一种氟代氧化吲哚类杂环化合物的制备方法
文献发布时间:2023-06-19 19:28:50
技术领域
本发明属于化工合成技术领域,具体地说是涉及一种氟代氧化吲哚类杂环化合物的制备方法。
背景技术
氧化吲哚类杂环化合物广泛存在于天然产物以及重要的药物分子结构当中,其独特生物学性质在药化领域颇受关注,已经被广泛应用于疾病的治疗,例如抗神经障碍、抗中风以及孕酮受体调节剂等药物。
传统制备3-氟氧化吲哚类衍生物方法主要可以分为两种,一种是对吲哚或者氧化吲哚类衍生物分别进行氟氧化或者亲核氟化,该方法需要较难制得的底物例如2-羟甲基吲哚,螺环环氧吲哚作为底物,以危险性较大的氟化试剂例如氢氟酸胺盐作为氟源,甚至额外添加氧化剂以及碱来促进反应进行,反应条件较为苛刻且步骤复杂;另一种常用的方法以难以制得的丙二酸酰胺或重氮乙酰胺作为环合底物,并加入昂贵的有机金属配合物以及其他添加剂来促进环合反应,因此增加了制备成本以及环境负担,不适合产业化。以上传统的制备方法因其较高的制备成本,繁琐的合成步骤,较低的安全性以及较多的废弃物而显著降低了其实用性,因此必须开发一种高效,环保且经济的氟代氧化吲哚类杂环化合物,尤其是3-氟氧化吲哚类衍生物的制备方法来更好地适用于实际应用领域。
发明内容
为了克服现有技术存在的不足,本发明提供了一种操作简便、效率较高的氟代氧化吲哚类杂环化合物(3-(3-氟-2-氧代吲哚-3-基)-喹啉酮类衍生物)的制备方法。
本发明采用的技术方案为:
一种氟代氧化吲哚类杂环化合物的制备方法,所述氟代氧化吲哚类杂环化合物的结构式如(I)所示:
其中,R
所述氟代氧化吲哚类杂环化合物的制备方法包括下述步骤:
(1)制备结构式如(II)所示化合物;
(2)制备结构式如(III)所示化合物;
(3)将结构式如(II)所示化合物、结构式如(III)所示化合物、铈催化剂加入反应容器中,加入溶剂,在室温蓝光照射条件下搅拌反应;随后加入N-氟代双苯磺酰胺,在35~65℃条件下继续搅拌反应;得到结构式如(I)所示产物。
本发明以光促进铈催化法制备氟代氧化吲哚类杂环化合物(3-(3-氟-2-氧代吲哚-3-基)-喹啉酮类衍生物),其反应过程为:
作为优选,R
作为优选,所述铈催化剂为无水氯化铈。
作为优选,所述溶剂为乙腈或二氯乙烷。
作为优选,结构式如(II)所示化合物、结构式如(III)所示化合物、N-氟代双苯磺酰胺、铈催化剂的摩尔比为1:1.1:1.5:0.005~0.01。
作为优选,继续搅拌反应的温度为50℃。
作为优选,反应完成后,旋干溶剂,以乙醇与水的混合溶液进行重结晶,随后抽滤,洗涤剂选择石油醚/乙酸乙酯的混合溶液洗涤三次,真空干燥得到产品。
本发明所提供的3-(3-氟-2-氧代吲哚-3-基)-喹啉酮类衍生物的制备方法科学合理,采用廉价以及小剂量的铈催化剂,以温和、高效的光促进铈催化法合成一系列3-(3-氟-2-氧代吲哚-3-基)-喹啉酮类衍生物。
本发明的有益效果在于:
(1)以廉价、高效的氯化铈以及N-氟代双苯磺酰胺作为催化剂以及氟化试剂,与传统合成3-氟氧化吲哚类杂环工艺相比,避免了贵金属的使用,且降低了金属的用量,反应条件更加温和、经济且高效,促进反应体系产业化可能;
(2)利用可见光促进偶联反应进行,避免了传统偶联反应强酸的使用,更加绿色环保;
(3)高效拓展了具有不同取代基的3-(3-氟-2-氧代吲哚-3-基)-喹啉酮类化合物,反应体系具有较好的官能团兼容性且提供较高的收率,目标产物的收率可以高达90%以上。
附图说明
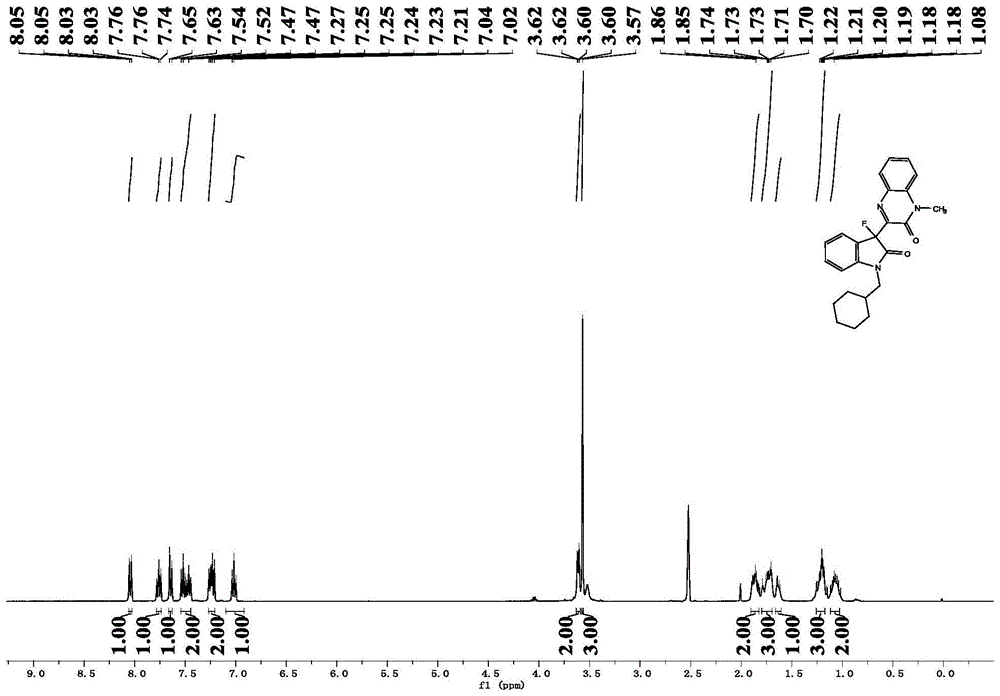
图1是实施例1制备得到的3-(3-氟-1-环己基甲基-2-氧代吲哚-3-基)-1-甲基喹啉-2(1H)-酮的核磁氢谱图。
具体实施方式
下面结合具体实施例对本发明作进一步说明,但本发明所要保护的范围并不限于此。本领域的普通技术人员可以且应当知晓任何基于本发明实质精神的简单变化或者替换均应属于本发明所要求的保护范围。
实施例1
N-环己基甲基吲哚的制备(结构式(II)中的R
在50mL圆底烧瓶中分别加入吲哚(13.2mmol,1.55g),氢氧化钾(46.2mmol,2.60g),15mL N,N-二甲基甲酰胺(DMF),于冰浴下搅拌,然后向圆底烧瓶中滴加(溴甲基)环己烷(15.84mmol,2.80g)的DMF溶液,滴加完毕后在室温下继续反应3个小时,反应用TLC检测。待反应完成后,将反应溶液用饱和氯化铵溶液洗涤,乙酸乙酯萃取,有机相再用食盐水洗涤,分出有机相,干燥,旋干,以石油醚和乙酸乙酯(80:1)比例为洗脱液过柱纯化,晾干后得到N-环己基甲基吲哚。
N-甲基喹喔啉酮的制备(结构式(III)中R
在100mL圆底烧瓶中分别加入2-羟基喹喔啉酮(12mmol,1.75g),碳酸钾(24mmol,3.31g),15mL N,N-二甲基甲酰胺(DMF),于冰浴下搅拌,然后向圆底烧瓶中滴加碘甲烷(14.4mmol,2g)的DMF溶液,滴加完毕后在室温下继续反应6个小时,反应用TLC检测。待反应完成后,将反应溶液用饱和氯化铵溶液洗涤,乙酸乙酯萃取,有机相再用食盐水洗涤,分出有机相,旋干得到粗品,粗品再用乙酸乙酯/石油醚(1:4)进行重结晶得到较纯的N-甲基喹喔啉酮。
3-(3-氟-1-环己基甲基-2-氧代吲哚-3-基)-1-甲基喹啉-2(1H)-酮的制备(结构式(I)中R
在25mL反应瓶中分别加入N-环己基甲基吲哚(0.2mmol,42.7mg),N-甲基喹喔啉酮(0.22mmol,35.2mg),无水氯化铈(0.001mmol,0.25mg),3mL乙腈,在室温蓝光照射条件下反应1小时;随后在反应体系当中缓慢加入N-氟代双苯磺酰胺(0.3mmol,94.6mg),在50℃条件下继续反应3h,反应用TLC检测。待反应完成后,旋干乙腈,加入乙醇溶解混合物后,缓慢滴加水直到反应液逐渐浑浊,降温至0℃继续搅拌20min析料,过滤,用石油醚/乙酸乙酯(3/1)的混合溶液洗涤滤饼三次,真空干燥得到目标产物3-(3-氟-1-环己基甲基-2-氧代吲哚-3-基)-1-甲基喹啉-2(1H)-酮,产率为95%。
3-(3-氟-1-环己基甲基-2-氧代吲哚-3-基)-1-甲基喹啉-2(1H)-酮的结构表征核磁共振数据如图1所示:
分析结果表明,获得的目标产物正确。
实施例2
以二氯乙烷代替乙腈,制备得到的目标产物的产率为90%。
实施例3
以N,N-二甲基甲酰胺(DMF)代替乙腈,制备得到的目标产物的产率为72%。
实施例4
5-溴-1-甲基-1H-吲哚的制备(结构式(II)中的R
在50mL圆底烧瓶中分别加入5-溴-1H-吲哚(13.2mmol,2.6g),氢氧化钾(46.2mmol,2.60g),15mL N,N-二甲基甲酰胺(DMF),于冰浴下搅拌,然后向圆底烧瓶中滴加碘甲烷(15.84mmol,2.2g)的DMF溶液,滴加完毕后在室温下继续反应3个小时,反应用TLC检测。待反应完成后,将反应溶液用饱和氯化铵溶液洗涤,乙酸乙酯萃取,有机相再用食盐水洗涤,分出有机相,干燥,旋干,以石油醚和乙酸乙酯(80:1)比例为洗脱液过柱纯化,晾干后得到5-溴-1-甲基-1H-吲哚。
3-(5-溴-3-氟-1-甲基-2-氧代吲哚啉-3-基)-1-甲基喹啉-2(1H)-酮的制备(结构式(I)中R
在25mL反应瓶中分别加入5-溴-1-甲基-1H-吲哚(0.2mmol,42mg),N-甲基喹喔啉酮(0.22mmol,35.2mg),无水氯化铈(0.002mmol,0.25mg),3mL乙腈,在室温蓝光照射条件下反应3小时;随后在反应体系当中缓慢加入N-氟代双苯磺酰胺(0.3mmol,94.6mg),在50℃条件下继续反应3h,反应用TLC检测。待反应完成后,旋干乙腈,加入乙醇溶解混合物后,缓慢滴加水直到反应液逐渐浑浊,降温至0℃继续搅拌20min析料,过滤,用石油醚/乙酸乙酯(3/1)的混合溶液洗涤滤饼三次,真空干燥得到目标产物3-(5-溴-3-氟-1-甲基-2-氧代吲哚啉-3-基)-1-甲基喹啉-2(1H)-酮,产率为92%。
3-(5-溴-3-氟-1-甲基-2-氧代吲哚啉-3-基)-1-甲基喹啉-2(1H)-酮的结构表征核磁共振数据:
分析结果表明,获得的目标产物正确。
实施例5
N-甲基吲哚的制备(结构式(II)中的R
在50mL圆底烧瓶中分别加入吲哚(13.2mmol,1.55g),氢氧化钾(46.2mmol,2.60g),15mL N,N-二甲基甲酰胺(DMF),于冰浴下搅拌,然后向圆底烧瓶中滴加碘甲烷(15.84mmol,2.25g)的DMF溶液,滴加完毕后在室温下继续反应3个小时,反应用TLC检测。待反应完成后,将反应溶液用饱和氯化铵溶液洗涤,乙酸乙酯萃取,有机相再用食盐水洗涤,分出有机相,干燥,旋干,以石油醚和乙酸乙酯(80:1)比例为洗脱液过柱纯化,晾干后得到N-甲基吲哚。
6-溴-1-甲基喹啉-2(1H)-酮的制备(结构式(III)中R
在100mL圆底烧瓶中分别加入6-溴喹啉-2(1H)-酮(12mmol,2.7g),碳酸钾(24mmol,3.31g),15mL N,N-二甲基甲酰胺(DMF),于冰浴下搅拌,然后向圆底烧瓶中滴加碘甲烷(14.4mmol,2g)的DMF溶液,滴加完毕后在室温下继续反应6个小时,反应用TLC检测。待反应完成后,将反应溶液用饱和氯化铵溶液洗涤,乙酸乙酯萃取,有机相再用食盐水洗涤,分出有机相,旋干得到粗品,粗品再用乙酸乙酯/石油醚(1:3)进行重结晶得到较纯的6-溴-1-甲基喹啉-2(1H)-酮。
6-溴-3-(3-氟-1-甲基-2-氧代吲哚啉-3-基)-1-甲基喹啉-2(1H)-酮的制备(结构式(I)中R
在25mL反应瓶中分别加入N-甲基吲哚(0.2mmol,26.2mg),6-溴-1-甲基喹啉-2(1H)-酮(0.22mmol,52.6mg),无水氯化铈(0.002mmol,0.25mg),3mL乙腈,在室温蓝光照射条件下反应3小时;随后在反应体系当中缓慢加入N-氟代双苯磺酰胺(0.3mmol,94.6mg),在50℃条件下继续反应3h,反应用TLC检测。待反应完成后,旋干乙腈,加入乙醇溶解混合物后,缓慢滴加水直到反应液逐渐浑浊,降温至0℃继续搅拌20min析料,过滤,用石油醚/乙酸乙酯(3/1)的混合溶液洗涤滤饼三次,真空干燥得到目标产物6-溴-3-(3-氟-1-甲基-2-氧代吲哚啉-3-基)-1-甲基喹啉-2(1H)-酮,产率为93%。
6-溴-3-(3-氟-1-甲基-2-氧代吲哚啉-3-基)-1-甲基喹啉-2(1H)-酮的结构表征核磁共振数据:
分析结果表明,获得的目标产物正确。
实施例6
N-炔丙基喹喔啉酮的制备(结构式(II)中R
在100mL圆底烧瓶中分别加入2-羟基喹喔啉酮(12mmol,1.75g),碳酸钾(24mmol,3.31g),15mL N,N-二甲基甲酰胺(DMF),于冰浴下搅拌,然后向圆底烧瓶中滴加3-溴丙炔(14.4mmol,1.71g)的DMF溶液,滴加完毕后在室温下继续反应6个小时,反应用TLC检测。待反应完成后,将反应溶液用饱和氯化铵溶液洗涤,乙酸乙酯萃取,有机相再用食盐水洗涤,分出有机相,干燥,旋干的到粗品,粗品再用乙酸乙酯/石油醚(1:4)进行重结晶得到较纯的N-炔丙基喹喔啉酮。
3-(3-氟-1-甲基-2-氧代吲哚-3-基)-1-炔丙基喹恶啉-2(1H)-酮的制备(结构式(I)中R
在25mL反应管中分别加入N-甲基吲哚(0.2mmol,26.2mg),N-炔丙基喹喔啉酮(0.22mmol,40.5mg),无水氯化铈(0.001mmol,0.25mg),3mL乙腈,在室温蓝光照射条件下反应1小时;随后在反应体系当中缓慢加入N-氟代双苯磺酰胺(0.3mmol,94.6mg),在50℃条件下继续反应3h,反应用TLC检测。待反应完成后,旋干乙腈,加入乙醇溶解混合物后,缓慢滴加水直到反应液逐渐浑浊,降温至0℃继续搅拌20min析料,过滤,用石油醚/乙酸乙酯(3/1)的混合溶液洗涤滤饼三次,真空干燥得到目标产物3-(3-氟-1-甲基-2-氧代吲哚-3-基)-1-炔丙基喹恶啉-2(1H)-酮,产率为92%。
3-(3-氟-1-甲基-2-氧代吲哚-3-基)-1-炔丙基喹恶啉-2(1H)-酮的结构表征核磁共振数据如下所示:
分析结果表明,获得的目标产物正确。
应当指出的是,对于本领域的普通技术人员来说,在不脱离本申请构思的前提下,还可以做出若干变形和改进,这些都属于本申请的保护范围。
- 一种4'-三氟甲基-3,5'-噁唑烷基螺环氧化吲哚化合物的手性制备方法
- 一种氮掺杂碳材料负载钴催化剂及其制备方法和在N-杂环化合物催化氧化中的应用
- 一种卤代吡啶类氮氧化物的制备方法
- 一种含三氟甲基吲哚衍生的α,β-氨基酸类似物的手性制备方法
- 二氢香豆素骨架拼接硫代吡咯啉酮螺环氧化吲哚化合物及其制备方法及应用
- 一种3‑氟烯基氧化吲哚‑螺‑3,3’‑三氟甲基氧化吲哚类化合物的制备方法
- 一种3-氟烯基氧化吲哚-螺-3,3’-三氟甲基氧化吲哚类化合物的制备方法