新型唾液酸苷及其治疗用途
文献发布时间:2024-04-18 20:01:55
技术领域
本发明涉及合成的含有唾液酸的碳水化合物,也称为唾液酸苷(sialoside)。这些新型化合物可以通过发酵产生。这些唾液酸苷能够与一些病毒的表面蛋白结合,特别是与流感病毒的血凝素结合。结果是这些唾液酸苷抑制了病毒与宿主细胞的粘附,因而是预防或治疗病毒感染的有用化合物。
背景技术
病毒已经进化到进入了来自生命的所有三个域(细菌、古细菌和真核生物)的细胞中。在超过3600种已知病毒中,有数百种可以感染人的细胞,其中大多数与疾病有关。
病毒进入动物细胞是通过附着于受体开始的,随后病毒蛋白发生重要的构象变化,穿透细胞膜或与细胞膜融合。
一些病毒(如流感病毒)可以非常迅速地穿过核内体膜,进入效率可以超过50%(即50%的附着病毒确实进入了细胞)。
预防或至少减少病原病毒对细胞的病毒感染的一种方法是抑制和/或阻断病毒与宿主细胞受体的相互作用。因此,对病毒/宿主细胞这些相互作用的研究至关重要。
在正粘病毒科(Orthomyxoviridae)病毒中,流感病毒是研究最多的病毒之一。流感病毒的感染周期由病毒血凝素蛋白(HA)与宿主细胞表面存在的糖蛋白和糖脂中掺入的含有唾液酸的聚糖(唾液酸苷)的相互作用启动。
已经提出,通过用包含唾液酸的合成配体使病毒血凝素HA的结合位点饱和,来抑制流感病毒与宿主细胞的粘附(Matrosovich和Klenk,2003)。
目前正在研究这一有前景的方法,特别是通过研究最合适的合成配体,即对每种流感病毒的血凝素都具有更好亲和力的合成配体。
对流感病毒的血凝素和唾液酸苷的这种相互作用的研究表明,流感病毒通过选择性结合特异性唾液酸苷而对流感病毒的宿主细胞产生特异性。
事实上,人和动物流感病毒具有的血凝素对宿主细胞表面表达的各种唾液酸苷具有不同的结合偏好。
禽流感病毒毒株表达的血凝素偏好结合通过α2-3键与聚糖链的其余部分连接的唾液酸。相比之下,来自人流感病毒毒株的血凝素与α2-6连接的唾液酸的结合增强。这与人上呼吸道中大量的α2-6连接的唾液酸以及在鸟类肠粘膜中存在α2-3连接的唾液酸相关,流感病毒的人毒株和禽毒株分别在所述上呼吸道和所述肠粘膜中进行复制。
在全球范围内,唾液酸连接的特异性影响着宿主的范围:流感病毒的禽毒株和马毒株识别以α2,3键连接至半乳糖的唾液酸,而人毒株偏好以α2,6键连接的唾液酸。然而,流感嗜性的决定因素比仅偏好结合α2-6和α2-3唾液酸更复杂。
人呼吸道上皮细胞被证明表达具有延伸的聚-N-乙酰基乳糖胺(聚-LacNAc)链的唾液酸化的N-聚糖,表明人HA偏好结合延伸的α2-6唾液酸苷。
这一点通过合成唾液酸苷的聚糖微阵列研究证实,表明来自亚型H1、H2和H3的人流感病毒HA与具有二LacNAc和三LacNAc延伸的直链唾液酸苷的结合效果最佳(Nycholat等人,2012)。
有趣的是,来自H3流感病毒亚型的HA的受体特异性似乎近年来发生了演变:“最早的”H3病毒偏好短的支链唾液酸苷,而“最近的”H3病毒仅与具有三LacNAc延伸的直链唾液酸苷结合(Yang等人,2015)。
现在已经明确,并非所有唾液酸苷都以相同的效率与HA结合,不同的病毒毒株根据其宿主嗜性表现出了不同的受体特异性。
合成唾液酸苷的生产
开发这些有前景的治疗分子的最大技术问题是这些特异性唾液酸苷配体的获得/生产。
结构多样的唾液酸苷的制备可以通过化学合成、酶法合成或天然唾液酸苷的修饰来实现。不考虑从天然来源中提取和纯化,因为通过这种方式只能获得非常少量。
长链唾液酸化聚乳糖胺似乎是通过诱饵策略(decoy strategy)有效预防或治疗流感感染的最佳候选物。然而,无法通过化学或酶法大规模生产长链唾液酸苷。因此,生产这些唾液酸苷的最佳可替选方法是微生物方法,即通过特定工程改造的微生物发酵进行生产。
国际申请WO 2007/101862中公开了通过发酵生产3'唾液乳糖和6'唾液乳糖的方法。
此外,发酵工艺已用于工业化大规模生产人乳寡聚糖用于婴儿配方奶粉市场(Bych等人,2019)。该技术基于在大肠杆菌菌株中表达特定的重组糖基转移酶基因,所述菌株经过代谢工程改造以过量生产在糖基化反应中使用的糖核苷酸。
尽管有报道称在代谢工程改造细菌中生产出了聚乳糖胺结构,但使用微生物生产长链唾液酸化聚乳糖胺似乎难以实现,原因是:
-分子结构的复杂性,以及
-预期副产品的数量,这些副产物的产生会大幅降低产量。
本发明的主要目的是公开新型结构的长链唾液酸苷,其有效地结合病原病毒,特别是人流感病毒,并且可以通过发酵工艺大量生产。
发明内容
本发明涉及具有式(I)的合成唾液酸苷:
Neu5Ac-α2-6-R1(R2)[GlcNAcβ1-4]n-GlcNAc式(I)
其中:
-GlcNAc是N-乙酰葡糖胺;
-GlcNAcβ1-4是以β1-4连接键连接的N-乙酰葡糖胺单元;
-n大于或等于1;
-R1是包含至少一个半乳糖(Gal)的聚糖结构;以及
-R2选自以下基团:H、以α1-3连接键连接的岩藻糖(Fucα1-3)和以α1-4连接键连接的岩藻糖(Fucα1-4)。
本发明还涉及多价唾液酸苷,其包含载体(support)与多个如上所述的合成唾液酸苷。
本发明的另一个目的是药物组合物,其包含在药学上可接受的载剂中的至少一种如上所述的多价唾液酸苷。
本发明的另一个目的是包含至少一种如上所述的多价唾液酸苷的医疗装置。
本发明还涉及本发明的多价唾液酸苷、或本发明的药物组合物作为药物的用途。
此外,本发明涉及本发明的多价唾液酸苷、或本发明的药物组合物在预防和/或治疗感染中的用途,所述感染由对唾液酸具有亲和力的病毒引起。所述病毒特别是流感病毒,优选人、马、猪或禽流感病毒。
附图说明
图1.使用SCH6菌株用于COV6S生物合成的基因工程改造代谢途径。
[10]壳五糖
[11]中间体六糖
[12]唾液酸化七糖COV6S
已表明壳寡糖(如壳五糖[10])可以被β1-4半乳糖基转移酶用作受体,以形成在其非还原端含有末端LacNAc基序的六糖[11]。然后,通过在代谢工程改造的大肠杆菌菌株中额外表达α2-6唾液酸转移酶,可以将[11]唾液酸化以形成唾液酸化七糖COV6S[12]。由于GlcNAC不是α2-6唾液酸转移酶的底物,因此防止了副产物的形成,COV6S[12]几乎是唯一的终产物。
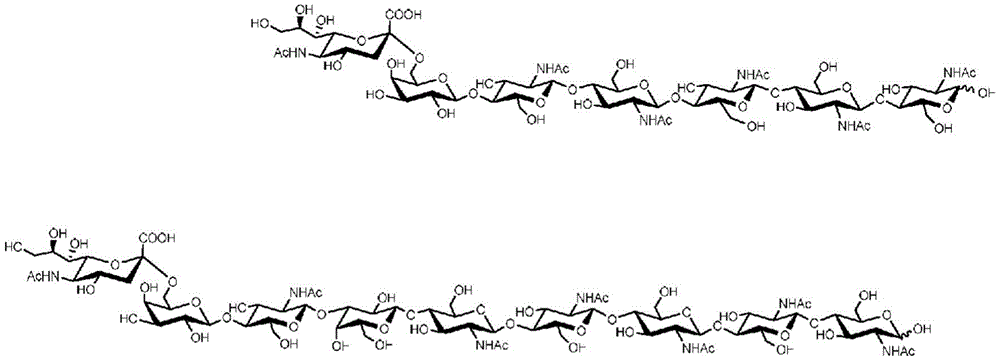
图2.本发明开发的两种唾液酸苷的结构
上部显示式(II)的COV6S;下部显示另一种唾液酸苷结构的示例(式(I),其中R1=Galβ1-4GlcNAcβ1-3Galβ1-4,R2=H,n=4)。
图3.血凝抑制测定(HI或HAI)的示意图
图4.微中和测定(MN)的示意图
图5.用在D0(感染后1小时)、D1和D2的三次感染后处理,测定ER15化合物的潜在治疗效果。
在37℃在细胞中孵育A/California/7/09H1N1病毒1小时。与未处理条件相比,ER15以5种不同的浓度(20;10;5;2.5和1.25μM)使用(NT:未处理;PL:递送递增量的聚赖氨酸)。在感染后D1、D2和D3收集上清液样本,以通过以下对病毒生产进行定量:(i)在MDCK细胞上每毫升中感染颗粒的数量的滴定(TCID50/ml)和(ii)病毒基因组的定量(RT-qPCR)。
图5A和图5C:测量产生的传染性颗粒随着时间(感染后小时数:hpi)的变化,表示为TCID50/ml;
图5B和图5D:通过RT-qPCR测量病毒mRNA随着时间(感染后小时数:hpi)的变化;
图5A和图5B:感染复数(MOI)=0.1
图5C和图5D:感染复数(MOI)=0.01
图6.用在D0(感染后1小时)、D1和D2的三次感染后处理,测定ER15化合物的潜在治疗效果
实验条件与(图5)相同,不同之处在于在37℃在细胞中孵育A/Texas/50/2012H3N2病毒1小时。
图6A和图6C:测量产生的传染性颗粒随着时间(感染后小时数:hpi)的变化,表示为TCID50/ml;
图6B和图6D:通过RT-qPCR测量病毒mRNA随着时间(感染后小时数:hpi)的变化;
图6A和图6B:感染复数(MOI)=0.1
图6C和图6D:感染复数(MOI)=0.01
图7.用在D0(感染后1小时)的单次感染后处理,测定ER15化合物针对A/California/7/09H1N1毒株的潜在治疗效果使
图7A:感染复数(MOI)=0.1
图7B:感染复数(MOI)=0.01
图8.ER15针对A/California/7/09H1N1毒株的治疗特性,仅在病毒感染时伴随施用一次
图8A:感染复数(MOI)=0.1
图8B:感染复数(MOI)=0.01
图9.ER15针对A/California/7/09H1N1毒株的预防特性,仅在感染前一天进行单次处理
图9A:感染复数(MOI)=0.1
图9B:感染复数(MOI)=0.01
图10.ER15针对H1N1 A/Lyon/969/09毒株的治疗特性
实验条件与图5相同,不同之处在于在37℃在细胞中孵育A/Lyon/969/09H1N1病毒1小时。
图10A:感染复数(MOI)=0.1
图10B:感染复数(MOI)=0.01
图11.在受感染的体外上皮模型上测试ER15的疗效并评价细胞毒性
用150μl的A/California/7/2009H1N1毒株以0.1的感染复数在顶级感染上皮细胞。
将冻干的化合物ER15与以下混合:
-水,
-磷酸盐缓冲液(2g/L甘油,9g/L NaCl,10mM,pH=7.9)和
-柠檬酸盐缓冲液(2g/L甘油,9g/L NaCl,10mM,pH=6.0)
浓度为0.1mM。
图11A–每日测量的经上皮细胞电阻(TEER)
一个阴性对照是“模拟NT”细胞:未感染、未处理的细胞。
另一个阴性对照是“PL”:用在水中稀释为0.1mM的聚赖氨酸处理细胞。
图11B-对用ER15、聚赖氨酸(PL)处理的或未处理(NT)的模拟(未感染)细胞的乳酸脱氢酶(LDH)释放进行定量。每天从基础介质中收集样本。
图11C-通过每毫升产生的感染性颗粒的滴定(TCID50/ml)来定量病毒的产生。在D1、D2和D3时在顶极收集样本。
图11D-通过RT-qPCR对相同的样本进行病毒基因组定量。
图12.ER61针对H1N1 A/California/7/09毒株的治疗特性
实验条件与图5相同,不同之处在于使用了ER61化合物。测量产生的传染性颗粒随着时间(感染后小时数:hpi)的变化,表示为TCID50/ml
图12A:感染复数(MOI)=0.1
图12B:感染复数(MOI)=0.01
图13.ER15和ER61化合物针对最近毒株的治疗效果。
在37℃在A549细胞上以MOI 0.1孵育测试的病毒毒株1小时。与未处理的条件相比,ER15和ER61以3种不同的浓度(20、5和1.25μM)使用(NT:未处理)。在D0(感染后1小时)、D1和D2进行3次感染后处理。在感染后的D1、D2和D3收集上清液样本,以通过每毫升感染颗粒的数量的滴定(TCID50/ml)来对病毒的生产进行定量。
测试的病毒毒株如下:
A)A/Michigan/45/2015H1N1
B)A/Kansas/14/2017H3N2。
C)B/Phuket/3073/2013。
图14.ER15化合物针对耐药H1N1病毒毒株的治疗效果。
实验条件与图13相同。用ER15在D0(感染后1小时)、D1和D2对用通过反向遗传获得的以下病毒毒株感染的细胞进行3次感染后处理:
A)RG H1N1 A/Lyon/969/09I38T
B)RG H1N1 A/Lyon/969/09H275Y
C)RG H1N1 A/Lyon/969/09I38T+H275Y
具体实施方式
本发明涉及具有式(I)的合成唾液酸苷:
Neu5Ac-α2-6-R1(R2)[GlcNAcβ1-4]
其中:
-GlcNAc是N-乙酰葡糖胺;
-GlcNAcβ1-4是以β1-4连接键连接的N-乙酰葡糖胺单元;
-n大于或等于1,特别是n在1和4之间;
-R1是包含至少一个半乳糖(Gal)的聚糖结构;以及
-R2选自以下基团:H、以α1-3连接键连接的岩藻糖(Fucα1-3)或以α1-4连接键连接的岩藻糖(Fucα1-4)。
除非另有说明,否则如本文所使用的以下术语和短语旨在具有以下含义。
在本发明的意义上,术语唾液酸苷是指唾液酸缀合物,其由唾液酸通过O-糖苷键与碳水化合物结合组成。
唾液酸是一个单糖家族,其包含43种天然存在的5-氨基-3,5-二脱氧-D-甘油-D-半乳糖-壬-2-酮糖酸(non-2-ulosonic acid)(神经氨酸)衍生物,神经氨酸是由九碳骨架组成的糖,在所有脊椎动物中普遍表达。最常见的唾液酸是5-乙酰氨基-2-酮-3,5-二脱氧-D-甘油-D-半乳糖醛酸,也称为N-乙酰神经氨酸或唾液酸,缩写为Neu5Ac。
唾液酸被认为是细菌毒素和一系列病毒的受体的关键成分。
术语“合成的”强调根据本发明的唾液酸苷的人工性质。事实上,这种唾液酸苷在自然界中并不存在,其结构是由发明人设计的。有利地,这种合成唾液酸苷是为了与对唾液酸具有亲和力的病毒表面蛋白(特别是与流感病毒毒株的血凝素)具有最佳的亲和力而设计的。本发明的每种唾液酸苷可以对各种血凝素具有不同的亲和力,特别是本发明的每种唾液酸苷可以偏好与一种特定血凝素结合。
其他术语和缩写是本领域技术人员众所周知的,特别是:
-N-乙酰葡糖胺(GlcNAc)是葡萄糖的酰胺衍生物。它是在葡糖胺和乙酸之间的仲酰胺。该分子在活生物体中广泛表达,因为它是构成细胞壁的生物聚合物的一部分。其CAS编号为7512-17-6。
-β1-4连接键是指将碳水化合物基团连接到另一个基团的β型共价糖苷键。
α-和β-糖苷键通过在糖中距C1最远的异头位置和立体中心的相对立体化学来区分。当两个碳具有相同的立体化学时,形成α-糖苷键,而当两个碳具有不同的立体化学时,形成β-糖苷键。
-“聚糖结构”是由糖苷键连接的单糖形成“链”的碳水化合物结构。
-半乳糖(Gal)是一种己醛糖,是葡萄糖的差向异构体。其CAS编号为59-23-4。
-H是指氢原子。
-岩藻糖(Fuc)是一种己糖脱氧糖,也称为6-脱氧-L-半乳糖。其CAS编号为2438-80-4。其可以以α1-3连接键结合(Fucα1-3)或以α1-4连接键结合(Fucα1-4)。
式(I)对应于发明人设计的新型长链唾液酸苷结构,其包含至少一个以β1-4连接键连接的N-乙酰葡糖胺单元,即n大于或等于1。在一些实施方案中,n等于2、3、4、5、6、7、8或更大。在优选的实施方案中,n在1和4之间。
在式(I)中,组分R1被定义为包含至少一个半乳糖(Gal)的聚糖结构。R1由一个或多个连接成一排的单糖的链组成,这定义了R1的“主链”。在该主链上,可以接枝单糖或包含至少两个单糖的次级链。
R1是一种可以化学修饰的聚糖结构,特别是通过在构成聚糖结构的一个或多个单糖上(特别是在主链的单糖上)添加一个或多个甲基基团(“甲基化”)或一个或多个硫酸酯(SO
在本发明的具体实施方案中,R1是主链包含最多五个单糖的聚糖结构。在该实施方案中,R1由一个、两个、三个、四个或五个连接的单糖组成,形成一条主聚糖链。该主聚糖链可以进一步用次级聚糖链接枝,在主链的一个或多个单糖上支化。
优选地,R1由包含一个、两个或三个连接的单糖的主链组成,形成一个主聚糖链。该主聚糖链可以进一步用次级聚糖链接枝,在主链的一个或多个单糖上支化。
在具体实施方案中,R1由包含三个连接的单糖的主链组成。
在另一个具体实施方案中,R1是由三个单糖组成的未接枝主链。
在另一个具体实施方案中,R1是由三个单糖组成的接枝主链。
在另一个具体实施方案中,R1由单个单糖组成,该单糖是半乳糖。
在本发明的另一个具体实施方案中,R1是聚糖结构,其中至少一个半乳糖连接在主链的一个末端处。
在第一构型中,R1在主聚糖链的一个末端包含一个半乳糖。在第二构型中,R1包含两个半乳糖,每个半乳糖连接在主聚糖链的每个末端。
在本发明的具体实施方案中,R1选自以下基团:
-Galβ1-4,
-Galβ1-3,
-Galβ1-4GlcNAcβ1-3Galβ1-4,和
-Galβ1-4(Fucα1-3)GlcNAcβ1-3Galβ1-4。
在本发明的另一个具体实施方案中,R2是H。根据该实施方案,具有如下所示的式(III)的结构是本发明的目的:
Neu5Ac-α2-6-R1-[GlcNAcβ 1-4]n-GlcNAc(式III)
其中:
-n大于或等于1,特别是n在1和4之间;以及
-R1是包含至少一个半乳糖(Gal)的聚糖结构。
下面列出了根据本发明的唾液酸苷结构的非详尽列表:
-Neu5Acα2-6Galβ1-4[GlcNAcβ1-4]n-GlcNAc
-Neu5Acα2-6Galβ1-4(Fuca1-3)[GlcNAcβ1-4]n-GlcNAc
-Neu5Acα2-6Galβ1-3[GlcNAcβ1-4]n-GlcNAc
-Neu5Acα2-6Galβ1-3(Fucα1-4)[GlcNAcβ1-4]n-GlcNAc
-Neu5Ac-α2-6Galβ1-4GlcNAcβ1-3Galβ1-4[GlcNAcβ1-4]n-GlcNAc
-Neu5Ac-α2-6Galβ1-4(Fucα1-3)GIcNAcβ1-3Galβ1-4[GlcNAcβ1-4]n-GlcNAc
-Neu5Ac-α2-6Galβ1-4GlcNAcβ1-3Galβ1-4(Fucα1-3)[GlcNAcβ1-4]n-GlcNAc
-Neu5Ac-α2-6Galβ1-4(Fucα1-3)GlcNAcβ1-3Galβ1-4(Fucα1-3)[GlcNAcβ1-4]n-
GlcNAc
根据本发明的具体实施方案,R1是以β1-4连接键连接的半乳糖(Galβ1-4)。在该实施方案中,合成唾液酸苷具有如下式(II):
Neu5Acα2-6Galβ 1-4GlcNAcβ 1-4GlcNAcβ 1-4GlcNAcβ 1-4GlcNAcβ 1-4GlcNAc
在本申请中,特别是在实施例部分中,式(II)的这种特定唾液酸苷也称为“COV6S”。其开发的结构如图2上部所示。
如实施例所示,已合成并测试了唾液酸苷的其他结构。对应于通式(I)和(III)的这些结构列于下表1中。
表1.实施例部分中所示的唾液酸苷
这些唾液酸苷COIII6S、COIV6S、1-COV6S和COV6S是根据本发明的合成唾液酸苷的代表,其含有壳寡糖末端链。这些新型唾液酸苷可以用于构建多价唾液酸苷化合物,其可以有效结合流感病毒,并用作抗流感感染的药物。
每种唾液酸苷可以对血凝素表现出不同的特异性。事实上,根据其结构,唾液酸苷可以表现出与各种病毒毒株的血凝素结合的特异性。
多价唾液酸苷
本发明还涉及多价唾液酸苷,其包含载体与多个合成唾液酸苷。
在本发明的意义上,“多价唾液酸苷”是指包含多个唾液酸苷的分子结构,与单个唾液酸苷的结合亲和力相比,所述结构显示出与其受体(如血凝素)的结合亲和力增加。
病毒最初附着在宿主细胞上受到多价相互作用的调节,其中多个HA三聚体参与细胞表面糖蛋白和糖脂上表达的多个唾液酸苷配体的相互作用。可溶性的单体唾液酸苷的结合太弱,无法竞争性地破坏这些强的多价相互作用。根据“多价效应”理论,基于抑制病毒/宿主细胞结合的合理药物设计必须包括合成多价唾液酸苷化合物。
在抑制流感病毒的情况下,已经表明“多价唾液酸苷”配体对血凝素具有更强的亲和力,通过阻断HA结合位点从而抑制HA与宿主细胞表面上存在的糖蛋白的相互作用,因此更适合作为拮抗剂。血凝素结合位点的这种饱和过程是基于多价结合,以获得稳定性并防止病毒解离。
已经开发了许多不同的策略来生产各种多价唾液酸苷结构。已经开发出流感病毒粘附的多种合成的多价唾液酸苷抑制剂。它们基于载体,如脂质体(Kingery-Wood等人,1992)、树枝状聚合物(Reuter等人,1999)、合成聚合物(Gambaryan等人,2005)、壳聚糖(Umemura等人,2010)、多糖(Li等人,2011)和金纳米颗粒(Papp等人,2010)。
如本文所用,短语“具有多个唾液酸苷的载体”、“带有多个唾液酸苷的载体”、“接枝在载体上的多个唾液酸苷”、“携带唾液酸苷的载体”和“负载在载体上的多个唾液酸苷”是等同的并且无区别地使用;它们均指根据本发明的多价唾液酸苷,其包含共价或非共价结合至本领域技术人员已知的任何载体的多条唾液酸苷链。
所述多个唾液酸苷通常全部相同;然而,也可以产生与单个载体结合的包含至少两种、三种或四种不同唾液酸苷的多价唾液酸苷。
在本发明的具体实施方案中,载体由脂质体、聚合物、树枝状聚合物或纳米颗粒组成。
脂质体可以用于包封药物、蛋白质、基因和荧光染料分子,用于成像和控制药物释放的应用。携带唾液酸苷的脂质体可以由唾液酸苷磷脂和两亲性前体制备。可以产生不同尺寸的脂质体。
使用聚合物产生唾液酸苷的聚合网络(polymeric network),是多价唾液酸苷的最早已知示例之一。已开发的获得带有唾液酸苷的聚合物的主要方法是:
·使用丙烯酸酯自由基聚合方法组装单体唾液酸苷配体;和
·将唾液酸苷配体偶联到市售的、结构明确的聚合物支架上。
树枝状聚合物是重要的多价支架,其用于以独特的均匀对称形式装饰感兴趣的配体。与聚合物相比,树枝状聚合物提供更可控的单分散性和持久的形状。此外,携带唾液酸苷的树枝状聚合物的3D几何结构非常类似于细胞表面的唾液酸苷基团的装饰。
纳米颗粒是组织良好、坚固的树枝状聚合物,其具有大表面积以及有固有的光学、电化学和磁学特性,有助于对特定相互作用进行传感和成像。文献中已经报道了数种类型的带有唾液酸苷的纳米颗粒,其中最常见的是金、银、二氧化硅、铁、镉、硒和病毒样纳米颗粒。
在本发明的具体实施方案中,载体是聚合物,优选天然聚合物,特别是由聚赖氨酸(ε-聚-L-赖氨酸)组成。
ε-聚-L-赖氨酸(ε-PL)是通过相邻赖氨酸分子的羧基和ε氨基之间的肽键连接的均聚物。它是天然存在的、水溶性的、可生物降解的、可食用的并且对人和环境无毒。ε-PL由25-35个L-赖氨酸残基组成。它是使用白色链霉菌(Streptomyces albulus)通过需氧发酵进行工业生产的。它具有抗菌活性,已在日本和美国被批准作为食品防腐剂。
多价唾液酸苷的结构特征在于其组分(载体和唾液酸苷)以及其接枝率。
接枝率,也称为偶联产率,定义为载体的反应基团被唾液酸苷取代的百分比。
本领域技术人员知道如何调节唾液酸苷在其载体上的接枝率。特别是,接枝率由反应条件控制,尤其是由载体和唾液酸苷的比例量控制,如实施例6所示。
接枝率通常为10%至100%。
在本发明的具体实施方案中,多价唾液酸苷具有20%至100%的接枝率。
优选地,接枝率在30%和100%之间、40%和100%之间、50%和100%之间、60%和100%之间、70%和100%之间、80%和100%之间或在90%和100%之间。
药物组合物
本发明还涉及药物组合物,其包含在药学上可接受的载剂中的至少一种如上所述的多价唾液酸苷。
本发明还涉及兽用组合物,其包含在药学上可接受的载剂中的至少一种如上所述的多价唾液酸苷。有利地,所述多价唾液酸苷被设计为用于抑制具有动物宿主细胞的病毒。
该药物组合物或兽医组合物包含有效量的根据本发明的至少一种多价唾液酸苷。
出于本发明的目的,“有效量”是指足以在被感染的生物体内抑制病毒增殖和/或复制和/或发生病毒感染的多价唾液酸苷的量。例如,可以通过测量病毒复制来对这种抑制进行定量。
例如,在体外,所谓的“有效”量在1μm和1mM之间,如实施例9至11所示。
根据本发明的药物组合物或兽医组合物可以包含一种、两种、三种、四种、五种或更多种多价唾液酸苷。有利地,组合物中的每种多价唾液酸苷对特定病毒毒株表现出独特的特异性。
根据本发明,术语“药学上可接受的载剂”是指向个体或动物施用时不伴有显著有害作用的一种或多种药学上可接受的载剂或赋形剂,并且其是本领域技术人员公知的。
根据本发明的药物组合物或兽医组合物适用于口服、舌下、吸入、皮下、肌肉内、静脉内、经皮、经眼或直肠施用。
根据本发明的优选的方面,所述药物组合物的特征在于其是适合通过吸入施用的盖伦(galenic)形式。
吸入是指通过呼吸道吸收。这特别是一种吸收治疗目的的化合物、吸收气体、悬浮剂中的微滴或粉末形式的某些物质的方法。
通过吸入(即通过经鼻和/或口服途径)施用药物组合物是技术人员公知的。
有两种类型的通过吸入施用:
-当组合物为粉末形式时,通过吹入施用,和
-当组合物为气溶胶(悬浮剂)形式或溶液(例如水溶液)形式时,在压力下通过雾化施用。然后将推荐使用雾化器或喷雾器来施用药物组合物或兽医组合物。
因此,此处考虑的盖伦形式选自:粉末、液滴的水性悬浮剂和加压溶液。优选通过滴鼻施用。
医疗装置
本发明还涉及包含至少一种如上所述的多价唾液酸苷的医疗装置。
在实施方案中,所述医疗装置由包被有至少一种本发明的多价唾液酸苷的载体组成。
在具体的实施方案中,载体包被有多种(至少两种)多价唾液酸苷,每种多价唾液酸苷对不同病毒毒株表面表达的血凝素具有特异性亲和力。
例如,载体包被有三种多价唾液酸苷,一种特异于H1N1流感病毒毒株,第二种特异于H2N2流感病毒毒株,第三种特异于H3N2流感病毒毒株。
所述载体可以是例如呼吸防护面罩或将插入面罩中的过滤器。随着重大流行病的出现,公众对防护口罩的需求越来越大。所述载体还可以是手套或任何其他个人防护装置。
所述载体包被有,例如由接枝了根据本发明的多个唾液酸苷的聚合物组成的多价唾液酸苷。每种唾液酸苷可以对不同的血凝素表现出不同的特异性。
有利地,该医疗装置可以用于捕获并保持(hold)至少一种对唾液酸具有亲和力的病毒,特别是人流感病毒或多种人流感病毒毒株。
在本发明的另一个实施方案中,医疗装置是鼻喷雾剂,其包含作为注射溶液的溶液,所述溶液包含至少一种根据本发明的多价唾液酸苷。
药物和治疗用途
本发明还涉及所述多价唾液酸苷、或含有至少一种多价唾液酸苷的药物组合物用作药物的用途。
本发明还涉及所述多价唾液酸苷、或含有至少一种多价唾液酸苷的药物组合物用于预防和/或治疗感染的用途,所述感染由对唾液酸具有亲和力的病毒引起。
本发明还涉及所述的多价唾液酸苷、或含有至少一种多价唾液酸苷的药物组合物用于预防病毒从一个宿主生物体传播至另一个宿主生物体的用途。
短语“对唾液酸具有亲和力的病毒”是指任何病毒,其具有致病作用,即引起活生物体中的疾病,表达唾液酸的受体,并因此能够结合唾液酸缀合物。
Matrosovich M等人(2015)的综述以非详尽的方式介绍了此类病毒。特别是,对唾液酸具有亲和力的此类病毒选自正粘病毒科(包括流感病毒)、冠状病毒科、副粘病毒科、杯状病毒科、小核糖核酸病毒科、呼肠孤病毒科、多瘤病毒科、腺病毒科和细小病毒科。
在本发明的具体实施方案中,该病毒是流感病毒,特别是人、马、猪或禽流感病毒。
流感病毒是导致流感疾病的原因。它们分为三种类型:甲型、乙型和丙型。
流感病毒也由其表面上蛋白质的类型来定义。根据甲型流感病毒表面上表达的血凝素(HA)和神经氨酸酶(NA)糖蛋白的性质,甲型流感病毒有多种亚型:在流行的甲型流感病毒中已鉴定出16种HA和9种NA类型。
在人中,最常见的甲型流感病毒是H1N1、H2N2和H3N2亚型,偶尔出现禽流感病毒H5N1、H7N7、H7N9、H5N2和H9N2的种间传播,尤其是由动物传播给人。
根据具体实施方案,流感病毒是甲型人病毒,其选自H1N1、H2N2、H3N2、H5N1、H7N7、H7N9、H5N2和H9N2亚型。
在本发明的另一个具体实施方案中,流感病毒是人流感病毒,并且选自H1N1、H3N2和乙型毒株。
在本发明的意义上,术语“流感病毒”包括流感病毒的所有类型(甲型或乙型)和亚型(H1N1、N3N2等)。该术语包括野生型流感毒株和突变的流感毒株,特别是具有导致所述毒株对抗病毒化合物具有耐药性的基因突变的毒株。
根据本发明的组合物特别是旨在用于预防流感病毒感染的用途。
有利地,这些组合物旨在用于预防流感突变毒株感染的用途,所述流感突变毒株对常用抗病毒化合物表现出耐药性,如奥司他韦耐药毒株和巴洛沙韦耐药毒株。实施例13详细介绍了这一方面。
术语“预防”是指预防或至少降低人或动物生物体中出现被至少一种流感病毒感染的可能性的事实。目的是用本发明的多价唾液酸苷使病毒表面上存在的血凝素的抗原位点饱和,从而防止病毒附着至宿主细胞。
根据本发明的组合物还可以旨在用于治疗流感病毒感染的用途。
术语“治疗”是指抵抗人或动物生物体中的至少一种流感病毒感染的事实。通过施用至少一种根据本发明的组合物,生物体中的病毒感染水平将逐渐降低,然后完全消失。术语“治疗”还指缓解与病毒感染相关的症状(发烧、疲劳等)的事实。
本发明还涉及包含至少一种多价唾液酸苷和至少一种其他抗病毒化合物的组合产品用于同时、单独或顺序用于预防和/或治疗流感病毒的病毒感染的用途。
在优选的方面,抗病毒化合物选自经典使用的预防或治疗流感的抗病毒化合物。此类抗病毒化合物可商购,并在参考书(如Vidal)中进行了描述。例如,奥司他韦和玛巴洛沙韦(Baloxavir marboxil)是抗病毒化合物,并可以与本发明的多价唾液酸苷组合使用。
本发明还涉及预防或治疗人的感染的方法,所述感染由对唾液酸具有亲和力的病毒引起,所述方法包括向所述人施用有效量的至少一种根据本发明的多价唾液酸苷。
实施例
尽管本文已经参考具体实施方案描述了本发明,但是将理解的是,这些实施方案仅是说明本发明的原理和应用。因此,将理解的是,在不脱离由所附权利要求限定的本发明的精神和范围的情况下,可以对说明性实施方案进行多种修改,也可以设计其他安排。
实施例1.COV6S唾液酸苷的生产
由于数种原因,壳寡糖是聚乳糖胺链的有意义替代品。首先,聚乳糖胺和壳寡糖都是通过β糖苷键连接的己糖长链。其次,两种结构都含有N-乙酰葡糖胺作为主要成分。第三,通过表达壳寡糖合酶nodC的代谢工程改造的大肠杆菌可以高产地生产壳寡糖(Samain等人,1997)。
此外,Bettler等人(1999)表明,壳寡糖(如壳五糖[10])可以被β1-4半乳糖基转移酶用作受体,以形成在其非还原端含有末端LacNAc基序的六糖[11],如图1所示。
然后,通过在如下所述获得的代谢工程化的大肠杆菌菌株SCH1中额外表达α2-6唾液酸转移酶,可以将[11]唾液酸化以形成唾液酸化七糖COV6S[12]。
菌株SCH1是通过用四种质粒pBSnodC、pBBR3-SS、pWKS-lgtB和pSU6ST转化宿主菌株ZLKA而构建的。
a)Fierfort和Samain(2008)描述了宿主菌株ZLKA的构建。
菌株ZLKA是大肠杆菌K12菌株DH1(endA1 recA1 gyrA96 thi-1glnV44relA1hsdR17)的衍生物,其获自德国微生物保藏中心(Deutsche Sammlung vonMikroorganismen)(参考号DSM 4235),在lacZ、lacA、nanA和nanK基因中携带额外的无效突变。
b)pBSnodC质粒携带来自茎瘤固氮根瘤菌(Azorhizobium caulinaudans)的壳寡糖合酶的nodC基因,如Cottaz和Samain(2005)所述构建。
c)Fierfort和Samain(2008)描述了pBBR3-SS质粒的构建。它携带来自空肠弯曲杆菌(Campylobacter jejuni)菌株ATCC 4343的三个基因neuC、neuB和neuA,分别编码N-乙酰葡糖胺-6-P差向异构酶、唾液酸合酶和CMP-NeuA合酶。
d)携带来自脑膜炎奈瑟菌(Neisseria meningitidis)的半乳糖基转移酶lgtB的pWKS-lgtB质粒的构建。该质粒如Cottaz和Samain(2005)所述构建。
e)携带来自发光杆菌属(Photobacterium sp.)JT-ISH-224的α2-6NeuAc转移酶的pSU6ST质粒的构建。该质粒如Richard等人(2017)所述。
该菌株SCH1如先前所述以高细胞密度生长(Priem等人,2002):在含有1.5升矿物培养介质的3升反应器中进行培养,温度维持在34℃,用14% NH4OH将pH调节至6.8。
高细胞密度培养由三个期组成:指数生长期,从发酵罐接种开始一直持续到碳底物(甘油17.5g.L-1)耗尽,5小时的补料分批(5g.L-1h-1的高甘油补料速率)和20小时的补料分批期(3g.L-1h-1的甘油补料速率)。
发酵阶段结束时,通过离心(7000g,30分钟)回收细菌细胞。将细胞沉淀重悬于蒸馏水中,并通过在100℃高压灭菌50分钟来透化细胞。将混合物离心(7000g,30分钟),并回收含有寡糖的上清液。
通过添加强阳离子交换树脂(AmberliteIR120 H+形式),将细胞外级分的pH降至3.0。这导致蛋白质沉淀,通过离心将其除去。然后,通过添加弱阴离子交换剂(Dowex 66游离碱形式)将澄清上清液的pH调节至6.0,倾析后,将上清液加载到Dowex 1(HCO3形式)色谱柱(5×20cm)上。用蒸馏水洗涤后,保留在Dowex1树脂上的酸性寡糖用0-500mM连续(NH4)
合并含有COV6S的洗脱级分,并通过冷冻干燥除去(NH
有趣的事实是,GlcNAC不是α2-6唾液酸转移酶的底物,这可以防止副产物的形成;因此,COV6S(图1中的产物[12])几乎是唯一的终产物。
COV6S的结构通过核磁共振(NMR)和质谱确认。
实施例2.1型COV6S唾液酸苷的生产
1型COV6S唾液酸苷(1-COV6S)具有下式:
式(IV)Neu5Acα2-6Galβ1-3GlcNAcβ1-4GlcNAcβ1-4GlcNAcβ1-4GlcNAcβ1-4GlcNAc
该式对应于通式(I),其中R1=Galβ1-3,R2=H且n=4。
它是通过菌株SCH11的发酵产生的,如实施例1中针对菌株SCH1所述。
菌株SCH11与SCH1相似,但没有含有b1-3半乳糖基转移酶基因的质粒pWKS-lgtB,并且pBBR3-SS被含有额外的β1-3半乳糖基转移酶基因的pBBR3-SS-β-3GalT替换。
质粒pBBR3的构建如下:使用幽门螺杆菌(Helicobacter pylori)ATCC43504的基因组DNA作为模板,通过PCR扩增含有β1-3半乳糖基转移酶基因序列的1.34kb的DNA。
SalI位点添加到左侧引物中:
5’GGTCGACGGTAAGGAGATATACATATGATTTCTGTTTATATCATTTCT TTAAAAG(SEQ IDNO.1)
以及将PstI位点添加到右侧引物中:
5’CTGCAGTTAAACCTCTTTAGGGGTTTTTAAAGG(SEQ ID NO.2)。
扩增的片段首先克隆到pCR4Blunt-TOPO载体中,然后亚克隆到pBBR3-SS质粒的XhoI和PstI位点中,以形成pBBR3-SS-β-3GalT。
实施例3.COIV6S唾液酸苷的产生
COIV6S唾液酸苷具有下式:
式(V)Neu5Acα2-6Galβ1-4GlcNAcβ1-4GlcNAcβ1-4GlcNAcβ1-4GlcNAc
该式对应于通式(I),其中R1=Galβ1-4,R2=H且n=3。
它是通过菌株SCH9的发酵产生的,如实施例1中针对菌株SCH1所述。
菌株SCH9与SCH1相似,不同之处在于编码来自茎瘤固氮根瘤菌(Azorhizobiumcaulinaudans)的壳寡糖合酶的质粒pBS-nodC被质粒pUC-nodC替换,pUC-nodC编码来自苜蓿中华根瘤菌(Sinorhizobium meliloti)的壳寡糖合酶,由Samain等人(1997)描述。
实施例4.COII6S和COIII6S唾液酸苷的生产
COII6S和COIII6S唾液酸苷具有下式:
式(VI)Neu5Acα2-6Galβ1-4GlcNAcβ1-4GlcNAc
式(VII)Neu5Acα2-6Galβ1-4GlcNAcβ1-4GlcNAcβ1-4GlcNAc
COII6S和COIII6S的式均对应于通式(I),其中分别是R1=Galβ1-4,R2=H且n=1或2。
如实施例1所述,通过发酵同时生产COII6S和COIII6S唾液酸苷,不同之处在于菌株SCH1被菌株SCH10替换。在Dowex1上进行纯化步骤后,在HW40色谱柱上通过尺寸排阻色谱法分离两种唾液酸苷。
菌株SCH10与菌株SCH1类似,不同之处在于质粒pWKS-lgtB被质粒pWKS-lgtB-ChiA替换,pWKS-lgtB-ChiA含有来自环状芽孢杆菌(Bacillus circulans)WL12的额外chiA基因。chiA基因先前已被证明编码一种壳多糖酶,其将壳五糖切割为壳二糖,同时瞬时积累壳三糖(Cottaz和Samain,2005)。
质粒pWKS-lgtB-ChiA的构建如下:从pBBR1-ChiA(Cottaz和Samain,2007)中通过KpnI notI消化切除含有chiA基因的1.3kb的DNA片段,在XbaI/kpnI接头存在下,将其克隆到pWKS-lgtB的XbaI notI位点中。
实施例5.LSTc唾液酸苷的生产(本发明中不包含的唾液酸苷)
LSTc是一种在人乳中发现的天然唾液酸苷,并用作参考化合物。其结构为:
Neu5Acα2-6Galβ1-4GlcNAcβ1-3Galβ1-4Glc
其可以如实施例1中所述通过发酵来生产,不同之处在于菌株SCH1被菌株LST1替换,并且在指数生长期结束时添加乳糖(5g/l)。菌株LST1与菌株SCH14相似,不同之处在于它不含pBS-nodC质粒。
实施例6.多价唾液酸苷的合成
使用基于天然聚合物(ε-聚-L-赖氨酸)的胺基团的还原性胺化的简单方法,连接实施例1至5中获得的不同唾液酸苷的末端还原端。
接枝率定义为ε-聚-L-赖氨酸的胺基团被唾液酸苷取代的百分比,可以通过反应条件来控制。如下表2所示,其变化范围为20%至100%。
下面介绍了获得ER15的简短实验方案:
在1.5ml微型管中,将0.4mmol的唾液酸苷(每胺1当量)溶解在500μl的去离子水中。需要超声处理以实现完美溶解(溶液1)。在另一个1.5ml微型管中,将50mg聚赖氨酸溶解在100μl的去离子水中(溶液2)。将溶液1和溶液2两者混合,并添加100μl的硼酸盐缓冲液(500mM,pH 8.5)。
在通风橱下,在1.5ml微型管中,将123.6mg的NaBH
将反应混合物(1ml)转移至15ml离心管中。用1ml去离子水洗涤微型管,然后将该去离子水转移至该15ml管中(总体积2ml)。添加4ml的96%乙醇(2体积)后,将管以9000rpm离心15分钟。除去上清液(约6ml),并用2ml的1%NaCl溶液溶解沉淀。添加4ml的96%乙醇并第二次离心(15分钟,9000rpm,4℃)后,将沉淀物收集在5ml去离子水中,并在制备型HW40立体色谱柱(5×90cm)上纯化。使用100mM碳酸铵以2ml/分钟的流速进行洗脱。
纯化后,合并含有产物的试管。将产物蒸发至干以除去碳酸铵,然后加入3ml的去离子冻干水。
为了获得化合物ER59、60和61,修改了寡糖当量数:分别为每胺0.75当量、每胺0.5当量和每胺0.25当量。
偶联率通过核磁共振(NMR)1H和凝胶渗透色谱(GPC)与多角度光散射检测(MALS)测定。
表2.具有ε-聚-L-赖氨酸骨架的多价唾液酸苷
实施例7.血凝抑制测定(HI或HAI)
HI技术在于将递减剂量的多价唾液酸苷(可能的病毒抑制剂)与固定量的病毒一起孵育。如果存在识别和结合,则病毒将自身附着在抑制剂上。
然后添加红细胞并孵育混合物。游离病毒将能够识别红细胞表面上存在的唾液酸,从而形成网络(红纱,代表血凝过程),而附着在多价唾液酸苷上的病毒将不能与红细胞结合,红细胞将沉积在孔底部,形成沉淀物(代表血凝抑制)。
血凝滴度对应于观察到血凝抑制的最高稀释度的倒数。稀释以比率为2进行,因此滴度<2意味着从第一次稀释到1/2时,就没有观察到血凝抑制,而例如滴度为4096意味着直到1/4096稀释时都存在血凝抑制。
图3显示了HI技术的概括方案。
对于这一HI测定,使用的病毒毒株是:A/California/7/2009H1N1、A/PortoRico/8/34H1N1、A/Texas/50/12H3N2、A/Moscow/10/99H3N2、B/Massachusetts/2/2012和B/Brisbane/60/08,剂量为每孔4HAU。使用的红细胞是0.5%的母鸡红细胞和0.8%的豚鼠红细胞。
下表3显示了用表2中列出的多价唾液酸苷获得的结果。ER10和ER13是不包括在式(I)中的多价唾液酸苷,因此不属于本发明。
表3.九种多价唾液酸苷的HI测定结果,其结构示于表2中
*C:来自鸡的红细胞;GP:来自豚鼠的红细胞。
显著结果以粗体显示。
表3中显示的结果表明,就H1N1病毒A/California/7/09和A/PR/8/34的HI而言,ER15是表现最佳的化合物,滴度分别为128和4096,而ER10或ER13化合物显示的滴度为2和16(ER10)以及<2和2(ER13)。
这些实验强调了GlcNAc基序的存在对于其HI特性的重要性,特别是ER15化合物的重要性。并非所有化合物都具有相同的血凝抑制活性,甚至没有活性,取决于病毒类型(甲型或乙型)和/或甲型病毒亚型。
实施例8.微中和测定
微中和测定允许对抗病毒化合物对传染性病毒的抑制潜力进行定量。该技术的示意图如图4所示。
该测定是在流感病毒的参考细胞系统上进行的:Madin-Darby犬肾细胞(MDCK),其在表面具有通过α2.3键或通过α2.6键与半乳糖结合的唾液酸。本次测试中使用的病毒毒株为A/California/7/2009H1N1,校准剂量为50μl中100TCID50。
该技术包括将递减剂量的测试化合物与固定量的病毒一起孵育1小时。如果被识别,病毒将与化合物结合。然后,将该病毒/化合物混合物添加至MDCK细胞,孵育72小时。未结合的病毒将能够识别细胞表面上存在的唾液酸,从而感染细胞(细胞病变效应的可视化),而与唾液酸化的化合物结合的病毒将无法自身附着至细胞,从而将不感染它们(“中和”感染)。
微中和滴度是最高稀释度的倒数,在该最高稀释度下,4个孔中至少有2个孔中完全没有观察到细胞病变效应。从1:5开始,以2的比率进行稀释,因此滴度<5意味着第一次以1:5稀释后没有中和感染,而例如,滴度160意味着稀释度高达1:160也中和了感染。
表4.微中和测定结果
ER15多价唾液酸苷对A/California/7/2009H1N1病毒毒株具有最佳中和活性。
实施例9.本发明的多价唾液酸苷在人细胞系A549上的测定
该测定的目的是评价ER15作为抗病毒剂以不同浓度和多种病毒剂量(剂量效应)、在不同处理条件(治疗/感染后、预防/感染前处理)下在人呼吸道细胞系A549(来自人肺癌)的性能。
在37℃和5% CO2的培养箱中,在DMEM+SVF 10%+L-谷氨酰胺2mM+青霉素(100U/ml)+链霉素(100μg/ml)中培养A549细胞。为进行感染和化合物评价,将细胞接种在12孔板中的DMEM+L-谷氨酰胺2mM+青霉素(100U/ml)+链霉素(100μg/ml)+牛胰蛋白酶(T6763Sigma)(0.5μg/ml)中。
A/California/7/2009H1N1病毒毒株以两种不同的感染复数(MOI):0.1和0.01使用。该毒株具有D225G突变的血凝素,使其与其细胞受体具有更大的亲和力。
另一种病毒毒株A/Lyon/969/09H1N1以三种不同的MOI:1;0.1和0.01使用。该菌株不具有D225G突变,并且对细胞受体的α2-6唾液酸具有较低的亲和力。
将冻干的化合物与水以0.1mM混合,在0.22μM上过滤并储存在-20℃。用作阴性对照的化合物是仅有聚赖氨酸(PL)。
A549细胞处理和感染后,在感染后D1、D2和D3采集上清液样本,通过以下对病毒生产进行定量:(i)在MDCK细胞上每毫升中感染颗粒的数量的滴定(TCID50/ml)和(ii)病毒基因组的定量(RT-qPCR)。
A-ER15与仅有聚赖氨酸(PL)针对H1N1毒株的治疗特性
通过三次感染后处理(在D0(感染后1小时)、D1和D2)测定ER15化合物的潜在治疗效果。在37℃在细胞中孵育A/California/7/09H1N1病毒1小时,然后除去。
添加含有ER15的介质,与未处理条件相比,ER15以5种不同的浓度(20;10;5;2.5和1.25μM)存在(NT:未处理;PL:递送递增量的聚赖氨酸)。在3个不同时间采集细胞上清液的数个样本,以进行病毒定量。
获得的结果如图5所示,表明在感染后1、24和48小时施用ER15处理使得A/California/7/09H1N1滴度降低5至6log。当病毒接种物的MOI为0.1时,观察到剂量依赖性效应取决于处理浓度。当MOI为0.01时,所有测试的ER15浓度(20;10;5;2.5和1.25μM)均允许在感染后24小时即最大程度地降低病毒载量。
RT-qPCR获得的结果是一致的,并表明显著抑制感染。
B-ER15与仅有聚赖氨酸(PL)针对H3N2毒株的治疗特性
实验条件与(A)相同,不同之处在于在37℃在细胞中孵育A/Texas/50/2012H3N2病毒1小时,然后除去。
获得的结果如图6所示,表明:
·在感染后1、24和48小时施用ER15处理,剂量为20μM,对于测试的2个MOI(即0.1和0.01),观察到感染滴度的显著降低(大约3log)。
·浓度为1.25至10μM的ER15处理不会显著降低该H3N2毒株的感染性病毒载量。
这些结果与HIA中获得的结果一致,其中滴度<2。
因此,观察到以剂量依赖性方式的针对H3N2毒株的显著效果。
在RT-qPCR中获得的结果是一致的,也显示仅20μM的处理显著降低了病毒载量。
总体而言,在A549细胞中的HIA和疗效结果显示,与A/California/7/09H1N1相比,ER15针对A/Texas/50/2012H3N2毒株的亲和力/性能较低。
C-ER15与仅有聚赖氨酸(PL)针对A/California/7/09H1N1毒株的治疗特性,感染后仅施用一次
实验条件与(A)相同,不同之处在于ER15仅在D0感染后1小时通过添加到介质中向被感染的细胞施用一次。测试了3种不同的浓度:20;5和1.25μM。
获得的结果如图7所示,表明无论起始MOI是多少(0.1或0.01),感染后1小时施用的ER15处理都足以大幅降低感染性病毒载量。在感染后24小时观察到至少2log的减少,在感染后72小时观察到多至超过5log的减少。因此,单次施用(感染后1小时)的效果是持久的,在MOI为0.1时观察到剂量效应。
D-ER15与仅有聚赖氨酸(PL)针对A/California/7/09H1N1毒株的治疗特性,仅在病毒感染时伴随施用一次
实验条件与(A)相同,不同之处在于ER15仅在D0通过添加到介质中与病毒一起向细胞施用一次。测试了3种不同的浓度:20;5和1.25μM。
获得的结果如图8所示,表明与病毒同时(在感染步骤期间)施用的ER15处理允许显著降低感染性病毒载量(多至3log),特别是在早期阶段(感染后24小时和48小时)。在MOI为0.01的条件下,效果会随着时间的推移而保持(在4和5log之间)。观察到ER15浓度的剂量依赖性效应以及MOI的剂量依赖性效应。
E-ER15与仅有聚赖氨酸(PL)针对A/California/7/09H1N1毒株的预防特性,
此处的目的是评价ER15化合物在感染前一天以3种不同的浓度(20;5和1.25μM)进行的单次处理的预防效果。
在感染时,将A/California/7/09H1N1病毒直接添加到已含有化合物的介质中。孵育5小时后,除去含有病毒和化合物的介质,更换为新鲜介质。
在D1、D2和D3采集样本,进行感染性病毒定量。
获得的结果如图9所示,表明在感染前24小时,使用ER15进行单次预防处理,随着时间的推移,显著降低了感染性病毒载量:感染后24小时降低在2和3log之间,以及降低多至5log,取决于MOI。效果会随着时间的推移而保持(在4和5log之间)。可以清楚地观察到ER15浓度和MOI的剂量依赖性效应。
F-ER15与仅有聚赖氨酸(PL)针对H1N1 A/Lyon/969/09毒株的治疗特性
实验条件与(A)相同,不同之处在于在37℃在细胞中孵育H1N1A/Lyon/969/09病毒毒株1小时,然后除去。
获得的结果如图10所示,表明ER15显著降低了A/Lyon/969/09H1N1毒株的感染性病毒载量。正如预期的那样,Lyon毒株的结果不如具有D225G突变的California毒株的结果好。
实施例10.在基于鼻源性重建人呼吸道上皮的体外生理模型中对ER15化合物的评价
上皮细胞生长在置于空气/液体界面处的24孔板中的插入物上。因此,构成基极的细胞与培养介质接触,而构成顶极的细胞与空气接触。培养介质是Mucilair(Epithelix),上皮细胞源自来自供体池的鼻拭子(Epithelix)。它们在37℃、5%CO2的培养箱中培养。
A/California/7/2009H1N1毒株的感染发生在顶级,在150μl的OptiMEM介质中,感染复数为0.1,在37℃和5% CO2进行1小时。然后,除去病毒悬浮液并进行洗涤。小心地除去顶极处的全部介质后,在感染后1小时以3种不同的浓度(50;20和5μM)在10μl中进行处理。
将冻干的化合物ER15与以下混合:
-水,
-磷酸盐缓冲液(2g/L甘油,9g/L NaCl,10mM,pH=7.9)和
-柠檬酸盐缓冲液(2g/L甘油,9g/L NaCl,10mM,pH=6.0)
浓度为0.1mM。
阴性对照是在水中稀释至0.1mM的聚赖氨酸(PL)。
一些细胞是未处理的(NT)和/或模拟的(未感染)。
每天进行经上皮细胞电阻(TEER)测量,以监测上皮的完整性。在D1、D2和D3在顶极收集样本,以通过每毫升感染性颗粒的数量的滴定(TCID50/ml-图11C)来对病毒产量进行定量,并通过RT-qPCR对病毒基因组进行定量(图11D)。
平行地,在相同处理条件下在模拟感染条件下评价ER15的细胞毒性。此处病毒接种物体积替换为OptiMEM介质。每天在基础介质中采集样本以对乳酸脱氢酶(LDH,在细胞毒性的情况下释放)的释放进行定量。获得的结果如图11B所示,表明ER15化合物在多达至少50μM时在上皮细胞上没有细胞毒性。
此外,50μM的ER15显著降低了上皮细胞中的感染性病毒载量(图11C和图11D)。与溶剂水相比,磷酸盐缓冲液和柠檬酸盐缓冲液似乎允许20μM的ER15具有更好的疗效。
这些结果在RT-qPCR中得到证实,如图11D所示。TEER测量值也与总体结果一致。
实施例11.ER61与仅有聚赖氨酸(PL)针对A/California/7/09H1N1毒株的治疗特性
通过三次感染后处理(在D0(感染后1小时)、D1和D2)测定ER61化合物的潜在治疗效果。在37℃在A549细胞上孵育A/California/7/09H1N1病毒1小时,感染复数为0.1或0.01,然后除去。
实验条件与实施例9(A)相同,不同之处在于使用ER61化合物。
与未处理条件相比,ER61以5种不同的浓度(20;10;5;2.5和1.25μM)测试(NT:未处理;PL:递送递增量的聚赖氨酸)。在3个不同时间采集细胞上清液的数个样本,以进行病毒定量。
获得的结果如图12所示,表明在感染后1、24和48小时施用ER61处理使得A/California/7/09H1N1滴度降低多至5log。当病毒接种物的MOI为0.1时,观察到剂量依赖性效应取决于处理浓度。当MOI为0.01时,所有测试的ER61浓度(20;10;5;2.5和1.25μM)均允许在感染后24小时即最大程度地降低病毒载量。
实施例12.ER15和ER61化合物针对最近毒株的治疗特性
进入疫苗组合物中的测试的“最近”毒株如下:A/Michigan/45/2015H1N1;A/Kansas/14/2017H3N2;和B/Phuket/3073/2013。
通过三次感染后处理(在D0(感染后1小时)、D1和D2)测定ER15和ER61化合物的潜在治疗效果。在37℃在A549细胞上孵育病毒1小时,感染复数为0.1,然后除去。
与未处理条件相比,在3种不同的浓度(20;5;和1.25μM)测试多价唾液酸苷ER15和ER61(NT:未处理)。在3个不同时间(感染后24、48和72小时)采集细胞上清液的数个样本用于病毒定量。
获得的结果如图13A、图13B和图13C所示,表明在感染后1、24和48小时施用ER15和ER61处理是等效的。观察到剂量依赖性效应取决于处理浓度。在浓度为20μM时,化合物ER15和ER61对三种病毒毒株均表现出显著的抗病毒效果。
实施例13.ER15化合物对重组的奥司他韦和巴洛沙韦耐药的H1N1病毒的治疗效果
使用A/Lyon/969/09H1N1遗传主链通过反向遗传生成三种重组H1N1病毒,其中所述遗传主链包含神经氨酸酶中的H275Y突变(赋予对奥司他韦的耐药性)或聚合酶PA亚基中的I38T突变(赋予对巴洛沙韦的耐药性),或两者(H275Y+I38T)。通过三次感染后处理(在D0(感染后1小时)、D1和D2)测定ER15化合物的潜在治疗效果。在37℃在A549细胞上孵育重组的耐药H1N1病毒1小时,感染复数为0.1,然后通过洗涤除去。实验条件与实施例9(A)相同,不同之处在于使用病毒。与未处理条件相比,ER15以5种不同的浓度(20;10;5;2.5和1.25μM)测试(NT:未处理;PL:递送递增量的聚赖氨酸)。在3个不同时间采集细胞上清液的数个样本用于病毒定量。
获得的结果如图14A所示,表明在感染后1、24和48小时施用ER15处理使得H1N1I38T滴度降低多至2log。观察到剂量依赖性效应取决于处理浓度。用H1N1 H275Y和双耐药H1N1(I28T+H275Y)病毒获得相似结果,分别如图14B和图14C所示。
参考文献
专利文献
WO 2007/101862
非专利文献
Matrosovich M,Klenk HD.Natural and synthetic sialic acid-containinginhibitors ofinfluenza virus receptor binding.Rev Med Virol.2003Mar-Apr;13(2):85-97.
Nycholat CM,McBride R,Ekiert DC,Xu R,Rangarajan J,Peng W,Razi N,Gilbert M,Wakarchuk W,Wilson IA,Pautson JC.Recognition ofsialylated poly-N-acetyllactosaminechains onN-and O-linked glycans by human and avianinfluenzaAvirus hemagglutinins.Angew Chem Int Ed Engl.2012May 14;51(20):4860-3.
Yang H,Carney PJ,Chang JC,Guo Z,Villanueva JM,Stevens J.Structure andreceptorbinding preferences Ofrecombinant human A(H3N2)virushemAgglutinins.Virology.2015Mar;477∶18-31.
Bych K,
JillE.Kingery-Wo0d,KeVin W.Williams,George B.Sigal,and GeorgeM.Whitesides.Theagglutination oferythrocytes by influenza virusis stronglyinhibited by liposomes incorporating ananalog ofsialyl gangliosides.Journalof the American ChemicalSociety1992114(18),7303-7305
Reuter JD,Myc A,Hayes MM,Gan Z,Roy R,Qin D,Yin R,Piehler LT,Esfand R,TomaliaDA,Baker JR Jr.lnhibitionof viral adhesionand infectionby sialic-acid-conjugateddendriticpolymers.Bioconjug Chem.1999Mar-Apr;10(2):271-8.
Gambaryan AS,Boravleva EY,Matrosovich TY,Matrosovich MN,Klenk HD,Moiseeva EV,Tuzikov AB,Chinarev AA,Pazynina GV,BoVin NV.Polymer-bound 6′sialyl-N-acetyllactosamine protects mice infected by influenzavirus.AntiviralRes.2005Dec;68(3):116-23.
Myco Umemura,Yutaka Makimura,Masae Itoh,Takeshi Yamamoto,ToshikiMine,SeijiMitani,Ichiro Simizu,Hisashi Ashida,Kenji Yamamoto.One-stepsynthesis ofefficientbinding-inhibitorforinfluenzayirus through multipleadditionof sialyloligosaccharideson chitosan.Carbohydrate Polymers,Volume 81,Issue 2,2010,Pages 330-334.
Li X,Wu P,Gao GF,Cheng S.Carbohydrate-functionalized chitosanfiberforinfluenzayirus capture.Biomacromolecules.2011Nov 14;12(11):3962-9.doi:10.1021/bm200970x.Epub 2011Oct 13.PMID:21978096.
Papp l,Sieben C,Ludwig K,Roskamp M,
Matrosovich M,Herrler G,Klenk HD.SialicAcidReceptorsofViruses.TopCurr Chem.2015;367:1-28
Samain E,Drouillard S,Heyraud A,Driguez H,Geremia RA.Gram-scalesynthesis ofrecombinantchitooligosaccharides inEscherichiacoli.CarbohydrRes.1997Jul 11;302(1-2):35-42.
Bettler E,Samain E,Chazalet V,Bosso C,Heyraud A,Joziasse DH,WakarchukWW,Imberty A,Geremia AR.The livingfactory:in viyo production ofN-acetyllactosaminecontaining carbohydrates inE.coli.Glycoconj J.1999Mar;16(3):205-12.
Cottaz S,Samain E.Geneticengineering ofEscherichiacolifor theproduction ofNl,NlI-diacetylchitobiose (chitinbiose)and itsutilizationas aprimerforthe synthesis ofcomplex carbohydrates.Metab Eng.2005Jul;7(4):311-7.
Fierfort N,Samain E.Geneticengineering ofEscherichiacolifortheeconomicalproduction ofsialylatedoligosaccharides.J Biotechnol.2008Apr 30;134(3-4):261-5.Richard E,Pifferi C,Fiore M,Samain E,Le
P riem B,Gilbert M,Wakarchuk WW,Heyraud A,Samain E.Anewferrnentationprocessallows large-scAle production of humarn milk oligosAccharides bymetabolicallyengineered bacteria.Glycobiology.2002Apr;12(4):235-40.
序列表
<110>国家科学研究中心
里昂高等师范学院
国家健康科学研究所
里昂第一大学
<120>新型唾液酸苷及其治疗用途
<130>B380778PCT D41145
<150>EP21305131.1
<151>2020-01-29
<160>2
<170>PatentIn version 3.5
<210>1
<211>55
<212>DNA
<213>人工序列
<220>
<223>包含SalI位点的寡核苷酸
<400>1
ggtcgacggt aaggagatat acatatgatt tctgtttata tcatttcttt aaaag 55
<210>2
<211>33
<212>DNA
<213>人工序列
<220>
<223>包含PstI位点的寡核苷酸
<400>2
ctgcagttaa acctctttag gggtttttaa agg 33