经基因组编辑的细胞的制造方法
文献发布时间:2023-06-19 11:32:36
技术领域
本发明涉及经基因组编辑的细胞的制造方法,主要涉及通过同源染色体之间的同源重组,从而杂合突变被置换成正常序列的细胞的制造方法。
背景技术
随着TALENs、CRISPR-Cas系统等可编程的核酸酶的出现,现在,基因组编辑技术正在呈现迅速的发展。Cas蛋白质与指导RNA形成复合体,该复合体与具有指导RNA的互补序列的基因组上的靶标位点结合,切割DNA双链的双方。作为上一代的基因组编辑技术的TALENs是以DNA为靶标的TALE蛋白质与切割DNA的核酸酶(主要为FokI)的融合蛋白质,但与CRISPR-Cas系统同样地,在基因组上的靶标位点产生DNA的双链切割。
该DNA的双链切割在通过非同源末端接合而被修复时,产生核苷酸的插入/缺失(insertion/deletion:indel(得失位)),通过移码等而可以进行基因敲除。另一方面,在从细胞外导入成为修复模板的供者DNA的情况下,通过基因组与供者DNA之间的同源重组,可以进行基因敲入。在该基因敲入中,不仅可以产生DNA的插入,还可以产生1~数核苷酸的置换、缺失。
然而,从基因组稳定性的观点考虑,使用可编程的核酸酶的基因组编辑存在大问题。其一是由DNA的双链切割造成的目的外的基因突变的发生。在体细胞中,与利用同源重组的修复相比,利用非同源末端接合的修复是优势的,因而在被基因组编辑系统进行了双链切割的靶标位点,与利用修复模板的敲入相比,更容易产生目的外的突变(indel,得失位)。因此,在实施了基因组编辑的细胞集团中,除了实现了按照目的的敲入的细胞、完全没有发生基因序列的变化的细胞之外,还包含不少发生了由非同源末端接合引起的目的外的突变的细胞。另外,如果从单细胞水平来看,则即使常染色体的等位基因中的1个按照计划被敲入,另一个也有可能发生了目的外的突变。另外,有可编程的核酸酶在与靶标序列类似性高的DNA序列(脱靶)中也产生DNA双链切割,从而产生基因组突变的报告。
于是,本发明者们通过使用使Cas9的2个核酸酶部位中的一个失活了的切口酶型Cas9,利用DNA的单链切割来诱导同源重组,从而开发了与现有的利用DNA的双链切割的方法相比,能够抑制由非同源末端接合造成的目的外的突变(indel,得失位)的发生的方法。其一是通过使用切口酶,在作为靶标的基因组导入2个位置的切口、在包含修复模板的供者质粒导入1个位置的切口,从而利用随机切口法的基因组编辑法(非专利文献1、专利文献1)。另外,本发明者们使该方法进一步发展,还开发了在作为靶标的基因组导入1个位置的切口、在包含修复模板的供者质粒导入1个位置的切口的SNGD法(a combination ofsingle nicks in the target gene and donor plasmid)(专利文献1)。
另一方面,在基因组编辑中,还存在向作为修复模板使用的供者DNA的基因组中的随机整合这样的问题。即,如果在细胞中导入大量的DNA,则DNA的一部分整合到基因组的任意位点的现象(随机整合)高频率地发生。然而,鉴定发生了随机整合的位点是困难的,在进行医疗应用等时,在安全性方面成为问题。
现有技术文献
专利文献
专利文献1:日本特开2018-11525号公报
非专利文献
非专利文献1:Nakajima K,et al.,Genome Res.28,223-230,2018
发明内容
发明要解决的课题
本发明是鉴于这样的上述现有技术存在的问题而做出的,其目的在于,不使用外来的供者DNA,特异性且高效地进行利用同源重组的基因组编辑的方法。
用于解决课题的手段
本发明者们为了实现上述目的而反复进行了深入研究,结果首先构思了,通过不从细胞外导入供者DNA,而是以细胞中本来存在的同源染色体作为修复模板进行基因组编辑,从而能够避免供者DNA的随机整合这样的问题。
在细胞内的染色体中存在杂合子突变或者复合杂合子突变的情况下,一方的等位基因(将此称为“等位基因A”)中存在的基因突变,在另一方的等位基因(将此称为“等位基因B”)中不存在。这里,假如能够在等位基因A与等位基因B之间诱导同源重组,则可以以等位基因B作为修复模板将等位基因A的突变修复成正常序列,或者相反以等位基因A作为模板在等位基因B的正常序列中导入突变。此时,现有法中作为模板使用的外来的供者DNA(人工合成DNA链、质粒等)变得不需要。然而,体细胞中发生同源重组修复的是姐妹染色单体之间,同源染色体之间的同源重组非常不易发生。
于是,本发明者们尝试了通过在同源染色体的靶标位点周围导入DNA切割来诱导同源染色体之间的同源重组的诱导。研究了各种切割模式,结果通过使成为受者的染色体上的作为修正对象的核苷酸的附近DNA区域的多个位置产生切口,并且对于成为供者的染色体,使与受者的染色体上产生切口的位置对应的位置的至少1个位置产生切口,从而成功地显著抑制非同源末端接合,特异性地诱导靶标位点的同源染色体之间的重组(将切口的导入例示于图1A~H)。并且,由此,成功地不产生目的外的突变地、高效地修复靶标的突变。进而,本发明者们根据该方法的原理发现,能够广泛地以在同源染色体之间不同的碱基作为靶标,统一成任一方的碱基,从而完成了本发明。
本发明涉及利用同源染色体之间的同源重组的基因组编辑,更详细地提供以下发明。
(1)经基因组编辑的细胞的制造方法,
该方法包括对同源染色体的特定位点具有在同源染色体之间不同的碱基的细胞,导入在该特定位点的附近DNA区域进行单链切割的位点特异性切口酶的组合,诱导以该同源染色体的一方为受者、另一方为供者的同源重组,将该特定位点的受者的碱基置换成供者的碱基,
该位点特异性切口酶的组合对于该受者的染色体,在该特定位点的附近DNA区域的多个位置进行单链切割,对于该供者的染色体,在与受者的染色体中被单链切割的位置对应的至少1个位置进行单链切割。
(2)根据(1)所述的方法,被置换的受者的碱基是突变碱基,供者的碱基是正常碱基。
(3)根据(1)或(2)所述的方法,位点特异性切口酶是CRISPR-Cas系统。
(4)用于在(1)~(3)的任一项所述的方法中使用的试剂盒,
该试剂盒包含对于同源染色体的特定位点具有在同源染色体之间不同的碱基的细胞,在该特定位点的附近DNA区域进行单链切割的位点特异性切口酶的组合,
该位点特异性切口酶的组合对于该受者的染色体,在该特定位点的附近DNA区域的多个位置进行单链切割,对于该供者的染色体,在与受者的染色体中被单链切割的位置对应的至少1个位置进行单链切割。
发明的效果
根据本发明,能够在显著地抑制由非同源末端接合造成的目的外的突变的发生的同时,通过同源染色体之间的同源重组而特异性且高效地进行基因组编辑。另外,由于同源重组中不利用外来的供者DNA,也不产生供者DNA的随机整合这样的问题,因而即使在进行基因治疗等医疗应用的情况下,也能够以高安全性进行基因组编辑。
附图说明
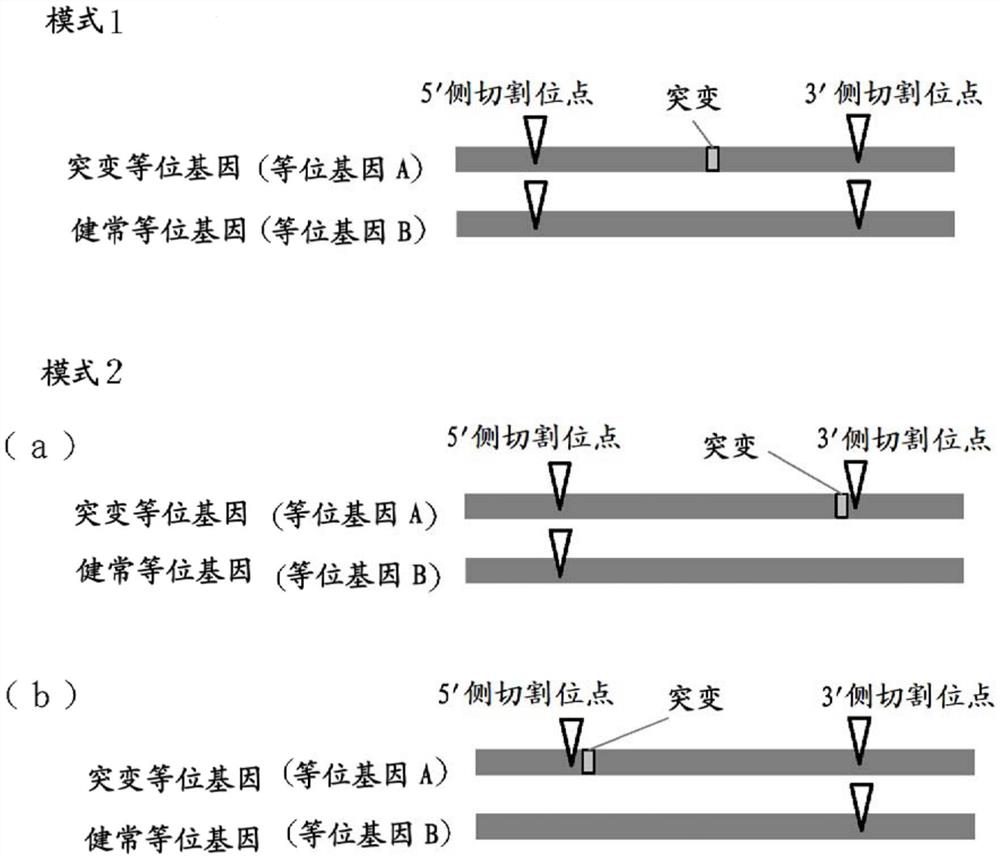
图1A显示本发明的方法中的、由位点特异性切口酶产生的同源染色体的单链切割的模式的例子。
图1B是图1A的继续的图。
图1C是图1B的继续的图。
图1D是图1C的继续的图。
图1E是图1D的继续的图。
图1F是图1E的继续的图。
图1G是图1F的继续的图。
图1H是图1G的继续的图。
图2A显示作为同源重组的供者的染色体的胸苷激酶1基因中的、本实施例中设计的crRNA(相当于sgRNA的5′侧区域)的靶标位点。大写字母表示外显子,小写字母表示内含子。用方框包围的碱基序列是PAM序列(以下、对于图2B~H也同样)。下划线自上起依次表示TSCER2_TK1(ex4)-322s的靶标位点、TSCER2_TK1(ex4)21s、TSCER2_TK1(ex4)29s的靶标位点。
图2B显示作为同源重组的受者的染色体的胸苷激酶1基因中的、本实施例中设计的crRNA(相当于sgRNA的5’侧区域)的靶标位点。下划线自上起依次表示TSCER2_TK1(ex4)-322s的靶标位点、TSCER2_TK1(ex4)21s的靶标位点、TSCER2_TK1(ex4)20s、TSCER2_TK1(ex4)29s的靶标位点。
图2C显示作为同源重组的受者的染色体的胸苷激酶1基因中的、本实施例中设计的crRNA(相当于sgRNA的5’侧区域)的靶标位点。下划线分别表示TSCER2_TK1(ex4)-S1(上图)和TSCER2_TK1(ex4)-S2(下图)的crRNA的靶标位点。
图2D显示作为同源重组的受者的染色体的胸苷激酶1基因中的、本实施例中设计的crRNA(相当于sgRNA的5’侧区域)的靶标位点。下划线分别表示TSCER2_TK1(ex4)-S3(上图)和TSCER2_TK1(ex4)-S4(下图)的crRNA的靶标位点。
图2E显示作为同源重组的受者的染色体的胸苷激酶1基因中的、本实施例中设计的crRNA(相当于sgRNA的5’侧区域)的靶标位点。下划线分别表示TSCER2_TK1(ex4)-S5(上图)和TSCER2_TK1(ex4)-S6(下图)的crRNA的靶标位点。
图2F显示作为同源重组的受者的染色体的胸苷激酶1基因中的、本实施例中设计的crRNA(相当于sgRNA的5’侧区域)的靶标位点。下划线分别表示TSCER2_TK1(ex4)-S7(上图)和TSCER2_TK1(ex4)-S8(下图)的crRNA的靶标位点。
图2G显示作为同源重组的受者的染色体的胸苷激酶1基因中的、本实施例中设计的crRNA(相当于sgRNA的5’侧区域)的靶标位点。下划线分别表示TSCER2_TK1(ex4)-S9(上图)和TSCER2_TK1(ex4)-S10(下图)的crRNA的靶标位点。
图2H显示作为同源重组的受者的染色体的胸苷激酶1基因中的、本实施例中设计的crRNA(相当于sgRNA的5’侧区域)的靶标位点。下划线分别表示TSCER2_TK1(ex4)-S11(上图)和TSCER2_TK1(ex4)-S12(下图)的crRNA的靶标位点。
图2I显示作为crRNA使用TSCER2_TK1(ex4)-322s与TSCER2_TK1(ex4)20s的组合的情况下的、靶标位点的单链切割。
图3显示本实施例的各样品中DNA的单链切割或双链切割发生的位置。
图4A显示对于图3所示的本实施例的样品以胸苷激酶活性的恢复为指标检测发生基因组编辑的细胞而得的结果。图下是将图上的样品#1~6的图放大而得的。
图4B是显示对于图3所示的本实施例的样品以胸苷激酶活性的恢复为指标检测发生基因组编辑的细胞而得的结果。与图4A在将表达切口酶型CRISPR-Cas系统的质粒导入细胞时的电穿孔的条件是不同的。
图5显示对于图3和4所示的本实施例的样品#2和#7分析基因组编辑后的靶标位点的碱基序列而得的结果。
图6显示图7所示的本实施例的样品中的DNA的单链切割发生的位置(图上)、和以胸苷激酶活性的恢复为指标检测发生基因组编辑的细胞而得的结果(图下)。
图7显示本实施例的各样品中DNA的单链切割发生的位置。
图8显示对于图6和7所示的本实施例的样品S3/20s和S12/20s分析基因组编辑后的靶标位点的碱基序列而得的结果。
图9显示图10所示的本实施例的各样品中的、DNA的单链切割发生的位置(图上)、和以胸苷激酶活性的恢复为指标检测发生基因组编辑的细胞而得的结果(图下)。
图10显示本实施例的各样品中DNA的单链切割发生的位置。
图11显示图12所示的本实施例的各样品中的DNA的单链切割发生的位置(图上)、和以胸苷激酶活性的恢复为指标检测发生基因组编辑的细胞而得的结果(图下)。
图12显示本实施例的各样品中DNA的单链切割发生的位置。
具体实施方式
本发明中的经基因组编辑的细胞的制造方法的原理是,利用通过由位点特异性切口酶产生的单链切割而被诱导的同源染色体之间的同源重组,将在同源染色体之间不同的碱基统一成任一方的碱基。
具体地,对同源染色体的特定位点具有在同源染色体之间不同的碱基的细胞,导入在该特定位点的附近DNA区域进行单链切割的位点特异性切口酶的组合,诱导以该同源染色体的一方为受者、以另一方为供者的同源重组,从而将该特定位点中的受者的碱基置换成供者的碱基。
作为成为本发明中的基因组编辑的对象的“细胞”,只要具有同源染色体,就不特别限定,可以以各种真核细胞作为对象。作为“真核细胞”,可列举例如,动物细胞、植物细胞、藻类细胞、真菌细胞。另外作为动物细胞,可列举例如,哺乳动物细胞,以及鱼类、鸟类、爬行类、两栖类、昆虫类的细胞。
“动物细胞”包括例如,构成动物的个体的细胞、构成由动物摘出的器官或组织的细胞、来源于动物的组织的培养细胞等。具体地,可列举例如,各阶段的胚的胚细胞(例如,1细胞期胚、2细胞期胚、4细胞期胚、8细胞期胚、16细胞期胚、桑葚期胚等);诱导多能性干(iPS)细胞、胚胎干(ES)细胞、造血干细胞等干细胞;成纤维细胞、造血细胞、神经元、肌肉细胞、骨细胞、肝细胞、胰脏细胞、脑细胞、肾细胞等体细胞等。基因组编辑动物的制成中可以使用受精后的卵母细胞、即受精卵。特别优选受精卵为原核期胚的。受精前的卵母细胞可以将冷冻保存的解冻使用。
本发明中“哺乳动物”是包含人和非人哺乳动物的概念。作为非人哺乳动物的例子,可列举牛、野猪、家猪、绵羊、山羊等偶蹄类、马等奇蹄类、小鼠、大鼠、豚鼠、仓鼠、松鼠等啮齿类、兔等兔目、狗、猫、鼬等食肉类等。上述非人哺乳动物可以是家畜或伴侣动物(宠物),也可以是野生动物。
作为“植物细胞”,可列举例如,谷物类、油料作物、饲料作物、水果、蔬菜类的细胞。“植物细胞”包含例如,构成植物的个体的细胞、构成从植物分离的器官或组织的细胞、来源于植物的组织的培养细胞等。作为植物的器官或组织,可列举例如,叶、茎、茎尖(生长点)、根、块茎、块根、种子、愈伤组织等。作为植物的例子,可列举稻、玉米、香蕉、花生、向日葵、番茄、拟南芥、烟草、小麦、大麦、马铃薯、大豆、棉花、康乃馨等。
同源染色体的特定位点中的“在同源染色体之间不同的碱基”可以是一个碱基,也可以是多个碱基(碱基序列)。另外,可以是突变,也可以是多态性。作为突变,可列举例如,置换、缺失、插入、或它们的组合,多态性可列举例如,单碱基多态性、微卫星多态性。
本发明中,同源染色体中在特定位点具有突变、多态性的染色体可以作为同源重组中的受者,也可以作为供者。即,通过本发明中的基因组编辑,可以使构成同源染色体的2条染色体的特定位点的碱基双方都成为正常序列,另外,也可以使双方都成为特定的突变序列、多态性序列。例如,对于HLA,也可以使杂合子的HLA变成纯合子HLA。
从医疗上的有用性的观点考虑的典型的本发明的利用方案是为了治疗或预防由杂合子突变引起的人的疾病,而将人细胞中的该突变修复成正常序列。这里作为“由杂合子突变引起的疾病”,是除了通过该杂合突变而直接产生的疾病(显性遗传疾病)之外,还包含通过2种不同的突变的组合(复合杂合子)产生的疾病(隐性遗传疾病)的含义。作为对象疾病,可列举例如,先天性免疫缺陷症中的OAS1异常症等采取常染色体显性遗传形式的通过常染色体杂合子突变而发病的疾病、ADA缺损症等采取常染色体隐性遗传形式的遗传性疾病、和女性的第VIII因子·第IX因子缺损的血友病等在女性中以X连锁伴性遗传形式发病的疾病,但不限于这些。
作为本发明中使用的“位点特异性切口酶”,只要是能够在基因组上位点特异性地对DNA进行单链切割的就不限制,但优选以切口酶型Cas蛋白质作为构成要素的CRISPR-Cas系统。Cas蛋白质通常包含参与靶标链的切割的结构域(RuvC结构域)和参与非靶标链的切割的结构域(HNH结构域),但切口酶型Cas蛋白质典型地通过这2个结构域的任一结构域的突变而丧失其切割活性。作为这样的突变,在spCas9蛋白质(酿脓链球菌(S.pyogenes)来源的Cas9蛋白质)的情况下,可列举例如,N末端起第10位的氨基酸(天冬氨酸)向丙氨酸的突变(D10A:RuvC结构域内的突变)、N末端起第840位的氨基酸(组氨酸)向丙氨酸的突变(H840A:HNH结构域内的突变)、N末端起第863位的氨基酸(天冬酰胺)向丙氨酸的突变(N863A:HNH结构域内的突变)、N末端起第762位的氨基酸(谷氨酸)向丙氨酸的突变(E762A:RuvCII结构域内的突变)、N末端起第986位的氨基酸(天冬氨酸)向丙氨酸的突变(D986A:RuvCIII结构域内的突变)。此外,各种来源的Cas9蛋白质是公知的(例如,WO2014/131833),可以利用它们的切口酶型。此外,Cas9蛋白质的氨基酸序列和碱基序列在公开的数据库、例如GenBank(http://www.ncbi.nlm.nih.gov)中登记(例如,登记号:Q99ZW2.1等),本发明中可以利用这些序列。
另外,本发明中还可以利用除Cas9以外的Cas蛋白质,例如,Cpf1(Cas12a)、Cas12b、CasX(Cas12e)、Cas14等。作为切口酶型Cpf1蛋白质中的突变,例如,在AsCpf1(Cas12)中,可列举N末端起第1226位的氨基酸(精氨酸)向丙氨酸的突变(R1226A:Nuc结构域内的突变)。Cpf1的氨基酸序列在公开的数据库、例如GenBank(http://www.ncbi.nlm.nih.gov)中登记(例如,登记号:WP_021736722、WP_035635841等)。
作为构成CRISPR-Cas系统的蛋白质,可以使用添加了核定位信号的。
在以切口酶型Cas蛋白质为构成要素的CRISPR-Cas系统中,切口酶型Cas蛋白质与指导RNA结合而形成复合体,以靶标DNA序列为靶标而将DNA单链切割。在CRISPR-Cas9系统中,指导RNA包含crRNA和tracrRNA,但在CRISPR-Cpf1系统中不需要tracrRNA。CRISPR-Cas9系统中的指导RNA可以是包含crRNA和tracrRNA的单分子指导RNA,也可以是由crRNA片段和tracrRNA片段组成的双分子指导RNA。
crRNA包含相对于靶标DNA序列互补的碱基序列。靶标DNA序列通常是由12~50碱基、优选17~30碱基、更优选17~25碱基组成的碱基序列,优选从与PAM(proto-spaceradjacent motif,前间区序列临近基序)序列邻接的区域选择。典型地,DNA的位点特异性切割在由crRNA与靶标DNA序列的之间的碱基对形成的互补性、和与其邻接而存在的PAM双方决定的位置产生。
在大量的CRISPR-Cas系统中,crRNA进一步在3’侧包含能与tracrRNA片段相互作用(杂交)的碱基序列。另一方面,tracrRNA在5’侧包含能与crRNA的一部分碱基序列相互作用(杂交)的碱基序列。通过这些碱基序列的相互作用,crRNA/tracrRNA(单分子或双分子)形成双链RNA,所形成的双链RNA与Cas蛋白质相互作用。
PAM根据Cas蛋白质的种类、来源不同而不同。典型的PAM序列例如,酿脓链球菌(S.pyogenes)来源的Cas9蛋白质(II型)中为“5′-NGG”,在硫矿硫化叶菌(S.solfataricus)来源的Cas9蛋白质(I-A1型)中为“5′-CCN”,在硫矿硫化叶菌(S.solfataricus)来源的Cas9蛋白质(I-A2型)中为“5′-TCN”,在H.walsbyl来源的Cas9蛋白质(I-B型)中为“5′-TTC”,在大肠杆菌(E.coli)来源的Cas9蛋白质(I-E型)中为“5′-AWG”,在大肠杆菌(E.coli)来源的Cas9蛋白质(I-F型)中为“5′-CC”,在绿脓杆菌(p.aeruginosa)来源的Cas9蛋白质(I-F型)中为“5′-CC”,在嗜热乳链球菌(S.Thermophilus)来源的Cas9蛋白质(II-A型)中为“5′-NNAGAA”,在无乳链球菌(S.agalactiae)来源的Cas9蛋白质(II-A型)中为“5′-NGG”,在金黄色葡萄球菌(S.aureus)来源的Cas9蛋白质中为“5′-NGRRT”或“5′-NGRRN”,在脑膜炎奈瑟菌(N.meningitidis)来源的Cas9蛋白质中为“5′-NNNNGATT”,在龋垢密螺旋体(T.denticola)来源的Cas9蛋白质中为“5′-NAAAAC”。在Cpf1中典型地为“5’-TTN”或“5’-TTTN”。此外,也可以通过改变蛋白质(例如,突变的导入),来改变PAM识别(Benjamin,P.等,Nature 523,481-485(2015)、Hirano,S.等,Molecular Cell 61,886-894(2016))。
本发明中,也可以利用除了CRISPR-Cas系统以外的位点特异性切口酶。作为这样的位点特异性切口酶,可列举例如,与具有切口酶活性的酶融合而成的人工核酸酶。作为人工核酸酶,可以利用例如,TALE(转录激活因子样效应物,transcription activator-likeeffector)、ZF(锌指,zinc finger)、PPR(三角状五肽重复,pentatricopeptide repeat)。作为能够通过与这些人工核酸酶的融合而发挥切口酶活性的酶,可列举例如,TevI(NatCommun.2013;4:1762.doi:10.1038/ncomms2782)。这些人工核酸酶通过与识别特定的碱基(或者特定的碱基序列)的模件(肽)连接而构建的DNA结合结构域,从而以靶标DNA序列作为靶标,通过融合于该DNA结合结构域的切口酶,将DNA单链切割。也可以在人工核酸酶中的DNA结合结构域与切口酶的之间导入适当的间隔肽。
本发明中,利用对于受者的染色体,在上述特定位点(在同源染色体之间不同的碱基)的附近DNA区域的多个位置进行单链切割,对于供者的染色体,在与受者的染色体中被单链切割的位置对应的位置的至少1个位置进行单链切割的位点特异性切口酶的组合。
这里“附近DNA区域”是指距离特定位点通常100000碱基以内、10000碱基以内、5000碱基以内、2000碱基以内、优选为1000碱基以内(例如,500碱基以内、400碱基以内、300碱基以内、200碱基以内、100碱基以内、50碱基以内、20碱基以内、10碱基以内)的区域。另外,“附近DNA区域的多个位置”可以在同一DNA链上,也可以在不同的DNA链上。
作为具体的方案的例子,可列举上述特定位点的5′侧附近DNA区域和3′侧附近DNA区域的各1个位置(图1A的模式1和模式2、图1C的模式1′和2(a)′、图1D的模式2(b)′)、上述特定位点的5′侧附近DNA区域的2个位置(图1B的模式3(a)、模式4(a)、图1D的模式3(a)′、图1E的模式4(a)′)、上述特定位点的3′侧附近DNA区域的2个位置(图1B的模式3(b)、模式4(b)、图1E的模式3(b)′、图1F的模式4(b)′)、以及上述特定位点的5′侧附近DNA区域和3′侧附近DNA区域的各至少1个位置的共计3个位置(图1G的模式5和模式6)。被切割的位置也可以是4个位置以上。另外,1个特定位点的附近DNA区域中也可以存在其他特定位点(在同源染色体之间不同的碱基)(图1H的模式7)。
在供者的染色体与受者的染色体被单链切割的位置的全部对应的方案(图1A的模式1、图1B的模式3、图1C的模式1′、图1D的模式3(a)′、图1E的模式3(b)′、图1G的模式6、图1H的模式7(d))中,与受者的染色体的靶标DNA序列结合的位点特异性切口酶可以设计成与供者的染色体的对应DNA序列也结合那样。该情况下,受者的染色体的靶标DNA序列与供者的染色体的对应DNA序列典型地为同一DNA序列。
另一方面,在供者的染色体与受者的染色体中被单链切割的位置的一部分不对应的方案(图1A的模式2、图1B的模式4、图1C的模式2(a)′、图1D的模式2(b)′、图1E的模式4(a)′、图1F的模式4(b)′、图1G的模式5、图1H的模式7(a)~(c))中,可以以与受者的染色体的靶标DNA序列结合的位点特异性切口酶的组合的一部分不与供者的染色体的对应DNA序列结合的方式进行设计。在该情况下、受者的染色体的靶标DNA序列和供者的染色体的对应DNA序列是不同的DNA序列。例如,如果以包含通过基因组编辑而进行置换的碱基(在同源染色体之间不同的碱基)的方式设定位点特异性切口酶的靶标DNA序列,则受者的染色体的靶标DNA序列与供者的染色体的对应DNA序列变成不同的DNA序列。在位点特异性切口酶是CRISPR-Cas系统的情况下,以对受者的靶标DNA序列具有结合特异性的方式设计指导RNA即可。另外,在位点特异性切口酶是与具有切口酶活性的酶融合而成的人工核酸酶的情况下,以对受者的靶标DNA序列具有结合特异性的方式设计DNA结合结构域即可。在该方案中,在位点特异性切口酶的设计上,被单链切割的位点距离上述特定位点(在同源染色体之间不同的碱基)通常为约100碱基位内、更优选为50碱基以内(例如,40碱基以内、30碱基以内、20碱基以内、10碱基以内)。
本发明中,在不同的DNA链上进行单链切割的情况下,如果单链切割的距离过近则会产生双链切割。因此,位于不同的DNA链上的单链切割位置的距离通常为100碱基以上、优选为200碱基以上,并且通常为2000碱基以内、优选为1000碱基以内、进一步优选为500碱基以内。
在本发明中,将上述位点特异性切口酶的组合导入细胞。在被导入细胞的“位点特异性切口酶”是CRISPR-Cas系统的情况下,例如,可以是指导RNA与Cas蛋白质的组合的方式,也可以是指导RNA与被翻译成Cas蛋白质的信使RNA的组合的方式,也可以是表达它们的载体的组合。指导RNA也可以进行用于抑制分解的修饰(化学修饰等)。在被导入细胞的“位点特异性切口酶”是与具有切口酶活性的酶融合而成的人工核酸酶的情况下,例如,可以是蛋白质的方式,也可以是被翻译成该蛋白质的信使RNA,也可以是表达该蛋白质的载体的方式。
在采用表达载体的方式的情况下,包含与要表达的DNA可工作地结合的1种以上的调节元件。这里,“可工作地结合”是指上述DNA能够表达地与调节元件结合。作为“调节元件”,可列举启动子、增强子、内部核糖体进入位点(IRES)、和其他表达控制元件(例如,转录终止信号、例如,多聚腺苷化信号和多聚U序列)。作为调节元件,根据目的,例如,可以是指向多样的宿主细胞中的DNA的结构性表达的,也可以是仅指向特定的细胞、组织、或者器官中的DNA的表达的。另外,可以是仅在特定的时期指向DNA的表达的,也可以是指向能人工地诱导的DNA的表达的。作为启动子,可列举例如,polIII启动子(例如,U6和H1启动子)、polII启动子(例如,作为反转录病毒的劳斯肉瘤病毒(RSV)LTR启动子、巨细胞病毒(CMV)启动子、SV40启动子、二氢叶酸还原酶启动子、β-肌动蛋白启动子、磷酸甘油酸激酶(PGK)启动子、和EF1α启动子)、polI启动子、或它们的组合。只要是本领域技术人员,就能够根据要导入的细胞的种类等选择适当的表达载体。
位点特异性切口酶向细胞的导入可以通过例如,电穿孔、显微注射、DEAE-葡聚糖处理、脂质体转染、纳米颗粒介导的转染、病毒介导的核酸递送等公知的方法来进行。
在向细胞的导入之后,位点特异性切口酶的组合对于受者的染色体,在靶标碱基的附近DNA区域的多个位置进行单链切割,对于供者的染色体,在与受者的染色体中被单链切割的位置对应的位置的至少1个位置进行单链切割。由此在显著抑制由非同源末端接合造成的目的外的突变的发生的同时,同源染色体之间的同源重组被诱导,靶标碱基特异性且高效地被置换成供者中的对应碱基。根据本发明,可以将由非同源末端接合造成的目的外的突变的发生抑制90%以上、优选为95%以上(例如,96%以上、97%以上、98%以上、99%以上、100%)。
另外,本发明提供用于在上述本发明的方法中使用的试剂盒,该试剂盒包含上述位点特异性切口酶的组合。有时该试剂盒进一步包含一种或多种追加的试剂,作为追加的试剂,可列举例如,稀释缓冲液、再构成溶液、洗涤缓冲液、核酸导入试剂、蛋白质导入试剂、对照试剂(例如,对照的指导RNA),但不限于这些。该试剂盒还可以包含用于实施本发明的方法的使用说明书。
实施例
以下,基于实施例更具体地说明本发明,但本发明不限于以下实施例。
[实施例1]多切口法的有效性
A.材料
(1)TSCER2细胞
是具有Thymidine Kinase(胸苷激酶)1基因(TK1)的杂合子突变的lymphoblast(淋巴母细胞)TK6细胞(第4外显子中单核苷酸插入。移码)来源的细胞。在健常等位基因的第4内含子中插入31碱基对(该插入自身与TK1基因功能的丧失无关)并在第5外显子的导入突变,改变成复合杂合子突变,使TK1基因的功能丧失。由于依赖于胸苷激酶的DNA合成补救(salvage)途径不发挥功能,因而如果通过氨基蝶呤阻断DNA从头(de novo)合成途径,则即使供给2-脱氧胞苷、次黄嘌呤、和胸苷也不能进行细胞增殖。在通过基因组编辑而胸苷激酶活性恢复了的情况下,在CHAT培养基(10μM 2-脱氧胞苷[Sigma]、200μM次黄嘌呤[Sigma],100nM氨基蝶呤[Sigma]、和17.5μM胸苷[Sigma])中也能够进行细胞增殖。
(2)切口酶型CRISPR-Cas9系统和靶标区域的结构
将成为供者的野生型的靶标区域(TK1第3内含子~第4外显子~第4内含子的序列)示于图2A(序列号:1)。大写字母表示外显子,小写字母表示内含子。用方框包围的碱基序列是PAM序列。下划线自上起依次表示TSCER2_TK1(ex4)-322s的靶标位点、TSCER2_TK1(ex4)21s的靶标位点、TSCER2_TK1(ex4)29s的靶标位点。
另外,将成为受者的突变型的靶标区域示于图2B(序列号:2)。大写字母表示外显子,小写字母表示内含子。用方框包围的碱基序列是PAM序列。下划线自上起依次表示TSCER2_TK1(ex4)-322s的靶标位点、TSCER2_TK1(ex4)21s的靶标位点、TSCER2_TK1(ex4)20s、TSCER2_TK1(ex4)29s的靶标位点。
另外,S1~S12的靶标区域示于图2C~2H(序列号:3~14)。大写字母表示外显子,小写字母表示其他序列。用方框包围的碱基序列是PAM序列。将靶标序列位点用下划线表示。
sgRNA中相当于crRNA的序列如下。此外,下划线是针对PAM的序列。
TSCER2_TK1(ex4)20s
CGTCTCGGAGCAGGCAGGCG
TSCER2_TK1(ex4)21s
ACGTCTCGGAGCAGGCAGGC
TSCER2_TK1(ex4)-322s
CCTCAGCCACAAGAGTAGCT
TSCER2_TK1(ex4)29s
CCTGGGCCACGTCTCGGAGC
TSCER2_TK1_S1
ACCTCTAGACCATGGATCTG
TSCER2_TK1_S2
CTGACAAAGAGCTCCTTCAC
TSCER2_TK1_S3
ATTCAAGGGAGGAGCACCCC
TSCER2_TK1_S4
CTTGTGATTTTCCACTGGAC
TSCER2_TK1_S5
GAAGTTGTACTTCCAACAGC
TSCER2_TK1_S6
CAGACTAGGCCAACTTCATC
TSCER2_TK1_S7
GATAACTTCCAAGTCAGCGA
TSCER2_TK1_S8
AGCTTCCCATCTATACCTCC
TSCER2_TK1_S9
CAACCGGCCTGGAACCACGT
TSCER2_TK1_S10
GATCTAGAACTGCTTGCAAT
TSCER2_TK1_S11
TCAATCATATCACTCTTAGC
TSCER2_TK1_S12
GGAGCTGTCCATGAGACCCAGGG(序列号:30)
预计在使用TSCER2_TK1(ex4)20s的情况下,在[CCCCGC]与[CTGCCTGCTCCGAGACG]之间产生DNA双链切割或者切口,在使用TSCER2_TK1(ex4)21s的情况下,在[CCCGCC]与[TGCCTGCTCCGAGACGT]之间产生DNA双链切割或者切口,在使用TSCER2_TK1(ex4)-322s的情况下,在[cccagc]与[tactcttgtggctgagg]之间产生DNA双链切割或者切口,在使用TSCER2_TK1(ex4)29s的情况下,在[CCTGCT]与[CCGAGACGTGGCCCAGG]之间产生DNA双链切割或者切口,在使用TSCER2_TK1_S1的情况下,在[CCTCAG]与[ATCCATGGTCTAGAGGT]之间产生DNA双链切割或者切口,在使用TSCER2_TK1_S2的情况下,在[CCAGTG]与[AAGGAGCTCTTTGTCAG]之间产生DNA双链切割或者切口,在使用TSCER2_TK1_S3的情况下,在[CCTGGG]与[GTGCTCCTCCCTTGAAT]之间产生DNA双链切割或者切口,在使用TSCER2_TK1_S4的情况下,在[CCTGTC]与[CAGTGGAAAATCACAAG]之间产生DNA双链切割或者切口,在使用TSCER2_TK1_S5的情况下,在[CCAGCT]与[GTTGGAAGTACAACTTC]之间产生DNA双链切割或者切口,在使用TSCER2_TK1_S6的情况下,在[CCTGAT]与[GAAGTTGGCCTAGTCTG]之间产生DNA双链切割或者切口,在使用TSCER2_TK1_S7的情况下,在[CCCTCG]与[CTGACTTGGAAGTTATC]之间产生DNA双链切割或者切口,在使用TSCER2_TK1_S8的情况下,在[CCAGGA]与[GGTATAGATGGGAAGCT]之间产生DNA双链切割或者切口,在使用TSCER2_TK1_S9的情况下,在[CCTACG]与[TGGTTCCAGGCCGGTTG]之间产生DNA双链切割或者切口,在使用TSCER2_TK1_S10的情况下,在[CCCATT]与[GCAAGCAGTTCTAGATC]之间产生DNA双链切割或者切口,在使用TSCER2_TK1_S11的情况下,在[CCAGCT]与[AAGAGTGATATGATTGA]之间产生DNA双链切割或者切口,在使用TSCER2_TK1_S12的情况下,在[CCCTGG]与[GTCTCATGGACAGCTCC]之间产生DNA双链切割或者切口(请参照图2A~H和序列号:1~14)。
突变阵列上的[CCC
使用表达Cas9和sgRNA两者的载体,使Cas9和sgRNA表达。将所利用的载体在以下显示。
V1:PX461(Cas9D10A-P2A-GFP)-TSCER2_TK1(ex4)20s
V2:PX461(Cas9D10A-P2A-GFP)-TSCER2_TK1(ex4)21s
V3:PX461(Cas9D10A-P2A-GFP)-empty
V4:PX462(Cas9D10A-P2A-PuroR)-TSCER2_TK1(ex4)-322s
V5:PX462(Cas9D10A-P2A-PuroR)-empty
V6:PX462(Cas9D10A-P2A-PuroR)-empty
V7:PX458(Cas9-P2A-GFP)-TSCER2_TK1(ex4)20s
V8:PX458(Cas9-P2A-GFP)-empty
V9:PX459(Cas9-P2A-PuroR)-empty
将上述的载体如下组合而导入TSCER2细胞。
样品#1:V1+V5
样品#2:V1+V4
样品#3:V2+V5
样品#4:V2+V4
样品#5:V3+V4
样品#6:V3+V5
样品#7:V7+V9
样品#8:V8+V9
将各样品中切口或者DNA双链切割产生的位置示于图3。
B.方法和结果
将2种质粒分别8μg与600×10
在CHAT培养基中每1孔接种A个细胞的情况下,将能够形成集落的孔的比例(%)设为B,将在通常培养基中每1孔接种C个细胞的情况下能够形成集落的孔的比例(%)设为D时,如下计算出编辑成功率。
(B/A)/(D/C)×100(%)
将结果示于图4A(电穿孔条件1)和图4B(电穿孔条件2)。在Cas9或Cas9切口酶不识别靶标基因的情况下,基因组编辑未发生(图3,4A,4B、样品#6,#8)。在作为修正对象的核苷酸附近发生了DNA双链切割的情况下,恢复胸苷激酶活性的细胞的比例达到5.43±0.77%,但其中由同源染色体之间重组带来的止于3.66%,96.3%是由核苷酸缺失造成的(图3,4A、样品#7和表1)。
表1
另一方面,如果使突变等位基因的作为修正对象的核苷酸附近(位点A)和远离的位点(位点B)的2个位置和健常等位基因的位点B的1个位置发生切口,则细胞的胸苷激酶活性以0.460±0.050%的比例恢复(图3,4A、样品#2)。如果改变电穿孔的条件,则该比例提高到1.04±0.105%(图4B)。这些与在突变等位基因的位点A加入1个位置切口的情况(0.0502±0.0113%、图3,4A、样品#1)相比修正效率高。在突变等位基因、健常等位基因均在位点A和位点B的2个位置发生切口的情况下,基因修正也以0.522±0.035%的比例进行(图3,4A、样品#4)。
接着,在样品#2和#7中,通过直接(direct)PCR扩增形成了集落的细胞中的TK1基因的第3内含子~第4内含子区域的DNA片段。直接PCR使用MightyAmp DNA PolymeraseVer.2(タカラバイオ)。作为引物使用[TCCTGAACAGTGGAAGAGTTTTTAG(序列号:31)][AACTTACAAACTGCCCCTCGTC(序列号:32)]。PCR片段以[TGAACACTGAGCCTGCTT(序列号:33)]作为引物利用Sanger测序法进行DNA序列分析。将编辑前的DNA测序结果示于图5A,将进行了利用同源染色体之间重组的修正的细胞的DNA测序结果示于图5B,变成了核苷酸缺失的DNA测序的例子示于图5C、D。突变等位基因被修正成野生型的细胞的比例在样品#2中为100%(111克隆/111克隆)(表1)。
[实施例2]切口之间的距离对基因组编辑效率的影响的检验
A.材料
本实施例中利用的载体在以下显示。
VN1:PX461(Cas9D10A-P2A-GFP)-TSCER2_TK1(ex4)20s
VN2:PX462(Cas9D10A-P2A-PuroR)-TSCER2_TK1(ex4)20s
VN3:PX462(Cas9D10A-P2A-PuroR)-TSCER2_TK1(ex4)-322s
VN4:PX461(Cas9D10A-P2A-GFP)-empty
VN5:PX462(Cas9D10A-P2A-PuroR)-empty
VS1:PX462(Cas9D10A-P2A-PuroR)-TSCER2_TK1-S1
VS2:PX462(Cas9D10A-P2A-PuroR)-TSCER2_TK1-S2
VS3:PX462(Cas9D10A-P2A-PuroR)-TSCER2_TK1-S3
VS4:PX462(Cas9D10A-P2A-PuroR)-TSCER2_TK1-S4
VS5:PX462(Cas9D10A-P2A-PuroR)-TSCER2_TK1-S5
VS6:PX462(Cas9D10A-P2A-PuroR)-TSCER2_TK1-S6
VS7:PX462(Cas9D10A-P2A-PuroR)-TSCER2_TK1-S7
VS8:PX462(Cas9D10A-P2A-PuroR)-TSCER2_TK1-S8
VS9:PX462(Cas9D10A-P2A-PuroR)-TSCER2_TK1-S9
VS10:PX462(Cas9D10A-P2A-PuroR)-TSCER2_TK1-S10
VS11:PX462(Cas9D10A-P2A-PuroR)-TSCER2_TK1-S11
VS12:PX462(Cas9D10A-P2A-PuroR)-TSCER2_TK1-S12
将上述载体如下组合而导入TSCER2细胞。
-322s/20s:VN1(1.5μg)+VN2(1.5μg)+VN3(3.0μg)
S1/20s:VN1(1.5μg)+VN2(1.5μg)+VS1(3.0μg)
S2/20s:VN1(1.5μg)+VN2(1.5μg)+VS2(3.0μg)
S3/20s:VN1(1.5μg)+VN2(1.5μg)+VS3(3.0μg)
S4/20s:VN1(1.5μg)+VN2(1.5μg)+VS4(3.0μg)
S5/20s:VN1(1.5μg)+VN2(1.5μg)+VS5(3.0μg)
S6/20s:VN1(1.5μg)+VN2(1.5μg)+VS6(3.0μg)
S7/20s:VN1(1.5μg)+VN2(1.5μg)+VS7(3.0μg)
S8/20s:VN1(1.5μg)+VN2(1.5μg)+VS8(3.0μg)
S9/20s:VN1(1.5μg)+VN2(1.5μg)+VS9(3.0μg)
S10/20s:VN1(1.5μg)+VN2(1.5μg)+VS10(3.0μg)
S11/20s:VN1(1.5μg)+VN2(1.5μg)+VS11(3.0μg)
S12/20s:VN1(1.5μg)+VN2(1.5μg)+VS12(3.0μg)
20s/emp:VN1(1.5μg)+VN2(1.5μg)+VN5(3.0μg)
emp/emp:VN4(1.5μg)+VN5(4.5μg)
此外,将成为各载体来源的指导RNA(gRNA)的靶标的基因组上的位置示于图6。另外,将各样品中切口产生的位置示于图7。在各样品中,预计的切口之间的距离(图7中的位点A与位点B之间的距离)和切割模式(参照图1A~H)如下。
-322s/20s:341nt、模式2(a)
S1/20s:8173nt、模式2(a)
S2/20s:5678nt、模式2(a)
S3/20s:3964nt、模式2(a)
S4/20s:2369nt、模式2(a)
S5/20s:1367nt、模式2(a)
S6/20s:608nt、模式2(a)
S7/20s:136nt、模式4(b)
S8/20s:1004nt、模式4(b)
S9/20s:2353nt、模式4(b)
S10/20s:4041nt、模式4(b)
S11/20s:6333nt、模式4(b)
S12/20s:8612nt、模式4(b)、模式7(a)
20s/emp.:切口仅在一个位置
emp./emp.:切口不产生
B.方法和结果
将上述列表中所示的量的各质粒、与150×10
将结果示于图6。在Cas9切口酶不识别靶标基因的情况下,基因组编辑未发生(图6和7的样品emp/emp)。如果使突变等位基因的作为修正对象的核苷酸附近(位点A)和远离的位点(位点B)的2个位置和健常等位基因的位点B的1个位置发生切口,则细胞的胸苷激酶活性以0.294±0.098%~2.82±0.010%的比例恢复(图6和7的样品-322s/20s、S1/20s、S2/20s、S3/20s、S4/20s、S5/20s、S6/20s、S7/20s、S8/20s、S9/20s、S10/20s、S11/20s、S12/20s),在全部样品中,都比仅在突变等位基因的作为修正对象的核苷酸附近(位点A)产生切口的情况(图6和7的样品20s/emp:细胞的胸苷激酶活性以0.0916±0.0498%的比例恢复)修正效率高。
接着,在样品S3/20s和S12/20s中,通过直接(direct)PCR或以提取出的基因组DNA作为模板的PCR,扩增形成了集落的细胞中的TK1基因的第3内含子~第4内含子区域的DNA片段和第4内含子~第5内含子区域的DNA片段。基因组DNA提取使用カネカ简易DNA提取试剂盒Version 2(Kaneka)。直接PCR使用MightyAmp DNA Polymerase Ver.2(タカラバイオ)。以基因组DNA作为模板的PCR使用KOD plus neo(TOYOBO)。作为第3内含子~第4内含子区域用的引物使用[TCCTGAACAGTGGAAGAGTTTTTAG(序列号:31)][AACTTACAAACTGCCCCTCGTC(序列号:32)]。作为第4内含子~第5内含子区域用的引物使用[AGTTGTGGATGTACCTGTCGTCT(序列号:34)][ATGCCCGGCTCTGTCCCTTT(序列号:35)]。第3内含子~第4内含子区域的PCR片段以[TGAACACTGAGCCTGCTT(序列号:33)]作为引物利用Sanger测序法进行DNA序列分析,第4内含子~第5内含子区域的PCR片段以[TAACCCTGTGGTGGCTGA(序列号:36)]作为引物利用Sanger测序法进行DNA序列分析。将第4内含子~第5内含子区域的编辑前的DNA测序结果示于图8(a),将通过同源染色体之间重组而两等位基因变成野生型的细胞中的DNA测序结果示于图8(b)。
样品S3/20s、S12/20s均没有在第4外显子和第5外显子中确认到伴随基因组编辑的新的突变的发生。在样品S3/20s中,第4外显子98.9%被修正,第5外显子1.06%被修正。没有第4外显子、第5外显子均被修正的细胞。样品S12/20s中,第4外显子83.2%被修正,第5外显子30.5%被修正。第4外显子、第5外显子均被修正的细胞为13.7%(表2)。在第5外显子被修正的全部细胞中,第4内含子中存在的31核苷酸的插入突变也被修正。
表2
通过S12/20s那样的模式(模式7(a)),能够同时修正第4外显子的单核苷酸插入、第4内含子的31核苷酸插入、第5外显子的单碱基置换和3个位置的突变。
[实施例3]追加的单链切割的导入对基因组编辑效率的影响的检验
A.材料
本实施例中利用的载体在以下显示。本实施例中,将载体如下组合而导入TSCER2细胞。
S3/20s/S8:VN1(1.5μg)+VN2(1.5μg)+VS3(3.0μg)+VS8(3.0μg)
S3/20s/S11:VN1(1.5μg)+VN2(1.5μg)+VS3(3.0μg)+VS11(3.0μg)
S6/20s/S8:VN1(1.5μg)+VN2(1.5μg)+VS6(3.0μg)+VS8(3.0μg)
S6/20s/S11:VN1(1.5μg)+VN2(1.5μg)+VS6(3.0μg)+VS11(3.0μg)
S3/20s/emp:VN1(1.5μg)+VN2(1.5μg)+VS3(3.0μg)+VN5(3.0μg)
S6/20s/emp:VN1(1.5μg)+VN2(1.5μg)+VS6(3.0μg)+VN5(3.0μg)
20s/S8/emp:VN1(1.5μg)+VN2(1.5μg)+VS8(3.0μg)+VN5(3.0μg)
20s/S11/emp:VN1(1.5μg)+VN2(1.5μg)+VS11(3.0μg)+VN5(3.0μg)
20s/emp/emp:VN1(1.5μg)+VN2(1.5μg)+VN5(6.0μg)
emp/emp/emp:VN4(1.5μg)+VN5(7.5μg)
此外,将作为各载体来源的指导RNA(gRNA)的靶标的基因组上的位置示于图9。另外,将各样品中切口产生的位置示于图10。各样品中的切割模式(参照图1A~H)如下。
S3/20s/S8:模式5
S3/20s/S11:模式5
S6/20s/S8:模式5
S6/20s/S11:模式5
S3/20s/emp:模式2(a)
S6/20s/emp:模式2(a)
20s/S8/emp:模式4(b)
20s/S11/emp:模式4(b)
20s/emp/emp:切口仅在一个位置
emp/emp/emp:切口不产生
B.方法和结果
将上述列表中所示的量的各质粒、与150×10
将结果示于图9。与在受者等位基因的2个位置、供者等位基因的1个位置产生切口的样品S3/20s/emp(2.68±0.37%)、样品20s/S8/emp(2.59±0.17%)相比,在受者等位基因的3个位置、供者等位基因的2个位置产生切口的样品S3/20s/S8中,是恢复胸苷激酶活性的细胞的比例为5.07±1.47%的高效率。显示样品S3/20s/S11中恢复胸苷激酶活性的细胞的比例也比样品S3/20s/emp和样品20s/S11/emp高,样品S6/20s/S8中恢复胸苷激酶活性的细胞的比例也比样品S6/20s/emp和样品20s/S8/emp高,另外,样品S6/20s/S11中恢复胸苷激酶活性的细胞的比例也比样品S6/20s/emp和样品20s/S11/emp高。
[实施例4]不利用外来性DNA的多切口法的基因组编辑效率的检验A.材料
将以图11所示的区域(S3、20s、29s、S8)作为靶标的sgRNA或人基因组中不存在靶标序列的sgRNA(no)如下组合,与Cas9D10A mRNA一起导入TSCER2细胞。
S3/20s/S8:分别100μM各0.3μL
S3/29s/S8:分别100μM各0.3μL
S3/20s:分别100μM各0.45μL
20s/S8:分别100μM各0.45μL
S3/S8:分别100μM各0.45μL
S3:100μM 0.9μL
20s:100μM 0.9μL
29s:100μM 0.9μL
S8:100μM 0.9μL
no:100μM 0.9μL
另外,将各样品中切口产生的位置示于图12。各样品中的切割模式(参照图1A~H)如下。
S3/20s/S8:模式5
S3/29s/S8:模式6
S3/20s:模式2(a)
20s/S8:模式4(b)
S3/S8:模式1
S3:切口在供者等位基因的碱基和受者等位基因的对应碱基中各一处
20s:切口仅为一处
29s:切口在供者等位基因的碱基和受者等位基因的对应碱基各一处
S8:切口在供者等位基因的碱基和受者等位基因的对应碱基各一处
no:切口不产生
B.方法和结果
在70×10
将结果示于图11。在不使用一切外来性DNA、并且不进行利用细胞分选仪的细胞选择的条件下,在受者等位基因的3个位置、供者等位基因的2个位置产生切口的样品S3/20s/S8(图1G模式5)中,胸苷激酶活性以3.46±0.34%的高效率恢复。在受者等位基因的3个位置、供者等位基因的3个位置产生切口的样品S3/29s/S8(图1G模式6)中,也在2.64±0.58%的细胞中胸苷激酶活性恢复。另外,在切口在距离靶标核苷酸1000bp以上的位置的情况下,在受者等位基因的2个位置、供者等位基因的2个位置产生切口的样品S3/S8(图1A模式1)中胸苷激酶活性以1.54±0.40%恢复,与在受者等位基因的1个位置、供者等位基因的1个位置产生切口的样品S3(0.133±0.026%)、29s(0.844±0.305%)、S8(0.773±0.221%)、在受者等位基因的1个位置产生切口、在供者等位基因中不产生切口的样品20s(0.147±0.022%)相比,胸苷激酶活性以高效率恢复。
产业可利用性
如以上说明的那样,根据本发明,利用通过由位点特异性切口酶产生的单链切割而被诱导的同源染色体之间的同源重组,能够将在同源染色体之间不同的碱基统一成任一方的碱基。不使用外来的供者DNA的本发明由于其安全性高,因而能够特别大地贡献于对由杂合突变引起的疾病的基因治疗。
序列表自由文本
序列号:15~36
·人工序列
- 基因组编辑方法、组合物、细胞、细胞制剂、以及细胞制剂的制造方法
- 经基因组编辑的细胞的制造方法