一种铋金属掺杂的氮化碳催化剂及其制备方法与应用
文献发布时间:2023-06-19 19:30:30
技术领域
本发明属于能源材料与电催化技术领域,更具体地,涉及一种铋金属掺杂的氮化碳催化剂及其制备方法与应用。
背景技术
石油、煤炭和天然气等化石燃料已成为人类和工业活动的主要能源,据估计,人类CO
在CO
因此,迫切需要提供一种高选择性、高活性、高稳定性的催化剂用于电催化CO
发明内容
本发明要解决的技术问题是克服现有电化学还原CO
本发明的目的是提供一种铋金属掺杂的氮化碳催化剂。
本发明的另一目的是提供一种铋金属掺杂的氮化碳催化剂的应用。
本发明上述目的通过以下技术方案实现:
氮化碳材料由于其制备路线简单、易大规模生产并具有良好的化学稳定性、热稳定性和机械稳定性,而且易于修饰,被普遍认为是一种颇具潜力的新材料。但是,氮化碳本身作为一类半导体材料,在电催化反应中其性能往往受到限制。利用金属对其进行修饰是提高其催化性能的有效办法,但是金属氮掺杂碳材料在制备和反应过程中纳米颗粒容易团聚、烧结而导致催化性能下降。
为了解决上述问题,本发明通过以下技术方案实现:
一种铋金属掺杂的氮化碳催化剂的制备方法,包括以下步骤:
S1.将尿素和铋源分散于水中,充分混合后,冷冻干燥得到前驱体;
S2.将步骤S1中所得前驱体在惰性气体的保护下500~1000℃进行煅烧,冷却后获得固体产物,酸洗,后处理,即得铋金属掺杂的氮化碳催化剂粉末。
优选地,步骤S1中,所述铋源为五水合硝酸铋、氯化铋的一种或多种。
优选地,步骤S1中,所述尿素和铋源的质量比为1.0:(0.025~0.5)。
更优选地,步骤S1中,所述尿素和铋源的质量比为1.0:(0.05~0.2)。
最优选地,步骤S1中,所述尿素和铋源的质量比为1.0:0.15。
优选地,步骤S1中所述冷冻干燥的条件为:温度为-10~-60℃,干燥时间为12~48h。
优选地,步骤S2中,所述煅烧的温度为550~1000℃。
进一步地,步骤S2中,所述煅烧的升温速率为2~10℃/min。
进一步地,步骤S2中,所述煅烧的时间为1~4h。
优选地,步骤S2中,所述惰性气体为氮气、氩气的一种或多种。
优选地,步骤S2中,所述惰性气体的流速为40.0~100.0mL/min。
具体地,步骤S2中,所述酸洗前先研磨,酸洗后干燥和再研磨。
进一步地,所述酸洗的条件为:将步骤S2中所得固体产物先用酸性溶液洗涤,再用水清洗至溶液呈中性。
优选地,所述酸性溶液为硝酸溶液。
更优选地,所述硝酸溶液的摩尔浓度为0.5M。
优选地,所述干燥的方式为真空干燥。
优选地,所述真空干燥的温度为50~100℃,干燥时间为12~48h。
更优选地,所述真空干燥的温度为60℃,干燥时间为24h。
由所述制备方法得到的铋金属掺杂的氮化碳催化剂。
进一步地,所述铋金属掺杂的氮化碳催化剂,具有多孔纳米片结构。
进一步地,所述铋金属掺杂的氮化碳催化剂由铋金属和氮化碳两部分组成,前者负载在后者上。
另外的,本发明还提供一种铋金属掺杂的氮化碳催化剂在电催化CO
具体地,所述应用的方法包括以下步骤:
采用气体扩散电极体系,以0.5M~1.0M KOH为电解液,先通入Ar排除空气后通入CO
优选地,所述电解液的摩尔浓度为1M。
本发明具有以下有益效果:
1.与常规金属掺杂催化剂相比,本发明所制备的铋金属掺杂的氮化碳催化剂为多孔片状结构,从而具有更多的催化剂活性位点和更高的稳定性。
2.基于冷冻干燥的方法提高了铋金属纳米颗粒在氮化碳载体上的分散度,提供了更大的电化学反应活性表面积与更丰富的反应活性位点,从而赋予了铋金属掺杂的氮化碳催化剂更高的催化活性。
3.本发明所制备的铋金属掺杂的氮化碳催化剂可以实现对其微观形貌、化学组成等的精细调控,操作步骤简单,过程绿色环保,易于规模化生产,不限于应用于电催化还原CO
附图说明
图1为实施例1~4中制备的铋金属掺杂的氮化碳催化剂的透射电镜图;a~d分别对应实施例1~4的透射电镜图;
图2为实施例4中制备的铋金属掺杂的氮化碳催化剂的扫描电镜图(左半图)和球差电镜图(右半图);
图3为实施例4中制备的铋金属掺杂的氮化碳催化剂的元素分析图;
图4为实施例1~5中制备的铋金属掺杂的氮化碳催化剂的XRD图谱;
图5为实施例1~5中制备的铋金属掺杂的氮化碳催化剂红外吸收光谱图;
图6为实施例4中制备的铋金属掺杂的氮化碳催化剂的X射线光电子能谱(XPS)图;
图7为实施例4中制备的铋金属掺杂的氮化碳催化剂电解时的电流密度随电解时间的变化曲线图;
图8为实施例1~4中制备的铋金属掺杂的氮化碳催化剂的线性扫描循环伏安曲线(LSV)图;
图9为实施例1~4中制备的铋金属掺杂的氮化碳催化剂的电化学交流阻抗谱(EIS)图;
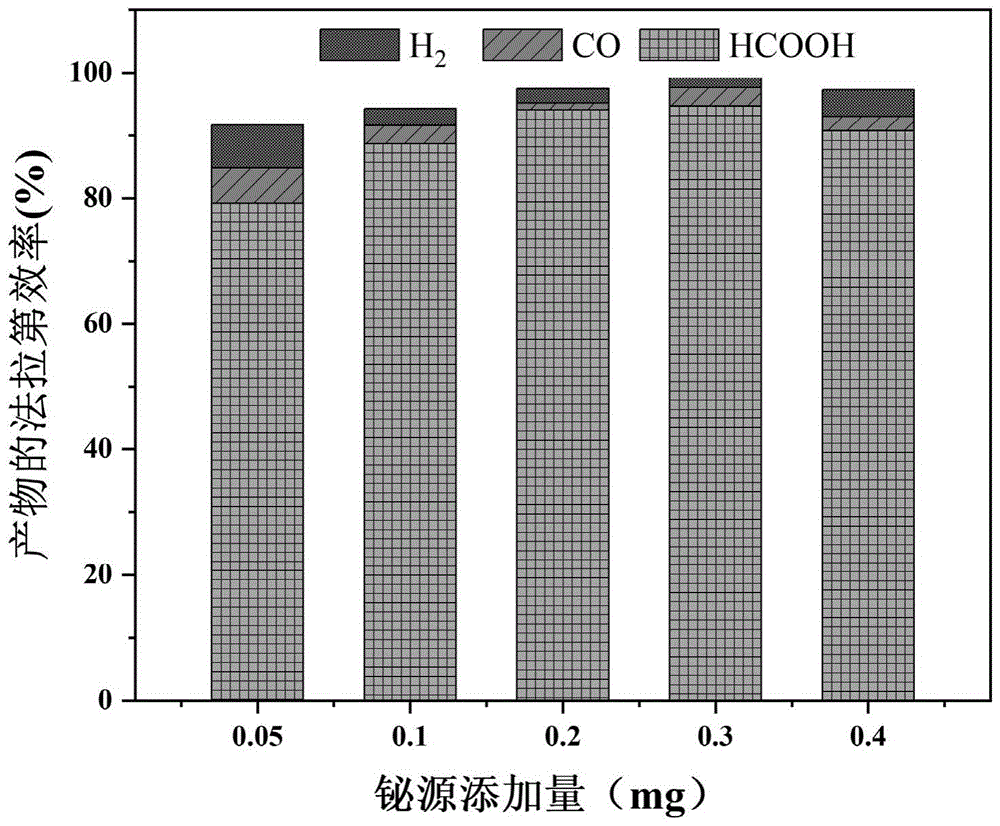
图10为实施例2~5中制备的铋金属掺杂的氮化碳催化剂电催化CO
图11为实施例1~5中制备的铋金属掺杂的氮化碳催化剂电催化CO
具体实施方式
以下结合说明书附图和具体实施例来进一步说明本发明,但实施例并不对本发明做任何形式的限定。除非特别说明,本发明采用的试剂、方法和设备为本技术领域常规试剂、方法和设备。
除非特别说明,以下实施例所用试剂和材料均为市购。
实施例1一种铋金属掺杂的氮化碳催化剂的制备
一种铋金属掺杂的氮化碳催化剂的制备,包括以下步骤:
S1.将2g尿素和0.05g五水合硝酸铋分散于10mL去离子水中,充分搅拌后,将混合溶液冷冻干燥,获得前驱体;
S2.将前驱体放入刚玉瓷舟内,在惰性气体氮气保护下,从室温升温至热解温度,升温速率为3℃/min,在550℃高温煅烧2h,自然冷却后得到固体产物;
S3.将固体产物用研钵研磨成均匀细小粉末,加入40mL 0.5M的硝酸常温下搅拌10h,离心,用超纯水洗4~5次,至溶液呈中性,随后在真空干燥箱中60℃干燥24h,再次研磨得到铋金属掺杂的氮化碳催化剂Bi-0.05-gCN。
实施例2~5一种铋金属掺杂的氮化碳催化剂的制备
实施例2与实施例1的区别在于:称量的五水合硝酸铋为0.1g,即尿素和五水合硝酸铋的质量比为2:0.1,得到铋金属掺杂的氮化碳催化剂Bi-0.1-gCN。
实施例3与实施例1的区别在于:称量的五水合硝酸铋为0.2g,即尿素和五水合硝酸铋的质量比为2:0.2,得到铋金属掺杂的氮化碳催化剂Bi-0.2-gCN。
实施例4与实施例1的区别在于:称量的五水合硝酸铋为0.3g,即尿素和五水合硝酸铋的质量比为2:0.3,得到铋金属掺杂的氮化碳催化剂Bi-0.3-gCN。
实施例5与实施例1的区别在于:称量的五水合硝酸铋为0.4g,即尿素和五水合硝酸铋的质量比为2:0.4,得到铋金属掺杂的氮化碳催化剂Bi-0.4-gCN。
其它参数及操作均参考实施例1。
对比例1~4一种铋金属掺杂的氮化碳催化剂的制备
对比例1与实施例4的区别在于:不添加硝酸进行酸洗,直接研磨得到铋金属掺杂的氮化碳催化剂。
对比例2与实施例4的区别在于:煅烧温度为500℃。
对比例3与实施例4的区别在于:称量的五水合硝酸铋为2g,即尿素和五水合硝酸铋的质量比为1:1。
对比例4与实施例4的区别在于:不采用冷冻干燥机而使用真空干燥剂进行干燥。
其它参数及操作均参考实施例1。
实验例
1、铋金属掺杂的氮化碳催化剂的物象表征与成分分析
测定实施例1~4制备的铋金属掺杂的氮化碳催化剂的透射电镜图,结果如图1(a)、(b)、(c)、(d)所示,可以观察到铋金属掺杂的氮化碳催化剂具有多孔片状结构;
测定实施例4中制备的铋金属掺杂的氮化碳催化剂的扫描电镜图,结果如图2的左半图所示,可以看到铋金属掺杂的氮化碳催化剂粉末表面的三维多层堆积形貌;
测定实施例4中制备的铋金属掺杂的氮化碳催化剂的球差电镜图,结果如图2的右半图所示,可以看Bi原子的高度均匀分散,其中单独的亮点为分散的Bi原子;
测定实施例4中制备的铋金属掺杂的氮化碳催化剂的元素分析图,结果如图3所示,可以观察到C,N,O和Bi元素都均匀的分布在催化剂中;
测定实施例1~5中制备的铋金属掺杂的氮化碳催化剂的XRD图谱,结果如图4所示,可以看到在2θ为13.1°和27.3°分别对应石墨相氮化碳的(100)晶面和(002)晶面;
测定实施例1~5中制备的铋金属掺杂的氮化碳催化剂的红外吸收光谱图,结果如图5所示,可以看到我们可以观察到3000cm
测定实施例4中制备的铋金属掺杂的氮化碳催化剂的X射线光电子能谱图,结果如图6所示,可以看到Bi 4f可以归结为两个明显的峰,分别为159.5eV和164.7eV,对应Bi
2、铋金属掺杂的氮化碳催化剂的性能测试
采用气体扩散电极体系,以Ag/AgCl参比电极,铂丝为对电极,将本发明所得铋金属掺杂的氮化碳催化剂制成工作电极,其中本发明制备的催化剂在碳布电极上的载量为0.025mg/cm
表1实施例1~5甲酸的法拉第效率
表2对比例1~4甲酸的法拉第效率
由表1可知,上述实施例1~5制备的铋金属掺杂的氮化碳催化剂在-1.0的外加电压下的电解产物甲酸的法拉第效率表现较差,明显低于相同条件下更低的外加电压-0.6V的产率,可以看出更低的外加电压下二氧化碳还原为甲酸的选择性更高。
测定实施例4中制备的铋金属掺杂的氮化碳催化剂电解时的电流密度随电解时间的变化曲线图,结果如图7所示,可以看到催化剂在不同电位电解一小时表现出相对稳定的电流密度;
测定实施例1~4中制备的铋金属掺杂的氮化碳催化剂的线性扫描循环伏安曲线图,结果如图8所示,可以看到在CO
测定实施例1~4中制备的铋金属掺杂的氮化碳催化剂的电化学交流阻抗(EIS)谱图,结果如图9所示,对于Bi-0.05-gCN、Bi-0.1-gCN、Bi-0.2-gCN和Bi-0.3-gCN,拟合阻抗分别为59、77、90和25Ω。EIS图显示Bi-0.3-gCN的电荷转移电阻是最小的,在CO
测定实施例2~5中制备的铋金属掺杂的氮化碳催化剂的电解液体产物核磁共振氢谱图,结果如图10所示,可以看到用氘代水作为溶剂,内标物为二甲基亚砜DMSO出峰位置在2.6ppm附近,主要产物甲酸出峰位置在8.4ppm附近;
测定实施例4中制备的铋金属掺杂的氮化碳催化剂电催化CO
由表2得出如下结论:
如对比例1的甲酸的法拉第效率的结果所示,当不采用硝酸酸洗时,所得催化剂的催化性能明显不如实施例4制备得到的催化剂的性能,酸洗的目的是清除杂质,不经过酸洗,其无法获得理想的形貌和催化的活性位点;
如对比例2的甲酸的法拉第效率的结果所示,当煅烧温度低于550℃时,催化剂的活性位不足,与实施例4所得催化剂相比,其催化性能明显降低,这是因为该温度下无法成功制备出拥有高分散铋金属掺杂的石墨相氮化碳gC3N4结构的催化剂;
如对比例3的甲酸的法拉第效率的结果所示,当金属盐添加量过量时,催化性能与实施例4所得催化剂相比明显下降,这是因为过量的铋金属纳米粒子容易发生团聚所导致;
如对比例4的甲酸的法拉第效率的结果所示,当不采用冷冻干燥方法干燥前驱体时,与实施例4所得催化剂相比,其催化性能大幅度降低,这是因为铋源不采取冷冻干燥方式干燥时容易发生水解生成碱式盐沉淀而无法形成高分散的金属掺杂催化剂所导致。
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
- 一种铋掺杂氧化锡/磷酸铋复合光催化剂的制备方法
- 一种氮掺杂碳包覆双纳米金属催化剂及其制备方法和用途
- 钴镍双金属氢氧化物纳米片/氮化碳包覆氮掺杂中空石墨烯球复合材料及其制备方法和应用
- 一种多孔氮掺杂碳负载氮化铁催化剂及其制备方法以及应用
- 一种铋/铈双金属掺杂氮化碳复合光催化剂及其制备方法和应用
- 一种无金属掺杂氮功能化碳催化剂及其制备方法和应用