一种聚合物夹心复合磁性微球及其制备方法
文献发布时间:2024-04-18 19:58:21
技术领域
本发明涉及高分子材料和磁性材料技术领域,具体涉及一种聚合物夹心复合磁性微球及其制备方法。
背景技术
磁性高分子微球是指磁性材料与高分子聚合物材料复合而形成的、表面带有功能性官能团的球形复合材料,粒径范围通常在100-100000nm之间。其中,磁性材料通常为Fe
磁性高分子微球通常有四种类型:核壳型,反核壳型,夹心型和弥散型。其中核壳型磁性微球通常是先合成磁性材料,再通过表面改性和聚合的方式在其表面形成高分子聚合物层;反核壳型和夹心型都是先合成聚合物微球,再使用原位浸渍的方法使磁性材料分布于聚合物微球表面,只是夹心型微球会在反核壳结构的基础上再包覆一层聚合物或无机材料层;弥散型磁性微球的合成则通常是先将磁性材料表面改性后稳定分散于聚合物单体中,再通过聚合形成。
磁性高分子微球通常在外界磁场作用下发生聚集,撤去磁场后能稳定分散于介质中,而其表面丰富的官能团又能与特定的有机或无机物质结合,因此磁性高分子微球广泛应用于物质的吸附与分离中。尤其是在生物医学领域,磁性高分子微球经表面改性后能与蛋白质、多肽、核酸等特异性结合,在免疫分析、固定化酶、核酸提取等需要借助磁分离的操作中起到重要作用。
磁性高分子微球的研究与应用在国内外均较为成熟,且有较多产品上市。然而,目前应用于生物磁分离的磁性高分子微球仍存在一些不足之处:市面上大部分磁性微球为核壳型或弥散型结构,其中核壳型微球磁含量较高,磁响应时间短;弥散型微球根据聚合方式不同可实现较为广泛的粒径调控范围,但磁性材料经亲油物质表面改性后,在聚合物单体中的分散能力仍然有限,因此该种微球磁含量通常不高。相比较而言,反核壳型或夹心型磁性微球中的高分子内核起到模板的作用,能保证复合微球的单分散性;内核经氨基或磺化处理后能对金属离子捕获与吸附,确保复合微球的磁含量较高;夹心型磁性微球的外层结构又能防止磁性组分溢出。
专利CN108467461A中,在一步种子溶胀聚合法制备多孔结构的PGMA微球和原位共沉淀合成磁性微球的基础上,使用含羧基的可聚合单体(丙烯酸或甲基丙烯酸)和交联剂(N,N’-亚甲基双丙烯酰胺),以蒸馏沉淀聚合的方式在微球表面引入羧基而形成复合微球,该方法所制备的微球饱和磁化强度高,单分散性好,但蒸馏沉淀聚合需要用到毒性较大的溶剂乙腈,且反应的高温可能导致微球中的Fe3O4组分氧化。
专利CN111205581A中,聚合物微球模板为PGMA,采用分散聚合法合成,磁性微球仍然通过原位共沉淀合成,羧基改性则是先在磁性微球表面引入酯基,然后加入多元醇产生大量羟基,再使用酸酐引入羧基,该方法合成出的微球具有表面亲水性高、特异性吸附强的特点,但流程复杂且反应过程仍需在高温下进行。
专利CN114093586A中,多孔聚合物微球模板为种子溶胀法制备的聚(苯乙烯-二乙烯基苯)微球,经磺化改性后浸渍铁盐,再使用蒸馏沉淀聚合法引入酯基,然后经氨基和羧基改性制得产品。该微球的制备流程仍然复杂,且会使用到有毒的乙腈和强腐蚀性的硫酸。
综上所述,有必要从反核壳或夹心型磁性微球的合成原理出发,通过更加简单、高效、环保的方法,研制磁含量较高、单分散性好、粒径可调范围大、磁响应时间短、介质中稳定性强的生物磁分离用的复合磁性微球。
发明内容
针对现有技术的不足,本发明提供一个新的磁性高分子聚合物PDDBP,再通过原位浸渍法合成磁性高分子夹心复合微球以避免Fe
为实现本发明的第一个目的,本发明公开了一种聚合物夹心复合磁性微球,内层为磁性高分子聚合物,夹心层为Fe
其中,M为稀土元素。
进一步的技术方案是:M为Sm
进一步的技术方案是:聚合物夹心复合磁性微球的粒径通过调控在300-500nm之间;其中,磁性高分子聚合物厚度250~350nm,Fe
进一步的技术方案是:聚合物夹心复合磁性微球中Fe
进一步的技术方案是:PDDBP平均分子量为15000~30000。
为实现本发明的第二个目的,本发明公开了聚合物夹心复合磁性微球的制备方法整个反应过程简单环保,产品可应用于免疫分析、核酸提取、蛋白质分离等生物医学分析检测中。本发明的一种聚合物夹心复合磁性微球的制备方法,包括以下制备步骤:
(1)在冰水浴中将4,4-二胺基-2,2-联苯并噻唑与乙二胺进行聚合反应,再与甲醛进行反应后加入稀土氯化物得到磁性高分子聚合物PDDBP微球;其中,4,4-二胺基-2,2-联苯并噻唑、乙二胺、甲醛与稀土氯化物的摩尔质量比为1:2:1.01:2;
(2)PDDBP微球在引发剂、交联剂、致孔剂的作用下于聚乙烯醇和十二烷基硫酸钠的混合溶液中形成分散液滴,再加入到以PDDBP微球为种子的聚乙烯醇溶液中进行溶胀聚合反应,得到溶胀微球;
(3)通过环氧基团的开环反应在溶胀微球表面修饰氨基,将其与二价和三价铁盐溶液混合,于真空条件下进行表面浸渍,之后在氮气保护下升高温度并加入表面活性剂和碱性沉淀剂,通过化学共沉淀法制备单分散、超顺磁性的PDDBP@Fe
(4)将PDDBP@Fe
(5)在氮气保护下将氨基化的PDDBP@Fe
进一步的技术方案是:步骤(2)中所述溶胀聚合引发剂为过氧化苯甲酰、偶氮二异丁腈、偶氮二异丁基脒盐酸盐中至少一种,使用浓度为0.1~0.3wt%;所述交联剂为二乙烯苯、乙二醇二甲基丙烯酸酯中至少一种,使用浓度为1~4wt%;所述致孔剂为甲苯、环己烷、正己烷、正庚烷中至少一种,使用浓度为1~2wt%;所述聚乙烯醇溶液浓度为0.5~1.0wt%;所述十二烷基硫酸钠溶液浓度为0.2~0.3wt%;所述溶胀聚合反应时间12~24h。
进一步的技术方案是:步骤(3)中所述修饰氨基所用的试剂为乙二胺、二乙烯三胺、三乙烯四胺、四乙烯五胺中至少一种,与溶胀微球的摩尔质量比为1:4;所述二价铁盐为氯化亚铁、硫酸亚铁中至少一种,三价铁盐为氯化铁、硫酸铁、硝酸铁中至少一种,二价和三价铁盐摩尔比为1:1.5~2.0,磁性微球与铁盐总质量的比例为1:1~3;所述表面浸渍温度为0~10℃;所述表面活性剂为柠檬酸钠或聚乙二醇,使用浓度为1~5wt%;所述碱性沉淀剂为浓氨水或NaOH溶液并调整反应体系pH为10~12;所述反应温度为70~80℃,加入沉淀剂后继续熟化1~1.5h。
进一步的技术方案是:步骤(4)中所述氨基化硅烷偶联剂为3-氨丙基三甲氧基硅烷、3-氨丙基三乙氧基硅烷中至少一种;所述
进一步的技术方案是:步骤(5)中所述酸酐为丁二酸酐、顺丁烯二酸酐、戊二酸酐、己二酸酐中至少一种;所述反应前酸酐的DMF溶液需磁力搅拌4~6h;所述磁性微球与酸酐的质量比为1:0.5~1.5,磁性微球的DMF溶液滴加时间为5~10min,酰胺化反应时间为10~15h。
本发明的有益效果:
1、聚双噻唑本身是抗磁体,但是双噻唑结构是一个良好的配体,能和众多的过渡金属离子配位生成螯合物。本发明在聚双噻唑分子链中引入过渡金属离子,在外加磁场的作用下,使螯合物中金属离子的未成对电子通过超交换作用和极化作用实现磁有序以具备良好的铁磁性能。
2、本发明通过调节反应参数:如单体体积、引发剂用量、分散剂用量、交联剂用量、反应介质类型等,控制微球的平均粒径为300-500nm,因此所制备的聚合物夹心复合磁性微球具有单分散性好、磁含量高、磁响应时间短、表面羧基密度高、在介质中分散稳定性强等特点。
3、本发明在磁性高分子微球表面包覆无机壳层SiO
附图说明
图1为聚1,3-二(4,4-二亚氨基-2,2-联苯并噻唑)亚苯基(PDDBP)的合成路线图。
图2为本发明实施例制备的聚合物夹心复合磁性微球的扫描电镜图;其中,(a)为实施例1的聚合物夹心复合磁性微球的扫描电镜图,(b)为实施例2的聚合物夹心复合磁性微球的扫描电镜图。
图3为本发明实施例制备的聚合物夹心复合磁性微球的透射电镜图;其中,(a)、(b)为本发明实施例3制备的聚合物夹心复合磁性微球的透射电镜图,(c)、(d)为本发明实施例4制备的聚合物夹心复合磁性微球的透射电镜图。
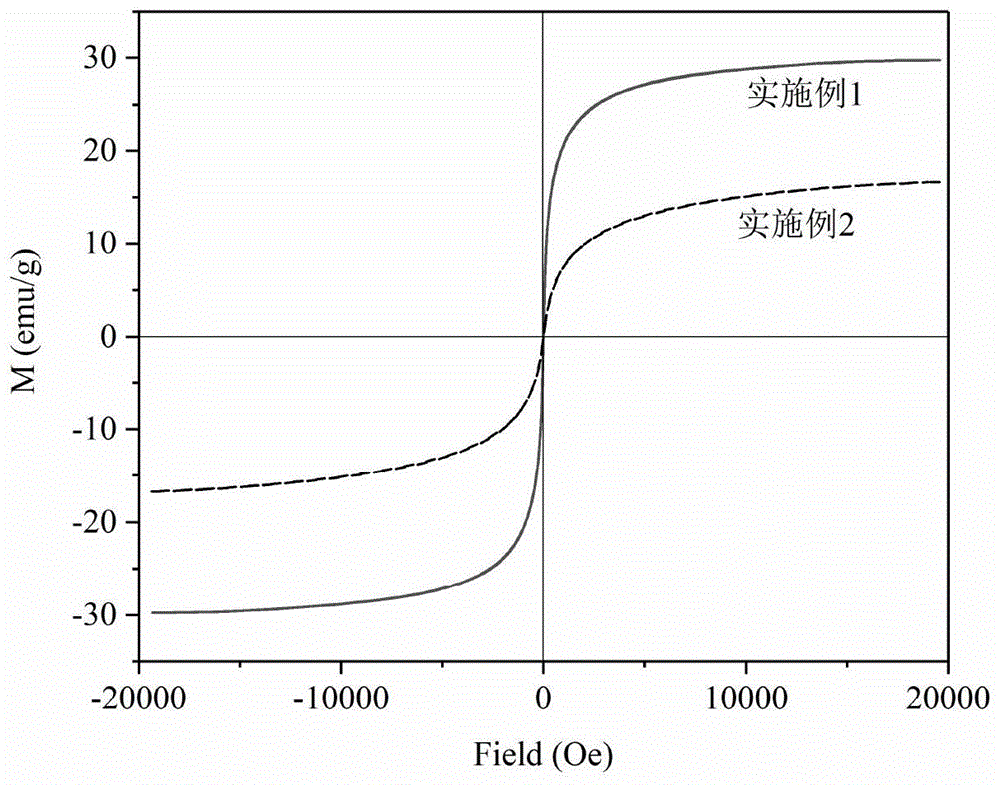
图4为本发明实施例1-2制备的聚合物夹心复合磁性微球的磁滞回线。
图5为本发明实施例1制备的聚合物夹心复合磁性微球的分散稳定性和磁响应性能测试图。
具体实施方式
为使本发明实施例的目的、技术方案和优点更加清楚,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
本申请实施例提供的聚合物夹心复合磁性微球具有单分散性好、磁含量高、磁响应时间短、表面羧基密度高、在介质中分散稳定性强等特点;其制备过程简单环保,制备的功能化磁性复合微球可应用于免疫分析、核酸提取、蛋白质分离等生物医学分析检测中。
为了更好的理解上述技术方案,下面将结合说明书附图以及具体的实施方式对上述技术方案进行详细的说明。
实施例1
(1)将1.49g的4,4-二胺基-2,2-联苯并噻唑加入到20ml DMF中并搅拌均匀,在氮气保护下加入0.6g乙二胺,并用冰水浴冷却。通过恒压液滴漏斗慢慢加入0.19ml甲醛与10mlDMF的混合液,在冰水浴中反应6h后倒入100ml甲醇中沉淀,用去离子水和甲醇反复洗涤沉淀物,置于80℃真空干燥箱中烘干。将10g得到的产物加入到50ml二甲基亚砜中,在氮气保护下加入0.17g氯化钐,80℃反应24h后倒入去离子水中沉淀,用去离子水和甲醇反复洗涤沉淀物,置于80℃真空干燥箱中烘干得到PDDBP,如图1所示为PDDBP的合成路线图。
(2)以0.2g PDDBP为种子微球,分散于20ml浓度为0.5wt%的聚乙烯醇溶液中,另取0.1g十二烷基硫酸钠和0.2g聚乙烯醇溶于50ml去离子水中形成混合溶液,再加入10gPDDBP、0.08g过氧化苯甲酰、0.8g二乙烯苯、0.8g甲苯,在高压均质机作用下形成粒径小于1μm的稳定分散液滴,将该液滴分散液加入到含有种子微球的聚乙烯醇溶液中,在氮气保护下室温溶胀12h,溶胀结束后聚合反应12h即得溶胀微球。
(3)将10g溶胀微球、0.08g过氧化苯甲酰、0.12g乙二胺加入到50ml甲醇中,于90℃下反应3h后反复离心洗涤得到沉淀物,将其与4.4g氯化亚铁、6.6g氯化铁一同加入50ml去离子水中混合,降温至0℃,于真空条件下进行表面浸渍,之后在氮气保护下升温至70℃并加入0.21g柠檬酸钠,加入浓氨水调节溶液pH为10,熟化1h。
(4)将11g步骤(3)的产物分散于10ml无水乙醇和15ml去离子水的混合溶液中,加入催化剂氨水,然后逐滴加入5.5g正硅酸四乙酯与10ml乙醇的混合溶液,并逐滴加入含有4.3g硅酸钠的乙醇溶液,室温下搅拌12h后在45min内逐滴加入含有2.75g 3-氨丙基三甲氧基硅烷的乙醇溶液,形成表面带氨基的PDDBP@Fe
(5)将16.5g丁二酸酐溶解于20ml DMF中,搅拌4h后在氮气保护下逐滴加入含有10g步骤(4)产物的DMF混合液,室温下继续搅拌10h后用甲醇与去离子水反复洗涤,60℃真空烘干后得到产物。
实施例2
(1)将1.49g的4,4-二胺基-2,2-联苯并噻唑加入到20ml DMF中并搅拌均匀,在氮气保护下加入0.6g乙二胺,并用冰水浴冷却。通过恒压液滴漏斗慢慢加入0.19ml甲醛与10mlDMF的混合液,在冰水浴中反应6h后倒入100ml甲醇中沉淀,用去离子水和甲醇反复洗涤沉淀物,置于80℃真空干燥箱中烘干。将10g得到的产物加入到50ml二甲基亚砜中,在氮气保护下加入0.13g氯化钐,80℃反应24h后倒入去离子水中沉淀,用去离子水和甲醇反复洗涤沉淀物,置于80℃真空干燥箱中烘干得到PDDBP。
(2)以0.2g PDDBP为种子微球,分散于20ml浓度为0.6wt%的聚乙烯醇溶液中,另取0.11g十二烷基硫酸钠和0.24g聚乙烯醇溶于50ml去离子水中形成混合溶液,再加入10gPDDBP、0.16g偶氮二异丁腈、1.6g二乙烯苯、0.96g环己烷,在高压均质机作用下形成粒径小于1μm的稳定分散液滴,将该液滴分散液加入到含有种子微球的聚乙烯醇溶液中,在氮气保护下室温溶胀14h,溶胀结束后聚合反应15h即得溶胀微球。
(3)将10g溶胀微球、0.16g偶氮二异丁腈、0.21g二乙烯三胺加入到50ml甲醇中,于90℃下反应3h后反复离心洗涤得到沉淀物,将其与8.5g氯化亚铁、13.5g硫酸铁一同加入50ml去离子水中混合,降温至3℃,于真空条件下进行表面浸渍,之后在氮气保护下升温至75℃并加入0.66g柠檬酸钠,加入浓氨水调节溶液pH为11,熟化1.5h。
(4)将11g步骤(3)的产物分散于10ml无水乙醇和15ml去离子水的混合溶液中,加入催化剂氨水,然后逐滴加入7.7g正硅酸四乙酯与10ml乙醇的混合溶液,并逐滴加入含有4.3g硅酸钠的乙醇溶液,室温下搅拌16h后在50min内逐滴加入含有3.3g 3-氨丙基三甲氧基硅烷的乙醇溶液,形成表面带氨基的PDDBP@Fe
(5)将12.1g丁二酸酐溶解于20ml DMF中,搅拌5h后在氮气保护下逐滴加入含有10g步骤(4)产物的DMF混合液,室温下继续搅拌11h后用甲醇与去离子水反复洗涤,60℃真空烘干后得到产物。
实施例3
(1)将1.49g的4,4-二胺基-2,2-联苯并噻唑加入到20ml DMF中并搅拌均匀,在氮气保护下加入0.6g乙二胺,并用冰水浴冷却。通过恒压液滴漏斗慢慢加入0.19ml甲醛与10mlDMF的混合液,在冰水浴中反应6h后倒入100ml甲醇中沉淀,用去离子水和甲醇反复洗涤沉淀物,置于80℃真空干燥箱中烘干。将10g得到的产物加入到50ml二甲基亚砜中,在氮气保护下加入0.09g氯化钐,80℃反应24h后倒入去离子水中沉淀,用去离子水和甲醇反复洗涤沉淀物,置于80℃真空干燥箱中烘干得到PDDBP。
(2)以0.2g PDDBP为种子微球,分散于20ml浓度为0.7wt%的聚乙烯醇溶液中,另取0.13g十二烷基硫酸钠和0.28g聚乙烯醇溶于50ml去离子水中形成混合溶液,再加入10gPDDBP、0.24g偶氮二异丁基脒盐酸盐、2.4g二乙烯苯、1.12g正己烷,在高压均质机作用下形成粒径小于1μm的稳定分散液滴,将该液滴分散液加入到含有种子微球的聚乙烯醇溶液中,在氮气保护下室温溶胀16h,溶胀结束后聚合反应17h即得溶胀微球。
(3)将10g溶胀微球、0.24g偶氮二异丁腈脒盐酸盐、0.3g三乙烯四胺加入到50ml甲醇中,于90℃下反应3h后反复离心洗涤得到沉淀物,将其与12g氯化亚铁、21g硝酸铁一同加入50ml去离子水中混合,降温至5℃,于真空条件下进行表面浸渍,之后在氮气保护下升温至80℃并加入1.32g柠檬酸钠,加入浓氨水调节溶液pH为12,熟化1h。
(4)将11g步骤(3)的产物分散于10ml无水乙醇和15ml去离子水的混合溶液中,加入催化剂氨水,然后逐滴加入9.9g正硅酸四乙酯与10ml乙醇的混合溶液,并逐滴加入含有4.3g硅酸钠的乙醇溶液,室温下搅拌18h后在55min内逐滴加入含有3.8g 3-氨丙基三甲氧基硅烷的乙醇溶液,形成表面带氨基的PDDBP@Fe
(5)将7.7g顺丁烯二酸酐溶解于20ml DMF中,搅拌6h后在氮气保护下逐滴加入含有10g步骤(4)产物的DMF混合液,室温下继续搅拌14h后用甲醇与去离子水反复洗涤,60℃真空烘干后得到产物。
实施例4
(1)将1.49g的4,4-二胺基-2,2-联苯并噻唑加入到20ml DMF中并搅拌均匀,在氮气保护下加入0.6g乙二胺,并用冰水浴冷却。通过恒压液滴漏斗慢慢加入0.19ml甲醛与10mlDMF的混合液,在冰水浴中反应6h后倒入100ml甲醇中沉淀,用去离子水和甲醇反复洗涤沉淀物,置于80℃真空干燥箱中烘干。将10g得到的产物加入到50ml二甲基亚砜中,在氮气保护下加入0.24g氯化钕,80℃反应24h后倒入去离子水中沉淀,用去离子水和甲醇反复洗涤沉淀物,置于80℃真空干燥箱中烘干得到PDDBP。
(2)以0.2g PDDBP为种子微球,分散于20ml浓度为0.8wt%的聚乙烯醇溶液中,另取0.14g十二烷基硫酸钠和0.32g聚乙烯醇溶于50ml去离子水中形成混合溶液,再加入10gPDDBP、0.12g偶氮二异丁腈、3.2g乙二醇二甲基丙烯酸酯、1.28g正庚烷,在高压均质机作用下形成粒径小于1μm的稳定分散液滴,将该液滴分散液加入到含有种子微球的聚乙烯醇溶液中,在氮气保护下室温溶胀20h,溶胀结束后聚合反应22h即得溶胀微球。
(3)将10g溶胀微球、0.12g偶氮二异丁腈、0.38g四乙烯五胺加入到50ml甲醇中,于90℃下反应3h后反复离心洗涤得到沉淀物,将其与5.7g硫酸亚铁、11.3g氯化铁一同加入50ml去离子水中混合,降温至7℃,于真空条件下进行表面浸渍,之后在氮气保护下升温至70℃并加入1.12g聚乙二醇,加入NaOH调节溶液pH为10,熟化1h。
(4)将11g步骤(3)的产物分散于10ml无水乙醇和15ml去离子水的混合溶液中,加入催化剂氨水,然后逐滴加入13.2g正硅酸四乙酯与10ml乙醇的混合溶液,并逐滴加入含有4.3g硅酸钠的乙醇溶液,室温下搅拌22h后在60min内逐滴加入含有4.9g 3-氨丙基三乙氧基硅烷的乙醇溶液,形成表面带氨基的PDDBP@Fe
(5)将5.5g戊二酸酐溶解于20ml DMF中,搅拌4.5h后在氮气保护下逐滴加入含有10g步骤(4)产物的DMF混合液,室温下继续搅拌13h后用甲醇与去离子水反复洗涤,60℃真空烘干后得到产物。
实施例5
(1)将1.49g的4,4-二胺基-2,2-联苯并噻唑加入到20ml DMF中并搅拌均匀,在氮气保护下加入0.6g乙二胺,并用冰水浴冷却。通过恒压液滴漏斗慢慢加入0.19ml甲醛与10mlDMF的混合液,在冰水浴中反应6h后倒入100ml甲醇中沉淀,用去离子水和甲醇反复洗涤沉淀物,置于80℃真空干燥箱中烘干。将10g得到的产物加入到50ml二甲基亚砜中,在氮气保护下加入0.18g氯化钕,80℃反应24h后倒入去离子水中沉淀,用去离子水和甲醇反复洗涤沉淀物,置于80℃真空干燥箱中烘干得到PDDBP。
(2)以0.2g PDDBP为种子微球,分散于20ml浓度为1.0wt%的聚乙烯醇溶液中,另取0.15g十二烷基硫酸钠和0.36g聚乙烯醇溶于50ml去离子水中形成混合溶液,再加入10gPDDBP、0.2g偶氮二异丁基脒盐酸盐、2g乙二醇二甲基丙烯酸酯、1.6g正庚烷,在高压均质机作用下形成粒径小于1μm的稳定分散液滴,将该液滴分散液加入到含有种子微球的聚乙烯醇溶液中,在氮气保护下室温溶胀24h,溶胀结束后聚合反应24h即得溶胀微球。
(3)将10g溶胀微球、0.2g偶氮二异丁基脒盐酸盐、0.24g乙二胺加入到50ml甲醇中,于90℃下反应3h后反复离心洗涤得到沉淀物,将其与10g硫酸亚铁、18g硫酸铁一同加入50ml去离子水中混合,降温至10℃,于真空条件下进行表面浸渍,之后在氮气保护下升温至80℃并加入1.95g聚乙二醇,加入NaOH调节溶液pH为12,熟化1.5h。
(4)将11g步骤(3)的产物分散于10ml无水乙醇和15ml去离子水的混合溶液中,加入催化剂氨水,然后逐滴加入16.5g正硅酸四乙酯与10ml乙醇的混合溶液,并逐滴加入含有4.3g硅酸钠的乙醇溶液,室温下搅拌24h后在55min内逐滴加入含有5.5g 3-氨丙基三乙氧基硅烷的乙醇溶液,形成表面带氨基的PDDBP@Fe
(5)将9.9g己二酸酐溶解于20ml DMF中,搅拌5.5h后在氮气保护下逐滴加入含有10g步骤(4)产物的DMF混合液,室温下继续搅拌15h后用甲醇与去离子水反复洗涤,60℃真空烘干后得到产物。
实施例6
将实施例1-6所得的聚合物夹心复合磁性微球,分别进行电镜扫描和电镜透视,以及进行性能验证。
其中,图2中(a)为实施例1的聚合物夹心复合磁性微球的扫描电镜图,(b)为实施例2的聚合物夹心复合磁性微球的扫描电镜图,可以得到微球粒径在300-500nm之间且单分散性好。
图3中(a)、(b)为本发明实施例3制备的聚合物夹心复合磁性微球的透射电镜图,(c)、(d)为本发明实施例4制备的聚合物夹心复合磁性微球的透射电镜图,从透射电镜图中可以明显地观察到微球有三层,证实合成的为夹心复合微球。
图4为本发明实施例1-2制备的聚合物夹心复合磁性微球的磁滞回线,由图片可看出本发明的合物夹心复合磁性微球呈超顺磁性,剩余磁化强度和矫顽力几乎为零,且具有较高的饱和磁化强度。
图5为本发明实施例1制备的聚合物夹心复合磁性微球的分散稳定性和磁响应性能测试图,反映了本发明的聚合物夹心复合磁性微球磁响应时间短、在介质中分散稳定性强。
- 一种磁性壳聚糖-蒙脱土纳米复合微球的制备方法
- 一种氨基和羧基功能化磁性微球复合吸附剂的制备方法
- 一种磁性复合微球及其制备方法和应用
- 一种用于污水处理的低成本复合磁性微球材料及制备方法
- 具有功能化壳层的夹心结构磁性复合微球及其制备方法
- 具有功能化壳层的夹心结构磁性复合微球、其制备方法及其应用