一种复合芯材相变微胶囊及其制备方法
文献发布时间:2023-06-19 19:28:50
技术领域
本发明属于相变储能材料技术领域,具体涉及一种复合芯材相变微胶囊及其制备方法。
背景技术
相变微胶囊,也称微胶囊相变储能材料(Microencapsulated Phase ChangeMaterial, MEPCM)是指应用微胶囊技术在固-液相变储能材料微粒表面包覆一层稳定的薄膜,构成具有核-壳结构的复合相变储能材料。相变微胶囊在发生相态转变过程中,内部发生固-液相变,而其外层的薄膜始终保持固态结构,因此这类材料在宏观上表现为固态颗粒。相变微胶囊不仅解决了相变储能材料的稳定性、热导率和体积膨胀等问题,而且简化了相变储能材料的使用工艺并降低其使用成本。
但是,传统的相变微胶囊多以有机相变材料(如石蜡、脂肪酸等)作为相变芯材,虽然相态转变性质稳定却存在着相变潜热值有限、成本高、导热系数低、易燃等不足,为此,人们将目光转向原料来源更加广泛的无机相变材料。水合盐作为一种无机相变材料,具有相变潜热值高、导热系数高、不易燃烧等优势,正在广泛应用于太阳能采暖、地板辐射式采暖、精密组件温控等技术领域。
CN102676124A公开了一种无机水合盐相变储能微胶囊及其制备方法。通过选用合适的有机溶剂,辅以表面活性剂,在超声作用下使芯材与囊材的高分子单体材料形成分散均匀的混合溶液;通过加入引发剂、润滑剂、稳定剂以及阻聚剂等功能性添加剂,在反应条件下使高分子单体在混合溶液中悬浮聚合固化后包覆芯材形成相变储能微胶囊。其中,无机水合盐芯材占微胶囊的质量百分比为50%~80%,乙烯基单体聚合物囊材占微胶囊的质量百分比为20%~50%。但是,该方法制备的相变储能微胶囊囊材为有机材质,造成其导热系数不高;而且,仅依靠囊材包覆,对于无机水合盐芯材过冷和相分离的缺陷并不能彻底解决,多次循环使用后,容易出现这两方面的问题。
CN106244117A公开了一种无机水合盐相变储能微胶囊及其制备方法。将芯材加入去离子水中,配制芯材饱和溶液。将乳化剂加入到芯材的饱和溶液中,超声乳化20min~50min,在35℃~60℃的水浴中磁力搅拌一段时间,得到分散均匀的芯材乳液A。将壁材预聚体加入到有机相中,磁力搅拌30min,得到透明预聚体溶液B。将分散均匀的芯材乳液A逐滴滴加到预聚体溶液B中,磁力搅拌形成分散均匀的溶液,滴加pH调节剂,将溶液调至pH值为9.5~11,恒温搅拌6h~10h反应完成,离心洗涤、干燥后得到无机相变储能微胶囊。该方法制备的相变储能微胶囊,由于无机芯材未经任何处理,多次使用后存在过冷和相分离的技术问题。
发明内容
针对现有技术的不足,本发明提供了一种复合芯材相变微胶囊及其制备方法。本发明的复合芯材相变微胶囊具有更广泛相变控温范围,改善了无机水合盐芯材的过冷和相分离问题;通过靶向性沉积反应获得无机壳材,既提高了包覆率和包覆完整性,也有效克服有机壳材导热系数低的缺点。
本发明的复合芯材相变微胶囊的制备方法,包括以下内容:
(1)将熔融态无机水合盐、糖醇和聚醚胺按比例混合反应得到复合相变芯材;然后复合相变芯材与水混合,得到水相物质;
(2)将一定量非水溶性有机溶剂与表面活性剂混合,得到油相物质;
(3)将水相物质缓慢滴加到油相物质中,剧烈搅拌反应,得到均质的油包水乳液;
(4)向油包水乳液中缓慢加入可溶性钡盐水溶液,一定温度下持续搅拌反应,反应结束后,反应物料过滤、洗涤、干燥,得到复合芯材相变微胶囊。
在步骤(1)中,所述的无机水合盐选自六水合硝酸锌、十二水磷酸氢二钠或十水硫酸钠中的一种或多种。所述的糖醇选自木糖醇、赤藓糖醇或山梨糖醇中的一种或多种。所述的聚醚胺的平均分子量为1000~2000的聚醚胺。
在步骤(1)中,所述的无机水合盐、糖醇、聚醚胺的质量比为1:0.01~0.08:0.001~0.008,优选1:0.03~0.06:0.003~0.006。
在步骤(1)中,无机水合盐、糖醇和聚醚胺混合反应条件为:反应温度为90℃~150℃,优选110℃~130℃,反应时间为15~45分钟,优选25~35分钟;反应一般在搅拌条件下进行。
在步骤(1)中,所述的复合相变芯材与水的质量比为1:0.5~1.2,优选1:0.7~1。
在步骤(1)中,所述的复合相变芯材与混合阶段,反应温度为60℃~95℃,优选75℃~85℃;反应时间为10~30分钟,优选15~25分钟。混合在搅拌条件下进行。
在步骤(2)中,所述的非水溶性有机溶剂选自环庚烷、环辛烷、环癸烷中的一种或多种,优选环癸烷。所述的表面活性剂选自阴离子表面活性剂和非离子表面活性剂的组合物,其中阴离子表面活性剂选自十二烷基硫酸钠、十二烷基苯磺酸钠、十二烷基萘磺酸钠中的一种或多种;非离子表面活性剂选自辛烷基酚聚氧乙烯醚-10、壬基酚聚氧乙烯醚-10、吐温20中的一种或几种。
在步骤(2)中,所述的非水溶性有机溶剂和表面活性剂的质量比为1:0.05~0.5,优选1:0.08~0.25。其中,阴离子表面活性剂和非离子表面活性剂的质量比为1:0.04~0.1,优选1:0.06~0.08。
在步骤(2)中,所述的混合温度为30℃~70℃,优选45℃~55℃,混合时间为5~25分钟,优选10~15分钟。混合在搅拌条件下进行。
在步骤(3)中,所述的水相物质的滴加速率为0.5g/min~3g/min,优选1g/min~2g/min。
在步骤(3)中,所述的剧烈搅拌反应温度为70℃~115℃,优选90℃~105℃,反应时间为25~55分钟,优选35~45分钟;搅拌转速为3000rpm~8000rpm,优选5000rpm~6000rpm。
在步骤(4)中,所述的可溶性钡盐选自氯化钡和/或硝酸钡。可溶性钡盐与阴离子表面活性剂的质量比为1:0.55~0.95,优选1:0.68~0.83。
在步骤(4)中,所述的可溶性钡盐水溶液质量百分比浓度为30%~70%,优选50%~60%。所述的滴加速率为0.5g/min~3g/min,优选1g/min~2g/min。
在步骤(4)中,反应温度为70℃~115℃,优选90℃~105℃;反应时间为2.5~8.5小时,优选5~6.5小时。
在步骤(4)中,洗涤采用乙醇水溶液,所述的乙醇水溶液的质量百分比浓度为30%~50%。干燥温度为50℃~100℃,优选70℃~80℃。干燥时间为10~30小时,优选15~20小时。
本发明同时提供一种复合芯材相变微胶囊,所述的复合芯材相变微胶囊的平均粒径尺寸为200nm~400nm,熔化潜热值为105J/g~122J/g,包覆率为65%~75%,导热系数为0.68W·m
本发明制备的复合芯材相变微胶囊可用于太阳能蓄热调温、功能型热流体和大型电子器件温控等多个技术领域。
与现有技术相比,本发明具有如下优点:
(1)本发明采用无机水合盐与糖醇复合后作为相变芯材,糖醇能与无机水合盐基体形成氢键作用,促进基体成核;加入聚醚胺可以增加基体稠度,使成核剂稳定分布在基体材料中,从而使成核剂有效发挥成核作用,有效克服无机水合盐作为相变芯材时易出现过冷和相分离的不利现象;同时,无机水合盐与糖醇通过调变二者间配比,可以获得不同相变温度的复合芯材,拓宽了产品相变温度的适用范围;
(2)本发明采用糖醇作为复合相变芯材的一部分,在水相物质滴定油相物质过程中,与阴离子型表面活性剂和非离子型表面活性剂共同形成复配表面活性剂协同作用,同时复配的表面活性剂得到的使溶液体系更加稳定性;
(3)本发明采用化学沉积方式获得无机壁材,在壁材沉积、预聚阶段分布在水相物质周围的表面活性剂组分可以起到靶向性导航作用,从而使获得的无机壁材薄厚均匀、质地优良,延长复合芯材相变微胶囊的使用周期。
附图说明
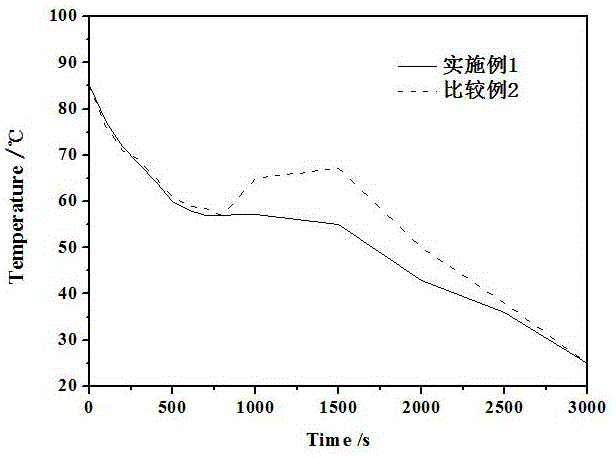
图1为实施例1和比较例2制备的相变微胶囊的100次循环后的步冷曲线。
图2为实施例1制备的相变微胶囊的扫描电镜照片(SEM)。
具体实施方式
下面通过实施例来进一步说明本发明复合芯材相变微胶囊的制备方法和效果。实施例在以本发明技术方案为前提下进行实施,给出了详细的实施方式和具体的操作过程,但本发明的保护范围不限于下述的实施例。
以下实施例中的实验方法,如无特殊说明,均为本领域常规方法。下述实施例中所用的实验材料,如无特殊说明,均从常规生化试剂商店购买得到。
实施例1
取50g六水合硝酸锌、2.5g木糖醇和0.25g聚醚胺D-2000混合,在120℃下搅拌反应30分钟,得到复合相变芯材。取10g复合相变芯材,向其中加入8g去离子水,在80℃下搅拌反应20分钟,得到水相物质。向50g环癸烷中加入7g十二烷基硫酸钠和0.5g辛烷基酚聚氧乙烯醚-10,在50℃下搅拌反应12分钟,得到油相物质。以1.5g/min的速率将水相物质滴加到油相物质中,在100℃下剧烈搅拌反应40分钟,得到均质的油包水乳液。以1.5g/min的速率向乳状液中滴加17.6g氯化钡水溶液,在100℃下搅拌反应6小时,所得悬浮液抽滤后使用质量浓度为40%的乙醇水溶液洗涤,75℃下干燥18小时,得到复合芯材相变微胶囊。
实施例2
取50g六水合硝酸锌、1.5g木糖醇和0.15g聚醚胺D-2000混合,在110℃下搅拌反应25分钟,得到复合相变芯材。取10g复合相变芯材,向其中加入7g去离子水,在75℃下搅拌反应15分钟,得到水相物质。向50g环癸烷中加入3.77g十二烷基硫酸钠和0.23g辛烷基酚聚氧乙烯醚-10,在45℃下搅拌反应10分钟,得到油相物质。以1g/min的速率将水相物质滴加到油相物质中,在90℃下剧烈搅拌反应35分钟,得到均质的油包水乳液。以1g/min的速率向乳状液中滴加10g氯化钡水溶液,在90℃下搅拌反应5小时,所得悬浮液抽滤后使用质量浓度为30%的乙醇水溶液洗涤,70℃下干燥15小时,得到复合芯材相变微胶囊。
实施例3
取50g六水合硝酸锌、3g木糖醇和0.3g聚醚胺D-2000混合,在130℃下搅拌反应35分钟,得到复合相变芯材。取10g复合相变芯材,向其中加入10g去离子水,在85℃下搅拌反应25分钟,得到水相物质。向50g环癸烷中加入11.57g十二烷基硫酸钠和0.93g辛烷基酚聚氧乙烯醚-10,在55℃下搅拌反应15分钟,得到油相物质。以2g/min的速率将水相物质滴加到油相物质中,在105℃下剧烈搅拌反应45分钟,得到均质的油包水乳液。以2g/min的速率向乳状液中滴加25.3g氯化钡水溶液,在105℃下搅拌反应6.5小时,所得悬浮液抽滤后使用质量浓度为50%的乙醇水溶液洗涤,80℃下干燥20小时,得到复合芯材相变微胶囊。
实施例4
同实施例1,不同在于采用十二水磷酸氢二钠代替六水合硝酸锌,其它反应条件和物料组成不变,得到复合芯材相变微胶囊。
实施例5
同实施例1,不同在于采用十水硫酸钠代替六水合硝酸锌,其它反应条件和物料组成不变,得到复合芯材相变微胶囊。
实施例6
同实施例1,不同在于采用赤藓糖醇代替木糖醇,其它反应条件和物料组成不变,得到复合芯材相变微胶囊。
实施例7
同实施例1,不同在于采用山梨糖醇代替木糖醇,其它反应条件和物料组成不变,得到复合芯材相变微胶囊。
实施例8
同实施例1,不同在于取10g复合相变芯材,向其中加入5g去离子水,其它反应条件和物料组成不变,得到复合芯材相变微胶囊。
实施例9
同实施例1,不同在于采用环庚烷代替环癸烷,其它反应条件和物料组成不变,得到复合芯材相变微胶囊。
实施例10
同实施例1,不同在于采用环辛烷代替环癸烷,其它反应条件和物料组成不变,得到复合芯材相变微胶囊。
实施例11
同实施例1,不同在于采用十二烷基苯磺酸钠代替十二烷基硫酸钠,其它反应条件和物料组成不变,得到复合芯材相变微胶囊。
实施例12
同实施例1,不同在于采用十二烷基萘磺酸钠代替十二烷基硫酸钠,其它反应条件和物料组成不变,得到复合芯材相变微胶囊。
实施例13
同实施例1,不同在于采用壬基酚聚氧乙烯醚-10代替辛烷基酚聚氧乙烯醚-10,其它反应条件和物料组成不变,得到复合芯材相变微胶囊。
实施例14
同实施例1,不同在于采用吐温20代替辛烷基酚聚氧乙烯醚-10,其它反应条件和物料组成不变,得到复合芯材相变微胶囊。
实施例15
同实施例1,不同在于将水相物质的滴加速率提高到3g/min,其它反应条件和物料组成不变,得到复合芯材相变微胶囊。
实施例16
同实施例1,不同在于采用硝酸钡代替氯化钡,其它反应条件和物料组成不变,得到复合芯材相变微胶囊。
实施例17
同实施例1,不同在于将氯化钡水溶液的滴加速率降低到0.5g/min,其它反应条件和物料组成不变,得到复合芯材相变微胶囊。
实施例18
同实施例1,不同在于将洗涤的乙醇水溶液的质量浓度提高到50%,干燥温度降低到50℃,干燥时间延长到30小时,其它反应条件和物料组成不变,得到复合芯材相变微胶囊。
比较例1
同实施例1,不同在于省去木糖醇和聚醚胺D-2000,其它反应条件和物料组成不变,得到复合芯材相变微胶囊。
比较例2
同实施例1,不同在于省去聚醚胺D-2000,其它反应条件和物料组成不变,得到复合芯材相变微胶囊。
比较例3
同实施例1,不同在于省去辛烷基酚聚氧乙烯醚-10,其它反应条件和物料组成不变,得到复合芯材相变微胶囊。
比较例4
同实施例1,不同在于将水相物质直接倒入油相物质中,其它反应条件和物料组成不变,得到复合芯材相变微胶囊。
比较例5
同实施例1,不同在于将可溶性钡盐水溶液直接倒入乳状液中,其它反应条件和物料组成不变,得到复合芯材相变微胶囊。
比较例6
同实施例1,不同在于将十二烷基硫酸钠和辛烷基酚聚氧乙烯醚-10的用量分别减少到0.93g和0.07g,其它反应条件和物料组成不变,得到复合芯材相变微胶囊。
比较例7
按照CN102676124A描述的方法,称取15g十二水磷酸氢二钠和4.5g去离子水盛于100mL烧杯中,置于37℃恒温水浴箱中。随后加入0.375g司班80和50mL甲苯,搅拌均匀并超声粉碎30min,形成乳浊液。量取12.5mL甲基丙烯酸甲酯、2.25mL丙烯酸乙酯、0.003g聚丙烯酸钠、0.0072g硬脂酸、0.0036g过氧化二苯甲酰与50mL丙酮混合,超声粉碎20min形成混合液。将混合液逐滴加入乳浊液,磁力搅拌,控制温度在80℃~90℃,反应4h~5h,形成乳液。向上述乳液添加0.0018g对苯二酚单甲醚,混合均匀,室温放置5h~6h,得到含微胶囊的乳液。对乳液洗涤,离心分离,干燥后得到微胶囊产品。
比较例8
按照CN106244117A描述的方法,称取15g五水合硫代硫酸钠,溶于20mL去离子水中,加入0.6g~1g十二烷基硫酸钠,超声分散40min,置于35℃恒温水浴中以800rpm搅拌反应30min,得到芯材乳液A。将6mL~8mL的正硅酸乙酯加入到25mL环己烷中,以600rpm~1000rpm磁力搅拌30min,滴加5mL戊醇,持续搅拌10min,得到预聚体溶液B。将A逐滴加入到B中,在60℃恒温下,以600rpm~1000rpm磁力搅拌30min,滴加3-氨丙基二乙氧基硅烷,将溶液pH值调至10,恒温搅拌8h~10h。反应后离心分离,以去离子水和无水乙醇分别洗涤,在40℃下干燥16h,得到无机相变储能微胶囊。
测试例1
测定实施例1-18和比较例1-8中的相变微胶囊的理化性质,具体结果见表1。采用日本Hitachi S-4700型场发射扫描电子显微镜(SEM)观测相变微胶囊的形貌和粒径。采用差示扫描量热仪(DSC)测试相变微胶囊熔化过程的相变潜热值,仪器型号为日本岛津公司的DSC-60 Plus。氮气氛围下、温度测试范围为-70℃~70℃,升温速率为10℃/min,样品重量约为3.5mg。采用湘仪仪器有限公司DRL-III-P型导热系数测试仪测定相变微胶囊样品的导热系数。采用广东越联仪器有限公司YL-C10K压力试验机测定相变微胶囊样品的抗压强度。过冷度测试过程中,首先将实施例1和比较例2样品分别插入热电偶传感器,放入水浴锅中,在85℃下加热30min,取出后在室温下冷却,平均室温为25℃,通过数据采集系统记录温度变化,绘制出步冷曲线,如此反复100次,通过对比不同样品过冷度的变化,来评估其热可靠性能。
相变微胶囊的包覆率计算公式为:
包覆率/% = (微胶囊熔化潜热值 / 相变材料熔化潜热值)×100%
表1 实施例和比较例制备的相变微胶囊的性能
表1(续) 实施例和比较例制备的相变微胶囊的性能
由表1、图1-图2可见,本发明制备的复合芯材相变微胶囊具有良好的理化性质和规则的微观形貌。由于采用无机水合盐与糖醇复合后作为相变芯材,糖醇能与无机水合盐基体形成氢键作用,促进基体成核;加入聚醚胺可以增加基体稠度,使成核剂稳定分布在基体材料中,从而使成核剂有效发挥成核作用,即有效克服无机水合盐作为相变芯材时易出现过冷和相分离的不利现象。实施例1样品的熔化潜热值为121.1J/g,连续使用100个周期后,其过冷度仅为0.25℃,明显低于比较例样品的过冷度,即本发明制备的复合芯材相变微胶囊具有较好的热稳定性和重复使用性能。