一种促进酿酒酵母木糖发酵的方法、所构建的重组酵母菌株及其应用
文献发布时间:2024-04-18 19:58:30
技术领域
本发明涉及基因工程技术领域,具体涉及一种促进酿酒酵母木糖发酵的方法、所构建的重组酵母菌株及其应用。
背景技术
木质纤维素生物质(简称LCB)是自然界中最丰富的可再生资源,包括如农作物的秸秆、甘蔗渣、柳枝稷、森林废弃物、芒草等等,不仅来源丰富且价格低廉,是生产生物燃料很有潜力的原料。酿酒酵母(Saccharomyces cerevisiae)由于其良好的发酵性能和抗逆性,是生产燃料乙醇的主要菌株,利用酿酒酵母发酵玉米秸秆等木质纤维素类生物质水解产生的单糖,生成乙醇,具有绿色环保、可再生等优点。
LCB经过纤维素酶水解后会释放葡萄糖、木糖、阿拉伯糖等单糖,其中葡萄糖和木糖分别占总糖的60%-70%和30%-40%,木糖是仅次于葡萄糖含量的碳源,作为可再生的底物容易从自然界中获得,可用作生物精炼的原料。目前基于木糖利用开发的菌株生产生物燃料和生物基化学品等研究受到国内外的普遍重视。酿酒酵母常用于LCB生物转化的细胞工厂,但是酿酒酵母天然菌株普遍不能很好利用木糖,需要对其进行代谢途径的改造。
虽然目前对木糖的代谢通路做了很多的优化,也通过木糖的适应性进化挖掘到一些靶点,但重组酵母的木糖代谢速率还不够理想。目前菌株改造技术更多集中在酶蛋白和转运蛋白,对表观水平的靶点还没有研究报道。染色质重塑是表观调控的一部分,染色质重塑是表观调控的一部分,酿酒酵母是真核生物,其具有高度动态变化的复杂的染色质结构。核小体是染色质的基本单位,由H2A、H2B、H3和H4各两个拷贝形成组蛋白八聚体并包裹着周围约150个碱基对DNA形成。DNA和核小体之间的相互作用不断动态变化,确保核小体可以从DNA中移动或松开,允许其他蛋白质结合DNA。参与基因转录的主要共调控蛋白复合物是染色质修饰复合体,分为两大类:改变核小体位置的复合物和共价修饰染色质中组蛋白的复合物。改变核小体位置的重塑复合物通过ATP水解提供的能量驱逐、组装或交换核小体组蛋白;另一类复合物可以共价修饰组蛋白或DNA,如翻译后修饰,包括蛋白乙酰化、磷酸化和泛素化,以及DNA的甲基化。目前对染色质重塑复合物对代谢的影响还了解很少,对木糖利用的影响未见报道。
除了提高木糖利用效率,在分步糖化发酵过程中,生物反应器由酶解最适温度50℃降低到酵母生长最适温度30℃会消耗大量的冷却水和电量,因此构建一株能够在高温下发酵木糖的酿酒酵母菌株对乙醇的工业化生产中成本的降低具有重要意义。
发明内容
为解决上述问题,本发明的目的之一是提供一种促进酿酒酵母木糖发酵的方法,通过过表达染色质重塑因子SWR1和SWC5,构建出能在高温条件下高效利用木糖的重组菌株,实现酿酒酵母对木糖利用率的提升。所述染色质重塑因子SWR1和SWC5为Swr1复合体的关键亚基;所述SWR1的序列如SEQ ID NO:7所示,所述SWC5的序列如SEQ ID NO:8所示。
具体的,本发明通过op58444基因导入、XKS1替换PHO13基因、TAL1和CiGXS1
S1.以酿酒酵母菌株为出发菌株,将密码子优化的木糖异构酶基因op58444整合到基因组的delta位点(delta位置信息在文献“Kim JM,Vanguri S,Boeke JD,Gabriel A,Voytas DF.Genome Research.1998,8(5):464-478.”中公开);在PHO13位点整合TEF1p-XKS1-CYC1t表达盒,在GRE3位点整合KGD1p-TAL1-TAL1t和GPDp-CiGXS1mut-CYC1t表达盒,通过实验室适应性进化,筛选出一株在40℃下高效利用木糖的菌株P10-5;
S2.在菌株P10-5的基础上,将SWR1p的序列替换为TEF1p,在HO位点整合PGK1p-SWC5-ADH1t表达盒,形成重组酵母菌株TWZ01。
优选的,步骤S1中,所述出发菌株为通过原生质体融合技术选育的工业酿酒酵母菌株SPSC01的非絮凝突变菌株PLY01,该菌株具有良好的耐高温性能。本课题组前期利用原生质体融合技术,获得了一株工业乙醇酵母菌株SPSC01,该菌株保存在中国微生物菌种管理委员会普通微生物中心,保藏地址为北京市朝阳区北辰西路1号院3号中国科学院微生物研究所,保藏编号为CGMCCNO.0587。在此基础上敲除絮凝基因FLO1,获得了突变菌株PLY01,该菌株保留了出发菌株SPSC01对高温的良好耐性,利用该菌株进一步通过染色质重塑因子的改造,获得了本发明的木糖利用性能提升的菌株。
本发明的目的之二是提供上述促进酿酒酵母木糖发酵的方法制得的重组酵母菌株TWZ01,其分类命名为Saccharomyces cerevisiae,保藏于中国微生物菌种管理委员会普通微生物中心,保藏地址为北京市朝阳区北辰西路1号院3号中国科学院微生物研究所,保藏编号为CGMCC NO.28009,保藏日期为2023.07.25。
本发明的目的之三是提供上述促进酿酒酵母木糖发酵的方法制得的重组酵母菌株TWZ01在木糖发酵生产燃料乙醇中的应用。所述应用是在含有木糖的发酵培养基或者木质纤维素生物质水解液发酵中的应用,所述木质纤维素生物质包括但不限于水稻秸秆、玉米秸秆、甘蔗渣、芒草、农林废弃物等。
本发明具有以下有益效果:
1、通过op58444基因导入、一次性改造5个靶标基因(XKS1替换PHO13基因,TAL1、CiGXS1
2、在菌株P10-5中过表达染色质重塑因子SWR1和SWC5,形成重组酵母菌株TWZ01,进一步提升了木糖利用率。本发明证明了染色质重塑因子SWR1和SWC5的过表达提高了酿酒酵母高温木糖发酵能力,为酿酒酵母的木糖利用提供新的研究靶点。
3、本发明所构建的重组菌株TWZ01在30℃木糖发酵72h消耗29.16g/L木糖,相较于对照菌株(P10-5)提升28.2%。同时,TWZ01菌株在40℃木糖发酵12h木糖消耗速率是2.07g/L/h,相比于对照菌株(1.83g/L/h)提升13.1%。
附图说明
图1:实施例1中PLY01、P2、P3、P10-5菌株在YPX培养基中30℃发酵72h的细胞浓度(A)以及木糖消耗量(B)。
图2:实施例1中P10-5菌株在YPX培养基40℃发酵22h的细胞浓度(A)以及木糖残余和乙醇的产量(B)。
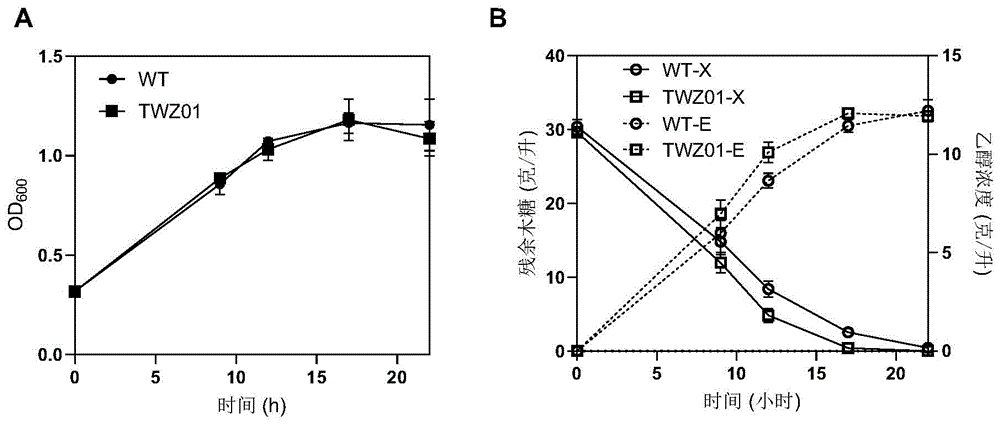
图3:实施例2中TWZ01菌株和对照菌株在YPX培养基30℃发酵72h的细胞浓度(A)以及木糖残余和乙醇的产量(B)。
图4:实施例2中TWZ01菌株和对照菌株在YPX培养基40℃发酵22h的细胞浓度(A)以及木糖残余和乙醇的产量(B)。
具体实施方式
以下结合附图和具体实施例对本发明进行进一步的说明。
实施例1:木糖利用重组酿酒酵母(P10-5)的构建
1.酿酒酵母木糖代谢菌株的构建
采用传统的以抗性基因G418为筛选标记的同源重组法对密码子优化后的木糖异构酶基因op58444(序列如SEQ ID NO:1所示,来源于Bos taurus fecal metagenome,在文献“Tang,R.,Ye,P.,Alper,H.S.et al.Applied Microbiology and Biotechnology.2019,103,9465–9477.”中公开),整合到酿酒酵母基因组的delta位点。
将载体pITy3(pITy3质粒在文献“Parekh R.N.,Shaw M.R.,Wittrup K.D.,Anintegrating vector for tunable,high copy,stable integration into thedispersed Tyδsites of Saccharomyces cerevisiae.Biotechnol prog,1996,12(1):16-21.”公开)用限制性内切酶Kpn I和Sac I在50μL体系中,37℃反应2h后,按照GeneJET琼脂糖凝胶回收试剂盒的说明进行胶回收,纯化酶切后的载体。同时,用引物pITy-XI-F/R扩增op58444基因,根据GeneJET PCR纯化试剂盒的说明书纯化PCR产物。随后,按插入片段op58444与线性化载体pITy3-的摩尔数之比为3:1的用量(通常载体质量设定为50ng左右),将片段和载体加入到7.5μL Gibson assembly mix中,50℃反应1h。随后,取10μL Gibsonassembly产物转化至100μL大肠杆菌感受态细胞中。待长出单克隆后进行菌落PCR验证,将初步验证正确的单克隆挑取到5mL LB培养基中,过夜培养后提取质粒,送测序,将测序正确的载体以及相应的菌株保存备用。
将5μg测序验证正确的表达载体pITy3-584,用限制性内切酶Xho I酶切后,用PCR纯化试剂盒纯化酶切产物,随后,用醋酸锂转化法将DNA转入酿酒酵母PLY01菌株中,涂布于含有300μg/mL G418的YPD固体培养基上,置于30℃培养箱中培养至长出单克隆(PLY01菌株在文献“Ye,P.-L.,Wang,X.-Q.,Yuan,B.,Liu,C.-G.,Zhao,X.-Q.2022.Manipulating cellflocculation-associated protein kinases in Saccharomyces cerevisiae enablesimproved stress tolerance and efficient cellulosic ethanolproduction.Bioresource Technology,348,126758.”中公开)。待长出单克隆后,提取基因组,具体步骤如下:挑取少量菌体到装有20μL去离子水的PCR管中,混匀后置于-80℃冷冻20min;将PCR管取出解冻后,置于微波炉高火加热3min;立即冰浴1min,再用微波炉高火加热3min,立即冰浴,取1μL用于PCR。用引物op58444-vF/vR进行菌落PCR,验证成功的重组菌株命名为P2。
2.木糖代谢途径的优化
为了提高改造效率,用拟过表达的基因替换拟敲除的基因,一次性改造所选定的五个靶标基因。敲除GRE3和PHO13,过表达TAL1、CiGXS1
(1)将实验室保存的CRISPR/Cas9的表达载体中URA3筛选标记替换为NAT筛选标记。以带有NAT抗性基因的Cas9-NAT(Addgene公司商业化质粒)为模板,用引物URA3up-NAT-F/URA3down-NAT-R扩增带有同源臂的NAT片段;以质粒pGAL-Cas9-URA3(Addgene公司商业化质粒)为模板,用引物URA3up-F/Amp-R和Amp-F/URA3down-R扩增出载体骨架pGAL-Cas9-URA-1和pGAL-Cas9-URA-2,最终将三个基因片段进行组装,转化至大肠杆菌,提质粒验证得到pGAL-Cas9-NAT载体。
(2)靶向GRE3和PHO13基因位点的sgRNA表达载体的构建步骤如下:用在线工具CRISPRdirect(http://crispr.dbcls.jp/)分别设计靶向GRE3和PHO13基因位点的sgRNA,根据选定的sgRNA序列,设计引物GRE3-sgRNA-F/R和PHO13-sgRNA-F/R,进行3个循环的PCR。随后,用2%琼脂糖凝胶回收大小约为100bp的条带,得到DNA片段GRE3-sgRNA和PHO13-sgRNA。同时,用限制性内切酶Spe I酶切3μg载体pGAL-Cas9-NAT(37℃反应2h),在反应体系中加入1μL虾碱性磷酸酶rSAP以防止单酶切后载体自连。随后,用0.5%琼脂糖凝胶回收酶切产物,得到线性化载体pGAL-Cas9-NAT。将载体和片段按照摩尔比1:3加入到Gibsonassembly mix中,50℃反应1h。将连接产物转化至大肠杆菌DH5α中。将阳性转化子转接至5mL LB液体培养基(含氨苄青霉素)中过夜培养,收集菌体后提取gRNA质粒。
将gRNA质粒进行测序验证。在验证正确的两个载体pGAL-Cas9-GRE3和pGAL-Cas9-PHO13的基础上构建了将GRE3-sgRNA和PHO13-sgRNA表达框串联的表达载体pGAL-Cas9-GRE3-PHO13。具体步骤如下:以质粒pGAL-Cas9-PHO13为模板,用引物T7p-F/PacI_arm-Sup4t-R扩增片段pSNR52-PHO13-sgRNA-Scaffold-Sup4t,其中pSNR52和Sup4t分别为sgRNA的启动子和终止子。胶回收纯化PCR产物后,用限制性内切酶Pac I酶切PCR产物,同时,用Pac I酶切3μg质粒pGAL-Cas9-GRE3,随后,用胶回收试剂盒分别回收酶切后的片段pSNR52-PHO13-sgRNA-Scaffold-Sup4t-PacI和载体骨架pGAL-Cas9-GRE3-PacI。将片段与载体骨架按摩尔比3:1进行T4酶连接。连接反应的体系为10μL,在16℃条件下过夜连接,随后,取5μL连接产物转化大肠杆菌,提质粒测序验证。
(3)Donor DNA的扩增,将TAL1基因表达框插入到转运蛋白表达载体p414-GPDp-CiGXS1
随后转化大肠杆菌,提质粒验证,将验证正确的载体命名为p414-KGD1p-TAL1-GPDp-CiGXS1mut,保存备用。通过引物KGD1p-cassette-F/CYC1t-R扩增出KGD1p-TAL1-TAL1t-GPDp-CiGXS1
(4)酵母转化。将带有op58444基因的PLY01酿酒酵母菌株(即P2菌株)在YPD固体平板上划线培养48h,挑取单克隆于YPD培养基中过夜活化。取1mL菌液至10mL的YPD培养基中,培养6h左右,3000g离心2min收集菌体,去除培养基后依次用水洗两遍、0.1M乙酸锂溶液洗一遍,弃去上清。向菌体中加入2mL 0.1M乙酸锂溶液,混合均匀后分装100μL至1.5mL离心管,至于30℃孵育10min。离心收集菌体,加入1μg gRNA质粒和1μg带有同源臂的Donor DNA,吹打均匀。向体系中依次加入5mg/mL煮沸后冷却的ssDNA(75μL)、50%PEG3350(360μL)、1M乙酸锂(55μL),混合均匀,置于30℃孵育30min后热击(42℃)30min。3000g离心2min,弃去上清,加入1mL YPD培养基,30℃、200rpm培养3h,离心浓缩菌体后涂布于含NAT的YPD固体平板上,30℃培养箱培养48h。
(5)转化子验证。从平板上随机挑取转化子至YPD培养基中活化24h,离心收集菌体后提取酿酒酵母基因组。取1μL基因组上清作为模板进行PCR验证,分别用引物PHO13-in-F/R和GRE3-in-F/R验证PHO13和GRE3基因的敲除,用XK-cassette-F/R和KGD1p-cassette-F/CYC1t-R验证XK表达框和TAL1-CiGXS1表达框的敲入,验证成功的重组菌株命名为P3。
3.实验室适应性进化
取P3重组酵母接种到含有2mL YPX培养基(YP:10g/L酵母提取物,20g/L蛋白胨;X:30g/L木糖)的培养管里,置于30℃、200rpm条件下培养。待菌液浓度不再变化,即生长到稳定期后,转接1μL菌液至新鲜的2mL YPX培养基中继续培养。随后,重复“转接-培养”的操作,每次转接1μL至2mL新鲜YPX培养基中,进行连续传代,有氧驯化2个月加半厌氧驯化1个月后,得到P10-5菌株。
4.实验结果
在不能利用木糖的PLY01工业菌株中通过木糖异构酶的外源引入、木糖代谢途径的优化和实验室适应性进化的组合操作,构建出了P10-5菌株。从图1看出P10-5菌株在72h内消耗22.74g/L木糖,相较于未驯化的重组菌株P3(木糖消耗7.55g/L)提升2.01倍,实验室适应性进化对提升重组菌株的木糖消耗具有很大的帮助;PLY01是一株能够耐高温和耐抑制物的工业菌株,从图2可以看出P10-5菌株具有优异的高温木糖发酵性能,其在40℃下22h内消耗29.94g/L木糖,乙醇/木糖收率是0.41g/g。
本实施例中用到以下引物:
实施例2:染色质重塑因子过表达重组菌株(TWZ01)的构建1.过表达SWR1基因重组菌株的构建
将SWR1基因(序列如SEQ ID NO:7所示)上游1000bp的序列输入到E-CRISP(http://www.e-crisp.org/E-CRISP/)网站,查找目标片段中PAM位点(NGG)上游的20bp靶向序列为:5’-GACTTGAAGAAAGAGTGCAA-3’。再将包含gRNA骨架上下游各23bp、22bp同源臂的gRNA片段进行合成,引物命名为gRNA-SWR1-F/R。将两条合成后的互补链按照体积1:1混合,加入8倍体积的蒸馏水后混匀,置于水浴锅中95℃孵育5min,冷却至室温后获得双链gRNA片段。以pRS42H-gRNA质粒为模板(Addgene公司的商业化质粒),以gRNA-SWR1-BB1/BB2引物按照普通PCR的方法扩增gRNA线性骨架。将获得的gRNA线性骨架和gRNA片段通过同源重组的方法进行连接,将连接产物转化至大肠杆菌DH5α中,提质粒测序验证。以菌株PLY01的基因组DNA为模板,通过引物TEF1-F/R扩增带有TEF1p的Donor片段。按照实施例1中醋酸锂转化的方法,将靶向SWR1启动子的gRNA质粒和Donor片段转到P10-5菌株中,待转化板上长出单克隆,挑取单克隆提取基因组,用引物v-TEF1-F/v-SWR1-R验证。
2.过表达SWC5重组菌株的构建
以pHO-yEGFP质粒为模板(该质粒在文献“He,L.Y.,Zhao,X.Q.,Bai,F.W.,Engineering industrial Saccharomyces cerevisiae strain with the FLO1-derivative gene isolated from the flocculating yeast SPSC01 for constitutiveflocculation and fuel ethanol production.Appl.Energy 2012,100,33–40.”已公开),通过HO-SWC5-BB1/HO-SWC5-BB2引物扩增线性片段;以PLY01菌株的基因组为模板,通过SWC5-F/SWC5-R引物扩增SWC5基因片段(SWC5基因序列如SEQ ID NO:8所示),将上述扩增得到的两个基因片段通过无缝克隆连接,连接产物转化至大肠杆菌DH5α中,提质粒测序验证。使用Not I限制性内切酶将验证正确的pHO-SWC5质粒在37℃酶切2h,随后通过醋酸锂转化的方法,将线性片段转化到过表达SWR1的P10-5菌株中,PGK1p-SWC5-ADH1t表达盒通过同源重组整合到HO位点,挑取单克隆提取基因组,用v-HO-F/SWC5-R验证转化子是否正确,验证成功的重组菌株命名为TWZ01。
3.重组菌株TWZ01的木糖发酵
挑取-80℃保存/平板活化的菌株,接种至5mL YPD(YP:10g/L酵母提取物,20g/L蛋白胨;D:20g/L葡萄糖)培养基中,30℃、200rpm培养24h。接着,将2mL菌液加入到100mL YPD培养基中,30℃、200rpm培养12h。随后,按照初始OD
4.实验结果
在P10-5菌株中过表达染色质重塑因子SWR1和SWC5基因构建TWZ01菌株,从图3可以看出TWZ01菌株进一步提升木糖利用,在72h内消耗29.16g/L木糖,相较于对照菌株(P10-5)提升28.2%;同时,TWZ01菌株在高温木糖发酵(图4)12h内的木糖消耗速率是2.07g/L/h,相比于对照菌株(1.83g/L/h)提升13.1%。
Swr1复合体的功能是将组蛋白H2A交换为组蛋白变体H2A.Z,从而调控转录的激活和抑制。从上述结构可得,过表达Swr1复合体的关键亚基SWR1和SWC5可以显著提高重组酿酒酵母P10-5的木糖利用,不仅为重组酿酒酵母的木糖代谢机制提供新的研究靶点,也提供一种通过染色质重塑因子的过表达进而提升菌株性能的改造策略。
本实施例中用到以下引物:
本具体实施方式仅仅是对本发明的解释,并不是对本发明的限制,本领域技术人员在阅读了本发明的说明书之后所做的任何改变,只要在本发明权利要求书的范围内,都将受到专利法的保护。