一种吡唑二硫的合成方法
文献发布时间:2023-06-19 12:13:22
技术领域
本发明涉及有机合成技术领域,尤其涉及一种吡唑二硫的合成方法。
背景技术
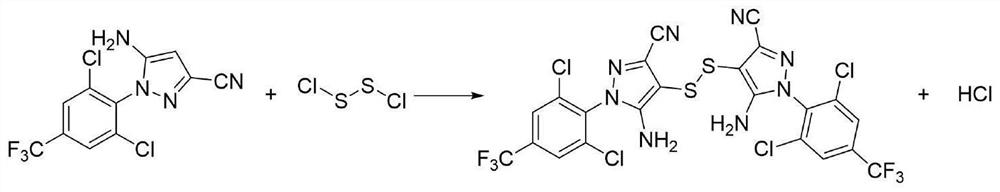
5-氨基-3-氰基-1-(2,6-二氯-4-三氟甲基-苯基)吡唑二硫化物又名1-(2,6-二氯-4-三氟甲基苯基)-3-氰基-5-氨基-吡唑-4-二硫醚,是氟虫腈、乙虫腈、丁烯氟虫腈等N-苯基吡唑类农药的重要中间体。
现有技术中的合成方法通常是采用将5-氨基-3-氰基-1-(2,6-二氯-4-三氟甲基-苯基)吡唑溶于氯苯/乙腈混合溶剂中,与一氯化硫反应后,用氨水中和至pH值6.5~7,然后过滤分离出含产物二硫化物的滤饼。滤饼再用氯苯、乙腈混合溶液洗涤后干燥,得到目标产物吡唑二硫。工业化生产是将5-氨基-3-氰基-1-(2,6-二氯-4-三氟甲基-苯基)-1-H吡唑与一氯化硫反应后,用氨水、氨气或三乙胺中和,压滤分离,滤饼用热水洗涤后干燥得到吡唑二硫。由于在合成5-氨基-3-氰基-1-(2,6-二氯-4-三氟甲基-苯基)吡唑二硫化物过程中,通常采用甲苯、四氯化碳、乙腈、氯苯以及等溶剂,使用上述溶剂进行合成时,取代反应比较剧烈,反应温度难以把控,容易造成氯化硫分解生成硫单质,在硫单质存在下吡唑二硫容易发生氧化反应生成吡唑三硫、吡唑四硫,影响产品纯度。同时该工艺处理后,产生溶剂中含有大量水,分离困难(乙腈、水共沸),分离后产生大量废水。废水中氨氮、COD含量较高,处理困难。
发明内容
本发明要解决的技术问题是如何减少取代反应过程中硫单质、吡唑三硫、吡唑四硫杂质的产生,并且提高溶剂回收效率,针对上述要解决的技术问题,现提出一种吡唑二硫的合成方法。
为实现上述目的,本发明提供如下技术方案:一种吡唑二硫的合成方法,通过如下步骤完成吡唑二硫的合成:
1)溶解吡唑环,将吡唑环原料溶解在醋酸丁酯中;并将预先称量好的还原剂加入到溶液中,充分分散混合;
2)溶解氯化硫,将氯化硫溶解在醋酸丁酯中;
3)取代反应,将步骤1)和步骤2)得到的溶解液混合后进行取代反应;
4)中和反应,向步骤3)反应后的混合液中加入碱性物质进行中和;
5)加热分层,将步骤4)中和后的反应液体加热至80~100℃,持续加热直至分层;
6)冷却结晶,将步骤5)中分层后的有机层进行冷却结晶;
7)干燥,将步骤6)中结晶液过滤得到的固体进行干燥得到固体吡唑二硫产品。
进一步的,所述步骤1)中的还原剂为四氢铝锂、硼氢化钠、硼氢化钾、亚硫酸钠、硫代硫酸钠中的一种,所述还原剂的添加量占氯化硫摩尔量的0.01~0.1。
进一步的,所述步骤1)和2)中,氯化硫与吡唑环的摩尔比为1:1.8~1:2.0。
进一步的,所述步骤3)中的取代反应温度为10~25℃。
进一步的,所述步骤4)中加入碱性物质中和至pH=6.5~7.5停止,所述碱性物质为氢氧化钠溶液、碳酸钠溶液、碳酸钾溶液、氨水中的一种或多种的混合物。
进一步的,所述步骤6)冷却结晶过程中过滤后的溶剂可用于重复套用。
与现有技术相比,本发明的有益效果是:
本发明用醋酸丁酯稀释氯化硫后再与吡唑环的醋酸丁酯溶液混合,使取代反应温度可控、反应更安全,并且反应的氯化硫配比过量,提高了生产效率,避免吡唑环原料反应不完全造成损失,减少了杂质硫单质的生成;通过在反应体系中加入催化量的还原剂,使吡唑三硫、吡唑四硫杂质减少,产品纯度高;另外,通过利用醋酸丁酯仅微溶于水的特性,溶剂回收时能通过简单的分层除去醋酸丁酯中的大部分水分,溶剂回收处理费用大大降低;并且本方法相较于现有技术中的传统方法,整体步骤减少,生产效率得到了大大提高。
附图说明
图1为本发明的化学合成路线图;
图2为本发明的方法流程示意图;
图3为吡唑环的高效液相色谱图。
具体实施方式
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
实施例1
本具体实施方式披露了一种吡唑二硫的合成方法,通过如下步骤完成吡唑二硫的合成:向干燥的装有回流冷凝管的1000ml四口瓶中加入吡唑环150g(98.7%,0.46mol)和600g的醋酸丁酯,同时加入0.95g(98.0%,0.10mol)硼氢化钠,充分搅拌混合,冷却至10℃备用;将一氯化硫34g(98.0%,0.25mol)与200g醋酸丁酯混合后转入250mL的恒压滴液漏斗中备用。控制温度,在30分钟内将滴液漏斗中的氯化硫溶液加入到吡唑环的醋酸丁酯溶液中,滴加完毕温度上升至23℃,在此温度下继续搅拌反应1小时,完成取代反应。在抽真空的条件下,通入空气吹脱30分钟,再加入10%的碳酸钠溶液,调节pH值到7.0。中和后的反应液加热至95℃分层;有机层冷却到10℃以下,保温2小时,抽滤,滤液蒸馏后作为溶剂继续套用到下一批,滤饼真空干燥后,得淡黄色结晶状粉末165.3g,高效液相色谱检测目标产物纯度99.84%,含量98.3%,从而确定收率为93.5%。具体的,高效液相色谱检测方法采用的是面积归一法,含量的测定采用外标法进行确定。可行的,在本发明的其他具体实施方式中,还原剂还可以为四氢铝锂、硼氢化钾、亚硫酸钠或硫代硫酸钠。在其他具体实施例中,用于中和的碱还可以采用氢氧化钠溶液、碳酸钠溶液、碳酸钾溶液、氨水中的一种或多种的混合物。
由于反应过程中,使用的醋酸丁酯仅微溶于水,溶剂回收时能通过简单的分层除去醋酸丁酯中的大部分水分,上述过滤母液只需通过简单的蒸馏工艺分出含水分高的前馏分,后面蒸出的几乎不含水的醋酸丁酯即可用于下一批次的合成,溶剂回收处理费用大大降低。
实施例2
本具体实施方式披露了一种吡唑二硫的合成方法,通过如下步骤完成吡唑二硫的合成:向干燥的装有回流冷凝管的1000ml四口瓶中加入吡唑环150g(98.7%,0.46mol)和600g的醋酸丁酯,同时加入1.59g(98.0%,0.05mol)亚硫酸钠,搅拌混合得到混悬液,冷却至10℃备用;将一氯化硫34g(98.0%,0.25mol)与200g醋酸丁酯混合后转入250mL的恒压滴液漏斗中备用。控制温度,在30分钟内将滴液漏斗中的氯化硫溶液加入到吡唑环的醋酸丁酯溶液中,滴加完毕温度上升至20℃,在此温度下继续搅拌反应1小时,完成取代反应。在抽真空的条件下,通入空气吹脱30分钟,再滴加10%的碳酸钠溶液,调节pH值到6.8。中和后的反应液体加热至98℃分层;有机层冷却到10℃以下,保温2小时,抽滤,滤液蒸馏后作为溶剂继续套用到下一批,滤饼真空干燥后,得淡黄色结晶状粉末164.4g,高效液相色谱检测目标产物纯度99.82%,含量98.1%,收率为92.8%。具体的,高效液相色谱检测方法采用的是面积归一法,含量的测定采用外标法进行确定。
实施例3
本具体实施例披露了针对实施列1和2的过滤母液进行循环套用合成吡唑二硫的方法。
实施列1和2的过滤母液,在70℃,-0.09MPa条件下进行溶剂回收,分走水分偏高的前馏分,得到含水量≤0.15%的醋酸丁酯用于实施列3。向干燥的装有回流冷凝管的1000ml四口瓶中加入吡唑环150g(98.7%,0.46mol)和600g的醋酸丁酯(水分≤0.15%),同时加入1.59g(98.0%,0.05mol)亚硫酸钠,搅拌混合得到混悬液,冷却至10℃备用;将一氯化硫34g(98.0%,0.25mol)与200g醋酸丁酯(水分≤0.15%)混合后转入250mL的恒压滴液漏斗中备用。控制温度,在35分钟内将滴液漏斗中的氯化硫溶液加入到吡唑环的醋酸丁酯溶液中,滴加完毕温度上升至18℃,在此温度下继续搅拌反应1小时,完成取代反应。在抽真空的条件下,通入空气吹脱30分钟,再滴加20%的氨水,调节pH值到6.5。中和后的反应液体加热至95℃分层;有机层冷却到10℃以下,保温2小时,抽滤,滤液蒸馏后作为溶剂继续套用到下一批,滤饼真空干燥后,得淡黄色结晶状粉末165.5g,高效液相色谱检测目标产物纯度99.85%,含量98.5%,得率93.8%。具体的,高效液相色谱检测方法采用的是面积归一法,含量的测定采用外标法进行确定。
实施例4
本具体实施方式披露了一种吡唑二硫的合成方法,通过如下步骤完成吡唑二硫的合成,与实施例1-3不同的是,本具体实施例中氯化硫与吡唑环的摩尔比,在本具体实施例中,氯化硫与吡唑环的摩尔比为1比2。
向干燥的装有回流冷凝管的1000ml四口瓶中加入吡唑环163g(98.7%,0.5mol)和600g的醋酸丁酯,同时加入0.95g(98.0%,0.10mol)硼氢化钠,充分搅拌混合,冷却至10℃备用;将一氯化硫34g(98.0%,0.25mol)与200g醋酸丁酯混合后转入250mL的恒压滴液漏斗中备用。控制温度,在30分钟内将滴液漏斗中的氯化硫溶液加入到吡唑环的醋酸丁酯溶液中,滴加完毕温度上升至23℃,在此温度下继续搅拌反应1小时,完成取代反应。在抽真空的条件下,通入空气吹脱30分钟,再加入10%的碳酸钠溶液,调节pH值到7.0。中和后的反应液体加热至95℃分层;有机层冷却到10℃以下,保温2小时,抽滤,滤液蒸馏后作为溶剂继续套用到下一批,滤饼真空干燥后,得淡黄色结晶状粉末178.7g,高效液相色谱检测目标产物纯度99.84%,含量98.5%,从而确定收率为93.7%。具体的,高效液相色谱检测方法采用的是面积归一法,含量的测定采用外标法进行确定。
此外,上述实施例中,得到的吡唑二硫产品高效液相色谱分析条件为:
高效液相色谱仪:Agilent 1260/ShimadzuLC-20A;色谱柱:250mm×4.6mm(i.d.)不锈钢柱,内装ZORBAX SB-CN、粒径为5μm的填料。流动相:乙腈+0.16%磷酸水=55+45;流速:1.0mL/min;波长:220nm;进样量:5μL;运行时间:50min。
具体操作步骤如下:
标样溶液:称取50.0mg吡唑二硫标准品,置于50mL容量瓶中,加40mL甲醇超声溶解,恢复至室温,用甲醇稀释至刻度,摇匀。
试样溶液:称取50.0mg吡唑二硫试样,置于50mL容量瓶中,加40mL甲醇超声溶解,恢复至室温,用甲醇稀释至刻度,摇匀。
在上述操作条件下,当仪器稳定后,进1针空白溶液,并连续注入数针标样溶液,其连续3针主峰峰面积的相对标准偏差RSD不大于2.0%后,按照标样溶液、试样溶液、试样溶液、标样溶液的顺序进行测定。
5-氨基-3-氰基-1-(2,6-二氯-4-三氟甲基苯基)吡唑的含量,按下述公式进行计算:
其中,式中:
C
r
r
P——标准品的含量,%。
单一杂质含量,按下式计算:
100×A
式中:
Ai,A——分别为试样图谱中其它单一杂质的峰面积与总峰面积。
本发明用醋酸丁酯稀释氯化硫后再与吡唑环的醋酸丁酯溶液混合,使取代反应温度可控、反应更安全,并且反应的氯化硫配比过量,提高了生产效率,避免吡唑环原料反应不完全造成损失;通过在反应体系中加入催化量的还原剂,使反应生成的硫单质、吡唑三硫、吡唑四硫杂质减少,产品纯度高;另外,通过利用醋酸丁酯仅微溶于水的特性,溶剂回收时能通过简单的分层除去醋酸丁酯中的大部分水分,溶剂回收处理费用大大降低;并且本方法相较于现有技术中的传统方法,整体步骤减少,生产效率得到了大大提高。
以上结合附图对本发明的实施方式作了详细说明,但本发明不限于所描述的实施方式。对于本领域的技术人员而言,在不脱离本发明原理和精神的情况下,对这些实施方式进行多种变化、修改、替换和变型,仍落入本发明的保护范围内。
- 一种吡唑二硫的合成方法
- 一种二氢二吡唑3,4-b:4′,3′-e吡啶类化合物的电化学合成方法