一种基于三代PacBio和Hi-C技术组装和注释贵德黑裘皮羊基因组的方法
文献发布时间:2023-06-19 11:34:14
技术领域
本发明属于生物信息技术领域,具体涉及一种基于三代PacBio和Hi-C技术组装和注释贵德黑裘皮羊基因组的方法。
背景技术
黑色素是一种生物多聚体,广泛的存在于动植物及微生物中。动物的毛发、皮肤和眼睛的颜色均由黑色素的相对数量、性质和分布所决定的。黑色素可以抑制紫外辐射产生的自由基,以颗粒形式,来吸收和反射非离子辐射,还可以作为弱阳离子交换多聚体,具有结合大量金属离子或药物的能力,可以保护蛋白质不被降解。但色素沉积是一个受多基因控制的复杂性状,已在哺乳动物中影响毛色的150多个基因中的鉴定出了300多个基因位点。
贵德黑裘皮羊(Guide BlackFur sheep)又名青海黑臧羊、贵德黑紫羔,属于裘皮用型绵羊地方品种,具有体质结实、抗寒、抗病力强、适应性好、善于登山远牧、夏季抓膘肥育迅速等特点。其所产黑紫羔皮皮板坚韧,毛色黝黑发亮,花形美观、卷花坚实,羊毛纤维类型比例适中,不易擀毡,保暖性好。贵德黑裘皮羊被毛为黑红色,部分为微黑红色,个别呈灰色,是研究动物黑色素沉积性状的理想素材。关于贵德黑裘皮羊的毛色色素沉积研究不仅有助于解析其毛色性状遗传机理,还可以辅助贵德黑裘皮羊育种。此外,贵德黑裘皮羊中心产区位于青海省海南藏族自治州的贵南县,平均海拔3100m,贵德黑裘皮羊也是研究动物高原低氧适应性状的理想素材。目前,关于贵德黑裘皮羊的基因组学研究中,缺乏贵德黑裘皮羊的高质量参考基因组。这制约了贵德黑裘皮羊关于其毛色的色素积淀机制研究和高寒低氧适应的分子机制的研究,同时不利于贵德黑裘皮羊品种资源的保存、挖掘和利用。
PacBio测序是基于光信号的三代测序技术,以单分子测序为特征,可以在在目标DNA分子复制过程中捕获序列信息,因其测序读长较长,对于基因组中复杂区域尤其是复杂结构变异的研究具较大的优势。Hi-C技术源于染色体构象捕获技术,利用高通量测序技术,结合生物信息分析方法,研究全基因组范围内整个染色质DNA在空间位置上的关系,获得高分辨率的染色质三维结构信息。Hi-C技术用于基因组组装,能够将杂乱的基因序列组装到染色体水平。专利CN201811068666.5公开了一种基因组的组装方法及装置,所述方法包括:获取纠错后的基因信息,所述基因信息包括基因组和比对信息;根据所述基因组和所述比对信息进行基因组组装;若判断获知组装结果未达到预设条件,对所述组装结果进行分区域聚类处理,并将处理结果进行重新组装。但是,上述专利并没有公开其可以应用于绵羊的基因组,并可以构建染色体水平的贵德黑裘皮羊的参考基因组。
本研究结合三代PacBio和Hi-C技术,并采用二代、三代转录组文库校正拼装序列,首次构建了染色体水平的贵德黑裘皮羊参考基因组,为贵德黑裘皮羊色素沉积的分子机制研究和高寒低氧适应的分子机制研究奠定了基础。同时也为其优良遗传资源保护和利用、选择育种和遗传改良等工作提供数据支撑。
发明内容
针对上述技术问题,本发明提供了一种基于三代PacBio和Hi-C技术组装和注释贵德黑裘皮羊基因组的方法,所述的方法包括如下步骤:
(1)采集贵德黑裘皮羊血液和组织样本:分别提取贵德黑裘皮羊血液和组织样本的DNA和RNA;
(2)构建基因组文库和转录组文库:针对步骤(1)提取得到的DNA片段分别构建二代DNA文库、三代DNA文库和Hi-C文库,获得贵德黑裘皮羊基因组文库;针对步骤(1)提取得到的RNA片段构建二代转录组文库和三代全长转录组文库,获得贵德黑裘皮羊转录组文库;并对构建所得的文库进行过滤;
(3)采用步骤(2)获得的二代DNA文库评估获得贵德黑裘皮羊的基因组大小和杂合率;
(4)基因组组装、纠错和评估:利用步骤(3)获得的贵德黑裘皮羊的基因组大小和杂合率结果,选择mecat2软件对步骤(2)所述的过滤后的三代DNA文库进行组装得到原始组装结果;使用smrtlink 7.0的纠错软件arrow基于步骤(2)得到的三代全长转录组文库对原始组装结果进行纠错,使用pilon软件基于步骤(2)得到的二代转录组文库进行再纠错;
(5)Hi-C辅助组装和评估:对步骤(4)进行再纠错后得到的组装结果进行Hi-C辅助组装,构建互作图谱,进行可视化纠错,得到贵德黑裘皮羊基因组;
(6)基因组注释和评估:重复序列的识别;非编码RNA的预测;编码基因结构预测和功能注释:使用De novo从头预测、homolog同源预测和转录组证据支持3种方法进行编码基因的结构预测,参数设置为默认参数;使用MAKER软件,将上述3种方法预测得到的基因集整合成一个基因集;最后借助于蛋白数据库InterPro、GO、KEGG_ALL、KEGG_KO、SwissProt、TrEMBL和NR对基因集中的蛋白进行功能注释。
优选地,步骤(4)所述的原始组装的mecat2软件参数设置为:MIN_READ_LENGTH=10000,CNS_OPTIONS="-r 0.6-a 1000-c 4-l 2000",ASM_OVLP_OPTIONS="-n 100-z 10-b 2000-e 0.5-j 1-u 0-a 400"。
优选地,步骤(4)所述的纠错软件arrow的版本为v2.2.2,参数设置为默认参数;pilon软件的版本为v1.22,参数设置为默认参数。
优选地,步骤(5)所述的Hi-C辅助组装是指使用ALLHi C软件将纠错后的组装结果进行组装;利用Juicer软件构建互作图谱,使用JucieBox软件对其进行可视化纠错。
优选地,步骤(5)所述的Hi-C辅助组装所需要的序列是将步骤(4)质控得到的clean data使用BWA软件进行比对,使用Lachesis软件将离酶切位点500bp以外的序列去除,得到的数据进行Hi-C辅助组装。
优选地,步骤(6)所述的重复序列的识别是结合基于RepBase库的同源预测方法、基于自身序列比对及重复序列特征的De novo从头预测方法检测重复序列;还利用了TRF软件寻找基因组中串联重复序列;非编码RNA的预测:非编码RNA的注释过程中,根据tRNA的结构特征,利用tRNAscan-SE软件来寻找基因组中的tRNA序列。
优选地,步骤(1)所述的DNA提取自贵德黑裘皮羊血液和肝脏组织;RNA提取自贵德黑裘皮羊组织,所述的贵德黑裘皮羊组织是指心脏、肝脏、肺脏、脾脏、瘤胃和肌肉。
优选地,步骤(2)所述的二代DNA文库的构建方法为:通过超声波破碎仪将提取获得的贵德黑裘皮羊DNA随机打断成长度为300-350bp的片段;DNA片段进行末端修复、加A尾、加测序接头、纯化、PCR扩增;
三代DNA文库的构建方法为:将基因组DNA剪切至20kb大小;DNA片段纯化和浓缩;DNA片段进行末端修复、加测序接头;目的片段筛选;杂交测序引物和DNA聚合酶绑定;Hi-C文库的构建方法为:使用多聚甲醛固定提取获得的贵德黑裘皮羊DNA的构象;限制性内切酶处理交联的DNA,产生粘性末端;DNA末端补平修复,并同时引入生物素,标记寡核苷酸末端;使用DNA连接酶连接DNA片段;蛋白酶消化解除与DNA的交联状态,纯化DNA并随机打断为300~500bp片段;使用亲和素磁珠捕获标记的DNA,对DNA片段进行末端修复、加A尾、加测序接头、纯化、PCR扩增;
二代转录组文库的构建方法为:从提取获得的贵德黑裘皮羊RNA中富集mRNA;通过超声波破碎仪将富集到的mRNA随机打断成200bp的片段;以片段化的mRNA为模板,利用随机引物反转录合成一链cDNA,合成第二链cDNA时dNTPs中的dTTP用dUTP代替;cDNA片段进行纯化、末端补平、加A尾、加测序接头;使用USER酶消化二链cDNA,使其文库中只含有一链cDNA;cDNA进行PCR富集,获得贵德黑裘皮羊的二代转录组文库;
三代全长转录组文库的构建方法为:合成mRNA的全长cDNA;纯化扩增的全长cDNA,去除1kb以下的小片段cDNA;对全长cDNA进行末端修复,连接SMRT哑铃型接头;进行核酸外切酶消化未连接接头的片段,再次使用PB磁珠进行纯化,获得贵德黑裘皮羊的三代转录组文库。
优选地,二代DNA文库的测序数据过滤条件为:(1)去除含有接头序列的reads;(2)去除重复reads;(3)当单端测序read中的一端含有的N的含量超过该条read长度比例的10%时,需要去除此对paired reads;(4)当单端测序read中的一端含有的Q≤5碱基数超过该条read长度比例的50%时,需要去除此对reads;
三代DNA文库的测序数据过滤条件为:(1)去除含有接头序列的reads;(2)去除长度短于1000bp的reads;(3)去除低质量reads;
Hi-C文库的测序数据过滤条件为:(1)去除含有接头序列的reads;(2)去除测序read两端连续质量小于20的碱基;(3)当测序read最终长度小于50bp时,去除此条reads;(4)仅保留成对reads;
二代转录组文库测序数据过滤条件为:(1)去除含有接头序列的reads;(2)去除3’端;(3)去除低质量reads;
三代全长转录组文库的测序数据过滤条件为:(1)使用SMRTlink软件对原始测序数据进行预处理,参数设置为最短Subreads长度=50,最大Subreads长度=15,000,最小测序循环数=3,最低预测准确性=0.99,对单分子测序的高质量reads进行拆分得到subreads,同一高质量reads得到的subreads经过自我纠错形成环化一致序列;(2)通过检测嵌合体序列、5’和3’端测序引物,对环化一致序列进行分类,找出全长非嵌合序列用于后续分析。
本发明的第二目的是提供所述的方法组装和注释得到的贵德黑裘皮羊基因组序列。
本发明的有益效果是:1.本发明提供了一种基于三代PacBio和Hi-C技术组装和注释贵德黑裘皮羊基因组的方法,所述的方法使得贵德黑裘皮羊基因组达到染色体级别,取得高质量的参考基因组,并对贵德黑裘皮羊的基因结构和基因功能进行了全面注释,对于贵德黑裘皮羊的遗传改良和物种资源保护至关重要,进一步为贵德黑裘皮羊的羊毛色素积淀的分子机制研究和高寒低氧适应的分子机制研究奠定了基础。2.本发明的组装方法获得了连续性更好的贵德黑裘皮羊参考基因组,为后续进行大规模基因组进化和功能研究提供保障。3.本发明构建组装获得的贵德黑裘皮羊基因组质量是现有文献中公开的最高水平,最终确定贵德黑裘皮羊基因组顺序及方向确定的染色体长度2.69Gb,contigs N50=20.30Mb,scaffoldN50=107.63Mb,contig长度锚定率为98.72%,contig数量锚定率为64.55%。PacBio组装和Hi-C辅助组装,并纠错后能完整比对BUSCO的基因占93.10%;4.采用本发明方法鉴定到的重复序列更多,基因注释的完整性更高,注释到的基因数目更加接近于绵羊的平均基因数量。
附图说明
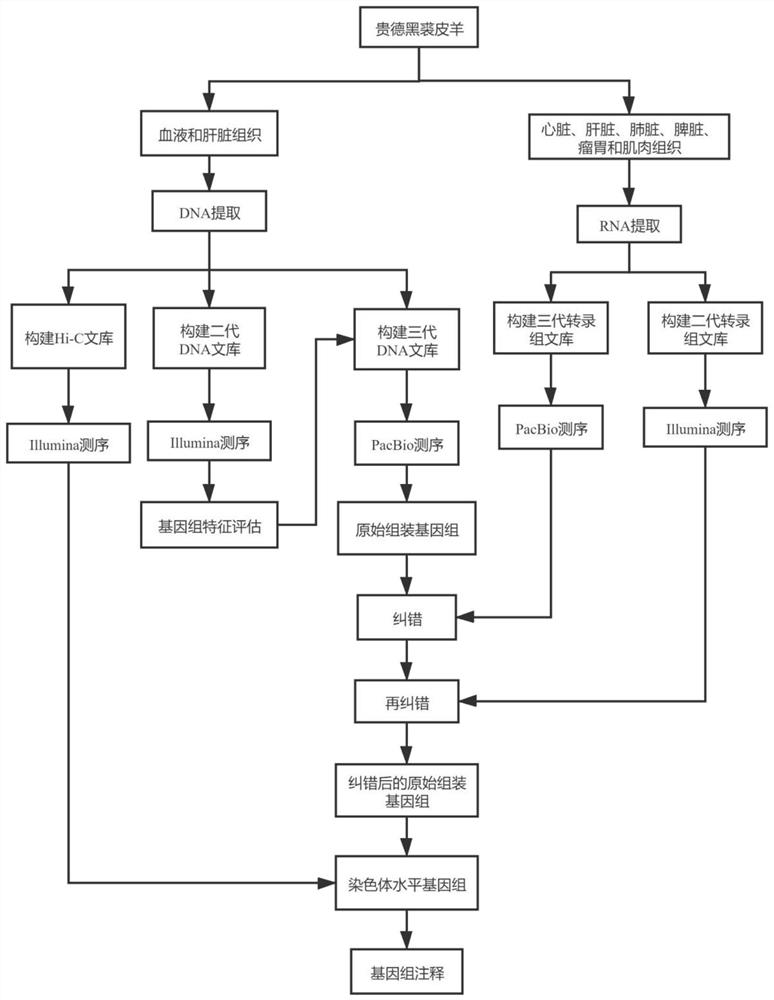
图1贵德黑裘皮羊基因组组装技术路线示意图
图2K-mer深度和K-mer种类数频率分布图
具体实施方式
为使本发明实施例的目的、技术方案和优点更加清楚,下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地说明,应当理解地是,以下实施例仅是本发明的一部分实施例而不是全部的实施例。在下述实施例的基础上,本领域技术人员在没有作出创造性劳动的前提下所获得的所有其他实施例,均属于本发明的保护范围。
名词解释:
以下实施例中所述的“reads”是高通量测序平台产生的序列标签。
以下实施例所述的“contig”是拼接软件基于reads之间的overlap区,拼接获得的序列。
以下实施例所述的“Contig N50”是reads拼接后会获得一些不同长度的Contigs。将所有的Contig长度相加,能获得—个Contig总长度。然后将所有的Contigs按照从长到短进行排序,如获得Contig 1,Contig 2,Contig3,……,Contig 25。
将Contig按照这个顺序依次相加,当相加的长度达到Contig总长度的一半时,最后一个加上的Contig长度即为ContigN50。举例:Contig l Contig 2Contig 3Contig 4=Contig总长度*1/2时,Contig 4的长度即为Contig N50。ContigN50可以作为基因组拼接的结果好坏的一个判断标准。
以下实施例所述的“Scaffold”是基因组de novo测序,通过reads拼接获得Contigs后,往往还需要构建454Paired-end库或llluminaMate-pair库,以获得一定大小片段(如3Kb、6Kb、10Kb、20Kb)两端的序列。基于这些序列,可以确定—些Contig之间的顺序关系,这些先后顺序已知的Contigs组成Scaffold。
以下实施例所述的“Scaffold N50”,Scaffold N50与Contig N50的定义类似。Contigs拼接组装获得—些不同长度的Scaffolds。将所有的Scaffold长度相加,能获得一个Scaffold总长度。然后将所有的Scaffolds按照从长到短进行排序,如获得Scaffold 1,Scaffold 2.Scaffold3,……,Scaffold 25。将Scaffold按照这个顺序依次相加,当相加的长度达到Scaffold总长度的一半时,最后一个加上的Scaffold长度即为Scaffold N50。举例:Scaffold l Scaffold 2Scaffold 3Scaffold 4Scaffold 5=Scaffold总长度*1/2时,Scaffold 5的长度即为Scaffold N50。ScaffoldN50可以作为基因组拼接的结果好坏的—个判断标准。
以下实施例所述的“smartlink软件”是深圳市华杰智控技术有限公司推出的实现PLC远程控制、远程下载、远程组网和监控的软件。同时支持PLC、触摸屏、伺服器、运动控制器、仪表仪器等设备远程控制。
实施例一、一种基于三代PacBio和Hi-C技术组装和注释贵德黑裘皮基因组的方法
1材料与方法
1.1样品采集
在青海省海南藏族自治州的贵南县贵德黑裘皮羊中心产区选择健康的成年公羊1只(海拔>3500m),早晨空腹通过颈静脉采血5ml加入EDTA-K2抗凝剂的采血管中,保存于-20℃冰箱。之后屠宰并立即分割心脏、肝肺、肺脏、脾脏、瘤胃和肌肉组织,迅速用预冷的生理盐水将组织表面的血液冲洗干净,切割成0.5cm
1.2文库构建及测序
采用血液基因组提取试剂盒(天根生化科技北京有限公司,北京)和CTAB(十六烷基三甲基溴化胺)法提取贵德黑裘皮羊血液和肝脏组织中的DNA并进行质检,分成三份用于二代、三代和Hi-C文库构建。使用TRlzol Reagent(Invitrogen,美国)提取贵德黑裘皮羊心脏、肝脏、肺脏、脾脏、瘤胃和肌肉组织中的总RNA并进行质检,用于二代转录组和三代全长转录组文库构建。
二代DNA文库构建:通过超声波破碎仪(Covaris,美国)随机打断成长度为300-350bp的片段;DNA片段进行末端修复、加A尾、加测序接头、纯化、PCR扩增;使用Qubit 2.0(Invitrogen,美国)和Agilent 2100(Agilent,美国)对构建文库的浓度和片段大小进行质量检测;构建好的文库通过IlluminaHiseq PE150测序。本实施例中的二代DNA文库用于预测基因组大小和杂合率,Illumina Hiseq PE150平台测序结果显示贵德黑裘皮羊基因组大小约为2669.83Mb,杂合率为0.39%,基因组杂合率较低,适宜于进行三代DNA文库的构建,并且该结果为后续选择组装策略提供了依据。
三代DNA文库构建:使用g-Tubes(Covaris,美国)将基因组DNA剪切至~20kb大小;DNA片段纯化和浓缩;DNA片段进行末端修复、加测序接头;目的片段筛选(20kb);杂交测序引物和DNA聚合酶绑定;使用Qubit 2.0(Invitrogen,美国)和Agilent 2100(Agilent,美国)对构建文库的浓度和片段大小进行质量检测;构建好的文库通过PacBio Sequel II平台进行测序。
Hi-C文库构建:使用多聚甲醛固定DNA构象;限制性内切酶处理交联的DNA,产生粘性末端;DNA末端补平修复,并同时引入生物素,标记寡核苷酸末端;使用DNA连接酶连接DNA片段;蛋白酶消化解除与DNA的交联状态,纯化DNA并随机打断为300~500bp片段;使用亲和素磁珠捕获标记的DNA,对DNA片段进行末端修复、加A尾、加测序接头、纯化、PCR扩增;使用Qubit 2.0(Invitrogen,美国)和Agilent 2100(Agilent,美国)对构建文库的浓度和片段大小进行质量检测;构建好的文库通过Illumina HiSeq PE150测序。
二代转录组文库构建:使用带有Oligo(dT)的磁珠进行mRNA富集;通过超声波破碎仪(Covaris,美国)将富集到的mRNA随机打断成约200bp的片段;以片段化的mRNA为模板,利用随机引物反转录合成一链cDNA,合成第二链cDNA时dNTPs中的dTTP用dUTP代替;cDNA片段进行纯化、末端补平、加A尾、加测序接头;使用USER酶消化二链cDNA,使其文库中只含有一链cDNA;cDNA进行PCR富集;使用Qubit 2.0(Invitrogen,美国)和Agilent 2100(Agilent,美国)对构建文库的浓度和片段大小进行质量检测;构建好的文库通过Illumina Hiseq2500平台进行双端测序。
三代全长转录组文库构建:使用Clonetech SMARTerTM PCR cDNA Synthesis Kit合成mRNA的全长cDNA;使用PB磁珠纯化扩增的全长cDNA,去除部分1kb以下的小片段cDNA;对全长cDNA进行末端修复,连接SMRT哑铃型接头;进行核酸外切酶消化未连接接头的片段,再次使用PB磁珠进行纯化,获得测序文库;使用Qubit 2.0(Invitrogen,美国)和Agilent2100(Agilent,美国)对构建文库的浓度和片段大小进行质量检测;构建好的文库通过PacBio测序仪进行全长转录组测序。
1.3测序数据质控
DNA和RNA测序结束后得到原始数据(Raw reads),使用Fast QC软件进行过滤获得高质量序列(Clean reads)。
二代DNA测序数据过滤条件为:1)去除含有接头序列的reads;2)去除重复reads;3)当单端测序read中的一端含有的N的含量超过该条read长度比例的10%时,需要去除此对paired reads;4)当单端测序read中的一端含有的低质量(<=5)碱基数超过该条read长度比例的50%时,需要去除此对paired reads。质控后共获得249998309340bp的cleandata。
三代DNA测序数据过滤条件为:1)去除含有接头序列的reads;2)去除长度短于1000bp的reads;3)去除低质量reads。质控后共获得2729705014bp的clean data。
Hi-C测序数据过滤条件为:1)去除含有接头序列的reads;2)去除测序read两端连续质量小于20的碱基;3)当测序read最终长度小于50bp时,去除此条reads。4)仅保留成对reads。质控后共获得2694746666bp的clean data。
二代转录组测序数据过滤条件为:1)去除含有接头序列的reads;2)去除3’端;3)去除低质量reads。质控后3个肝脏样品平均获得9151750600bp的clean data。
三代全长转录组测序数据过滤条件为:1)使用SMRTlink软件(参数设置为最短Subreads长度=50,最大Subreads长度=15,000,最小测序循环数=3,最低预测准确性=0.99)对原始测序数据进行预处理,对单分子测序的高质量reads进行拆分得到subreads,同一高质量reads得到的subreads经过自我纠错形成环化一致序列;2)通过检测嵌合体序列、5’和3’端测序引物,对环化一致序列进行分类,找出全长非嵌合序列用于后续分析。质控后具有polyA的全长非嵌合序列为289585775bp。
1.4基因组大小和杂合率评估
获得的高质量序列(Clean reads),采用基于K-mer的分析方法来估计基因组大小、杂合率和重复序列信息。其主要目的是了解该基因组的复杂程度。通过Illumina HiseqPE150平台测序后获得贵德黑裘皮羊二代基因文库,共包括249998309340bp的clean data,测序质量正常,测序错误率正常。选择K=17,可以产生的K-mer种类数为4
1.5基因组组装、纠错和评估
利用1.4获得的贵德黑裘皮羊的基因组大小和杂合率结果,使用mecat2软件得到原始组装结果,参数设置为:MIN_READ_LENGTH=10000,CNS_OPTIONS="-r 0.6-a 1000-c4-l2000",ASM_OVLP_OPTIONS="-n 100-z 10-b 2000-e 0.5-j 1-u 0-a 400"。
使用smrtlink 7.0的纠错软件arrow(v2.2.2,默认参数)基于三代全长转录组文库对原始组装结果进行纠错,使用pilon软件(v1.22,默认参数)基于二代转录组文库再进行纠错。
原始组装并纠错后获得2729705014bp的基因组序列,Contig number为1972,Contigs N50为20303496bp,Scaffoldnumber为726,ScaffoldN50为100767354bp。
组装和纠错后对基因组进行评估,统计A、G、C、T和N在基因组中的占比和GC含量;选择绵羊基因组中CLR(Continuous Long Reads)subreads,使用minimap2软件(默认参数)比对到组装好的基因组,统计reads的比对率、覆盖基因组程度和深度分布情况,由此评估组装的完整性和测序覆盖的均匀性;基于OrthoDB中的单拷贝同源基因集,使用BUSCO预测这些基因并统计其完整度、碎片化程度和可能的丢失率,由此评估整个组装结果中基因区的完整性;用BWA将reads比对到参考基因组,用GATK进行SNP calling并过滤,统计纯合和杂合SNP个数,根据比对结果统计insersize。
1.6 Hi-C辅助组装和评估
利用纠错后的组装结果进行Hi-C辅助组装,得到最终的基因组组装结果。质控得到的clean data使用BWA软件进行比对,使用Lachesis软件将离酶切位点500bp以外的序列去除,得到的数据进行辅助组装。基于顺式互作(同一染色体内的互作)远大于反式互作(不同染色体间的互作),且顺式互作中线性距离越近则互作越强的原理,将contigs或者scaffolds进行聚类、排序、定向,得到染色体水平基因组。将辅助组装后的基因组,利用Juicer软件构建互作图谱,使用JucieBox软件对其进行可视化纠错。基于OrthoDB中的单拷贝同源基因集,使用BUSCO软件预测这些基因并统计其完整度,碎片化程度及可能的丢失率。Hi-C辅助组装后获得2694746666bp的基因组序列,Contig number为1273,Contigs N50为20303496bp,Scaffoldnumber为27,ScaffoldN50为107633389bp,能完整比对BUSCO的基因占93.10%。
构建流程如图1所示。
1.7基因组注释和评估
重复序列的识别:结合基于RepBase库(http://www.girinst.org/repbase)的同源预测方法(RepeatMasker和RepeatProteinMask)、基于自身序列比对(RepeatModeler)及重复序列特征(LTR-FINDER)的De novo从头预测方法检测重复序列。此外,De novo从头预测方法还利用了TRF软件寻找基因组中串联重复序列。
非编码RNA的预测:非编码RNA的注释过程中,根据tRNA的结构特征,利用tRNAscan-SE软件来寻找基因组中的tRNA序列。由于rRNA具有高度的保守性,因此可以选择近缘物种的rRNA序列作为参考序列,通过BLASTN比对来寻找基因组中的rRNA。利用Rfam家族的协方差模型,采用Rfam自带的INFERNAL软件预测基因组上的miRNA和snRNA序列信息。
基因结构预测和功能注释:使用De novo从头预测(Augustus和Genscan软件)、homolog同源预测(选择C.hircus、H.sapiens、O.aries_rambouillet_v1.0和O.aries_Oar_v4.0作为同源物种)和转录组证据支持(trans.orf/ISOseq)3种方法进行编码基因的结构预测,参数设置为默认参数。使用MAKER软件(默认参数),将上述3种方法预测得到的基因集整合成一个非冗余的、更加完整的基因集。最后借助于外源蛋白数据库(InterPro、GO、KEGG_ALL、KEGG_KO、SwissProt、TrEMBL和NR)对基因集中的蛋白进行功能注释。
2结果
2.1贵德黑裘皮羊基因组大小和杂合率评估
通过IlluminaHiseq PE150平台测序后,共获得249998309340bp的clean data,测序质量正常,测序错误率正常。随机抽取10000对reads数据,通过Blast软件比对NCBI核苷酸数据库(NT库),比对结果显示文库数据中不含有明显的外源污染,建库测序成功。K-mer分析结果显示,K=17时可以产生的K-mer总数为223031778170,K-mer深度为81(图2)。因此预估贵德黑裘皮羊基因组大小约为2669.83Mb,杂合率为0.39%,重复序列比例为60.76%,基因组GC含量约为42%。
2.2贵德黑裘皮羊基因组组装结果评估
通过PacBio组装贵德黑裘皮羊基因组序列长度为2.73Gb,contig N50=20.30Mbp,共1972条contigs(表1)。通过Hi-C辅助组装,最终确定贵德黑裘皮羊基因组顺序及方向确定的染色体长度2.69Gb,contigs N50=20.30Mb,scaffold N50=107.63Mb,contig长度锚定率为98.72%,contig数量锚定率为64.55%(表1)。贵德黑裘皮羊基因组GC含量为42.42%(1158123872bp)。PacBio组装和Hi-C辅助组装后能完整比对BUSCO的基因占93.10%。
表1贵德黑裘皮羊基因组PacBio组装Hi-C辅助组装情况统计
2.3贵德黑裘皮羊基因组注释
通过TRF、Repeatmasker、Proteinmask和De novo方法进行贵德黑裘皮羊基因组重复序列注释。去掉4种方法的重叠部分结果,重复序列大小为1443323654bp,占贵德黑裘皮羊基因组的52.86%。非编码RNA中注释到tRNA、rRNA、miRNA和snRNA个数分别为254044(0.6816%)、231(0.0055%)、523(0.0016%)和2025(0.0084%)。
通过MAKER软件整合De novo从头预测、homolog同源预测和转录组证据支持的结果后,在贵德黑裘皮羊基因组中共预测到20504个编码蛋白质的基因,平均基因长度为44767.90bp,平均CDS序列长度为1464.19bp,平均每个基因外显子数目为12.81个,平均外显子长度为216.89bp,平均内含长度为4568.52bp(表2)。
表2贵德黑裘皮羊基因组预测结果统计
借助外源蛋白数据库InterPro、GO、KEGG_ALL、KEGG_KO、SwissProt、TrEMBL和NR对贵德黑裘皮羊基因集中的蛋白进行功能注释,共注释到20226个编码蛋白质的基因,占上述7种蛋白数据库的98.64%(表3)。使用BUSCO软件进行贵德黑裘皮羊基因组注释评估,能完整比对BUSCO的基因有3695个,占比为90.0%。
表3贵德黑裘皮羊基因组注释结果统计
2.4与已报道的绵羊基因组比较分析
通过Illumina、PacBio和Hi-C技术组装出了高质量染色体水平的贵德黑裘皮羊基因组。本发明所述的贵德黑裘皮羊基因组组装中Contigs N50为20.30Mb,ScaffoldN50为107.63Mb,相较于现有技术公开的湖羊、朗布依埃羊(Rambouillet sheep)、马可波罗羊(Marco Polo sheep)、特克赛尔羊(Texel)、雪羊(Snow sheep)以及摩弗仑羊(mouflon),本基因组序列长度更长,质量明显提高,鉴定到的重复序列更多,基因注释的序列完整性更高,注释基因数目更加接近于绵羊的平均基因数量,且本发明首次完成了贵德黑裘皮羊的基因组注释。具体数据见表4。
表4各品种绵羊基因组组装和注释效果比较分析
- 一种基于三代PacBio和Hi-C技术组装和注释贵德黑裘皮羊基因组的方法
- 一种基于三代PacBio和Hi-C技术组装和注释湖羊基因组的方法