相比,本申请的氨基酸共聚物采用精氨酸作为树突,其末端胍基在分子平面中拥有三个氨基,与核酸分子的络合能力更强,核酸负载率更高。
本申请的一种实现方式中,氨基酸共聚物为式(I)所示通式的聚合物的药学上可接受的盐、互变异构体或立体异构体。
可以理解,本申请的氨基酸共聚物除了式(I)所示通式的聚合物以外,其药学上可接受的盐、互变异构体或立体异构体同样具有相同或类似的功能,能够用于核酸药物递送,并具有稳定性好、生物相容性高、安全性高、与核酸分子络合能力强、核酸负载率高等优点。
本申请的一种实现方式中,本申请的氨基酸共聚物为PEG
本申请的一种实现方式中,PEG
需要说明的是,式(II)、式(III)、式(Ⅳ)和式(Ⅳ)所示分子结构的氨基酸共聚物只是本申请的一种实现方式中采用的氨基酸共聚物;可以理解,在此基础上,PEG分子的聚合度、赖氨酸的代数以及末端树突精氨酸的个数都可以根据需求进行调整。
本申请的第二方面公开了本申请的氨基酸共聚物在核酸药物递送中的应用。
本申请的一种实现方式中,核酸药物包括但不仅限于DNA、反义核酸、mRNA、环状RNA、siRNA、microRNA、LncRNA和saRNA中的至少一种。
需要说明的是,本申请的关键在于,研究发现了一种新的核酸药物递送材料,即本申请的氨基酸共聚物;采用本申请的氨基酸共聚物进行核酸递送,稳定性好、生物相容性高、安全性高、与核酸分子络合能力强、核酸负载率高,能够更好的满足临床使用需求。
本申请的第三方面公开了一种核酸纳米药物,该核酸纳米药物包括本申请的氨基酸共聚物和疾病治疗共轭能的核酸药物。
本申请核酸纳米药物中的核酸药物包括但不仅限于DNA、反义核酸、mRNA、环状RNA、siRNA、microRNA、LncRNA和saRNA中的至少一种。
本申请的一种实现方式中,核酸纳米药物还包括治疗相关的佐剂和/或药学上可接受的辅料。
本申请的一种实现方式中,核酸纳米药物还包括治疗效果监测的成像剂。
需要说明的是,本申请的核酸纳米药物,由于采用本申请的氨基酸共聚物,具有稳定性好、生物相容性高、安全性高、与核酸分子络合能力强、核酸负载率高等优点,具有更好的临床应用前景。
还需要说明的是,本申请的核酸纳米药物,可以采用本申请的氨基酸共聚物单一组分进行核酸的封装递送,也可以根据需求与其他组分一起,对核酸进行封装,在此不作具体限定。
本申请的第四方面公开了本申请的核酸纳米药物的制备方法,包括本申请的氨基酸共聚物在水溶液中自组装,通过其末端氨基的电离形成带有正点基团,与负电性的核酸药物通过静电相互作用封装核酸药物,通过氨基酸共聚物的树枝状分子间两亲性疏水相互作用自组装形成纳米颗粒,即获得本申请的核酸纳米药物。
本申请的第五方面公开了一种核酸药物递送的试剂盒,包括以下组分中的至少一种:
(a)本申请的氨基酸共聚物;
(b)本申请的核酸纳米药物。
需要说明的是,本申请的核酸药物递送的试剂盒,可以是已经包埋好某种特定核酸药物、具有治疗功能的试剂盒;也可以是本申请的氨基酸共聚物,使用者根据需求自行设计和包埋相应的核酸药物。可以理解,本申请试剂盒的关键在于含有本申请的氨基酸共聚物或核酸纳米药物,至于核酸递送所需的其他常规试剂,例如一些脂质或聚合物材料等,可以参考现有技术或市售购买获得。当然,为了使用方便,也可以将部分试剂组合到本申请的试剂盒中,在此不作具体限定。
由于采用以上技术方案,本申请的有益效果在于:
本申请的氨基酸共聚物,稳定性好、生物相容性高、安全性高、与核酸分子络合能力强、核酸负载率高;用于核酸纳米药物时,能够在水中自组装并封装核酸药物,制备工艺简单方便,能够很好的满足临床使用需求。
附图说明
图1是本申请实施例中PEG原料(化合物1)的MALDI-TOF图谱;
图2是本申请实施例中化合物2的MALDI-TOF图谱;
图3是本申请实施例中化合物3的MALDI-TOF图谱;
图4是本申请实施例中化合物4的MALDI-TOF图谱;
图5是本申请实施例中化合物5的MALDI-TOF图谱;
图6是本申请实施例中化合物6的MALDI-TOF图谱;
图7是本申请实施例中化合物1的GPC图谱;
图8是本申请实施例中化合物5的GPC图谱;
图9是本申请实施例中化合物6的GPC图谱;
图10是本申请实施例中包封率测试的琼脂糖凝胶电泳图;
图11是本申请实施例中氨基酸共聚物负载荧光标记的mRNA的细胞内吞荧光测试结果;
图12是本申请实施例中氨基酸共聚物的细胞毒性测试结果;
图13是本申请实施例中氨基酸共聚物负载的mRNA制剂的细胞毒性测试结果。
具体实施方式
下面通过具体实施例对本申请作进一步详细说明。以下实施例仅对本申请进行进一步说明,不应理解为对本申请的限制。在以下的实施方式中,很多细节描述是为了使得本申请能被更好的理解。然而,本领域技术人员可以毫不费力的认识到,其中部分特征在不同情况下可以省略,或者可以由其他试剂盒、材料、方法所替代。在某些情况下,本申请相关的一些操作并没有在说明书中显示或者描述,这是为了避免本申请的核心部分被过多的描述所淹没,而对于本领域技术人员而言,详细描述这些相关操作并不是必要的,根据说明书中的描述以及本领域的一般技术知识即可完整了解相关操作。以下实施例中,所用试剂或仪器未注明生产厂商者,均为可以通过市场购买获得的常规产品。
实施例1
本例分别采用不同的方案合成树形分子结构的氨基酸共聚物,具体如下:
方案(1):各步骤反应过程及具体参数条件如下:
/>
步骤1):称取原料赖氨酸Lys 1.2mM于茄型瓶中,加入10mL DMF溶剂充分溶解,依次加入1.2mM Oxyma消旋抑制剂和1.2mM DIC缩合剂,置于冰上进行10min的低温活化;再加入0.2mM氨基修饰的PEG原料BocNH-PEG5k-NH
步骤2):将全部所得化合物2用4-甲基哌啶:DMF为1:4的混合溶液20mL溶解于100mL茄型瓶中进行脱保护处理,室温反应16h;加入冷甲基叔丁基醚25mL反应20min,静置20min;弃上清加入冷甲基叔丁基醚25mL重复洗涤3次,旋干后加入原料赖氨酸2.4mM及缩合剂继续反应16h,其中缩合剂由2.4mM Oxyma消旋抑制剂和2.4mM DIC缩合剂组成,通过TCL点板验证化合物2完全反应后进行后处理除去过量未反应的赖氨酸得到化合物3,即PEG
步骤3):将上述所得化合物3全部用4甲基哌啶:DMF为1:4的溶液20mL溶解于100mL茄型瓶中进行脱保护处理,室温反应16h;加入冷甲基叔丁基醚25mL反应20min,静置20min;弃上清加入冷甲基叔丁基醚25mL重复洗涤3次,旋干后加入原料赖氨酸4.8mM及缩合剂(即4.8mM Oxyma消旋抑制剂和4.8mM DIC缩合剂)继续反应16h,通过TCL点板验证化合物3完全反应后进行后处理除去过量未反应的赖氨酸得到化合物4,即PEG
步骤4):将上述所得化合物4用4-甲基哌啶:DMF为1:4的溶液20mL溶解于100mL茄型瓶中进行脱保护处理,室温反应16h;加入冷甲基叔丁基醚25mL反应20min,静置20min;弃上清加入冷甲基叔丁基醚25mL重复洗涤3次,旋干后加入原料精氨酸9.6mM及缩合剂(即9.6mM Oxyma消旋抑制剂和9.6mM DIC缩合剂)继续反应16h,通过TCL点板验证化合物4完全反应后进行后处理,除去过量未反应的赖氨酸得到化合物5,即PEG
步骤5):将化合物5进行一次脱保护处理,首先将其溶解于TFA:TIS:H
方案(2):在上述方案(1)的基础上,进行方案(2)的各步反应过程如下:
/>
步骤6)在上述步骤4)得到化合物4(PEG
步骤7)将化合物6用4-甲基哌啶:DMF为1:4的溶液25mL溶解于100mL茄型瓶中进行赖氨酸上的Fmoc脱保护处理,具体包括,室温反应16h;加入25mL冷甲基叔丁基醚反应20min,静置20min;弃上清加入25mL冷甲基叔丁基醚重复洗涤3次,旋干后获得终产物1g左右冻于-20℃储存。
理化性质表征:本例对末端树枝状氨基酸共聚物分子的MALDI-TOF质谱表征,具体操作包括,配置共聚物分子浓度为5mg/mL的DMF溶液,将样品置于岛津瓶中,送样进行MALDI-TOF检测得到表征结果。同时,通过凝胶渗透色谱GPC对化合物1、化合物5和化合物6进行了分子量表征,三者的结果依序如图7、图8和图9所示,GPC是表征聚合物平均分子量的一种常用方法,结果显示4代氨基酸分子的成功合成,其平均分子量略大于MALDI-TOF的结果,但都一致证明了树枝状氨基酸共聚物的成功合成。
本例合成的氨基酸共聚物中,PEG
式(II):
式(III):
式(Ⅳ):
式(Ⅴ):
式(2):
式(3):
式(4):
式(5):
实施例2
采用实施例1制备的末端树枝状氨基酸共聚物进行核酸递送,本例具体作为mRNA药物递送试验。
一、mRNA/PEG
取2.5mg的PEG
二、mRNA/PEG
步骤1)采用制备的mRNA/PEG
三、mRNA/PEG
步骤1)细胞培养:人胚肾上皮细胞(293T)以含10%胎牛血清(FBS)的培基培养,并在37℃下含5%二氧化碳的条件下正常培养。
步骤2)细胞铺板:转染前将细胞接种在细胞培养板中,并在100μL含有10%FBS的新鲜培养基中培养24h。
步骤3)细胞内吞:弃除培养板中的培养基,用无血清的基础培养基润洗两遍细胞,将制备的mRNA/PEG
步骤4)固定细胞:取出培养板,用无血清的基础培养基润洗细胞2次,分别加入100μL的4%多聚甲醛室温固定10min,吸弃固定液后加入100μL细胞缓冲液PBS润洗2次,加入100μL PBS。
步骤5)荧光成像检测:将上述步骤4)中固定的细胞至于倒置荧光显微镜下进行荧光成像并记录图像,得到如图11所示的结果,图11结果显示,树枝状分子具有与商业化转染试剂LipoMAX、PEI及商业化的4代赖氨酸分子类似的递送荧光标记的mRNA进入细胞的能力,说明该树枝状分子具有转染及递送mRNA的潜力。
四、末端树枝状氨基酸共聚物的细胞毒性试验
步骤1)制备树形分子的储备液:在无菌条件下操作,将树形氨基酸共聚物10mg溶于无菌水1mL中,超声,静置,制备成储备液;
步骤2)将末端树枝状氨基酸共聚物分子溶液制备成不同浓度,本例具体制备了8梯度浓度,各浓度具体是0,0.32μg/mL,1.6μg/mL,8μg/mL,40μg/mL,200μg/mL,1000μg/mL,5000μg/mL,各浓度取200μL加入至接种有293T细胞的培养板中共孵育24h。
步骤3)在无菌条件下进行细胞活性监测,用基础培养基稀释CCK-8试剂,按试剂盒说明以培养基:CCK-8试剂为9:1的比例混匀,每个孔各加入100μL混合液至培养板中后放回培养箱中进行孵育2h,在酶标仪下监测细胞活性。细胞活性如图12所示。
图12的结果显示,PEG
五、mRNA/末端树枝状氨基酸分子纳米药物的细胞毒性试验
步骤1)细胞铺板:将293T细胞以6000个/孔的密度接种于96孔板中,37℃下培养24h。
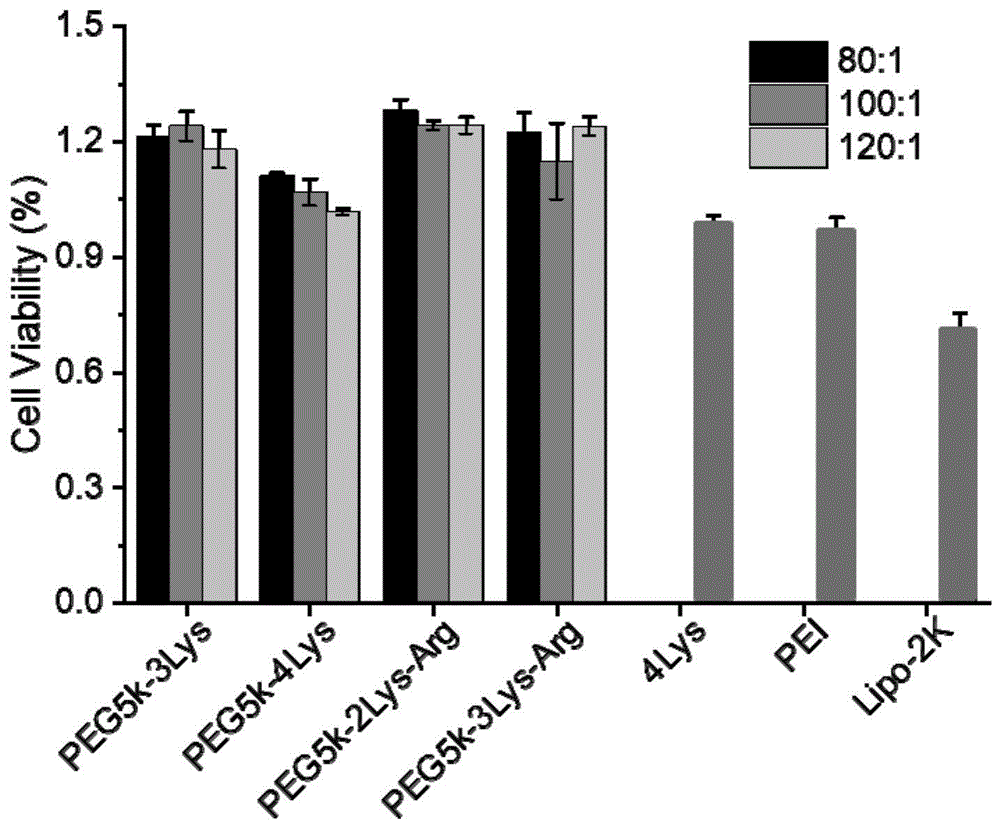
步骤2)分别加入10ng mRNA含量的三代或四代的赖氨酸末端及精氨酸末端的分子,同时,商业化的四代赖氨酸及PEI和转染试剂Lipo2K作为对照组,每组样品五个复孔,在37℃下孵育24h后,利用如上所述的CCK-8检测细胞活力。如图13所示,不同代数及末端的树枝状氨基酸共聚物分子没有诱导细胞毒性的产生,并且细胞活力比商业化转染试剂Lipo2K更高,说明树枝状分子作在为核酸递送载体方面具有巨大的优势。
以上内容是结合具体的实施方式对本申请所作的进一步详细说明,不能认定本申请的具体实施只局限于这些说明。对于本申请所属技术领域的普通技术人员来说,在不脱离本申请构思的前提下,还可以做出若干简单推演或替换。