含乙二醇单元的烯基封端的四芳基金属卟啉类化合物及其制备方法和应用
文献发布时间:2023-06-19 19:30:30
技术领域
本发明属于功能材料技术领域,具体涉及含乙二醇单元的烯基封端的四芳基金属卟啉类化合物及其制备方法和应用,本发明是申请号为2022113813391,发明名称为含乙二醇单元的烯基封端的四芳基卟啉类化合物及其制备方法和应用的分案申请。
背景技术
近年来,电子产品的广泛使用使眼睛接收过量的有害蓝光(波段385~445nm的光线),长期使用会抑制褪黑激素、造成眼睛黄斑部病变及影响儿童眼部发育等危害(GB/T20145-2006)。为减少有害蓝光对眼睛造成的伤害,防蓝光材料应运而生。其中,吸收型防蓝光材料通过有机蓝光吸收剂(如金属络合、偶氮类)(Advanced Optical Materials,2020,8(20):2000695)、有机-无机杂化蓝光吸收剂(如烷基镍)、无机蓝光吸收剂(如偏铝酸锌、偏锡酸锌)等达到滤除蓝光的效果;转换型防蓝光材料通过稀土荧光类、有机金属配合物等光致发光材料,使蓝光主波长红移,避开有害蓝光(Chemistry-A European Journal,2020,26(70):16568-16581);反射型防蓝光材料通过在基材表面镀制或沉积多层膜,设置各膜层材料的折射率、厚度等参数,实现对蓝光波段的相干相消。然而,这些材料有些因“过度”切割蓝光,严重影响了视物时的画面色调、饱和度及亮度;有些在阻挡有害蓝光的同时,也阻挡了有益蓝光(波段446~505nm的光线)及可见光,造成物体颜色的显示失真,不利于人体对有益蓝光的吸收。
卟啉类结构因其良好的生物相容性且兼具优异的光吸收特性(ACS AppliedMaterials&Interfaces,2016,8(41),27438-27443),引起研究人员的关注。目前已报道的几种卟啉类防蓝光材料主要为单纯卟啉(CN 200780029228.1)或金属络合卟啉(CN201580036787.X),未充分考虑环上取代基类型对吸收光谱的影响,且因溶解度较低等问题,导致添加量较少、有害蓝光滤除效果较弱(阻隔率5%~25%),而有益蓝光和可见光透过率也仅为80%左右。
因此,提升该类结构的整体溶解性能,对于提升其抗蓝光效果及应用范围具有重要意义。
发明内容
发明目的:本发明所要解决的技术问题是提供一种溶解性能优异的含乙二醇单元的烯基封端的四芳基(金属)卟啉类化合物、制备方法及其在制备防有害蓝光材料和抗蓝光角膜接触镜方面的应用。
技术方案:为了解决上述技术问题,本发明提供了两种含乙二醇单元的烯基封端的四芳基(金属)卟啉类化合物,结构分别如通式I和I-M所示:
其中:X选自氢、甲基、氟或氯中的一种;R选自H或甲基;n=0或1或2或3;m=2或3或4;M选自锌、镁、铜等二价金属元素。
其中,具体地,所述化合物I结构式为:
/>
其中,具体地,所述化合物I-M结构式为:
/>
/>
本发明中,含乙二醇单元的烯基封端的四芳基卟啉类化合物I的制备方法如下:先在有机碱存在下,以丙烯酸羟基酯III与酸酐IV反应,制备含乙二醇单元的烯酸II,再与四[(4-羟基)芳基]卟啉V经缩合反应,制备化合物I,反应式为:
通式II所示的含乙二醇单元的烯酸的制备方法,丙烯酸羟基酯III、酸酐IV与有机碱的摩尔比为1∶1~1.5∶3~4,溶于有机溶剂,20~80℃搅拌反应,经柱层析提纯制备烯酸II。其中:有机碱选自二乙胺、三乙胺、吡啶、4-二甲氨基吡啶(DMAP)、N,N-二甲基苯胺或N,N-二异丙基乙胺(DIEA);溶剂选自二氯甲烷、三氯甲烷、四氢呋喃、2-甲基四氢呋喃或甲苯。
通式I所示的含乙二醇单元的烯基封端的四芳基卟啉类化合物的制备方法,包括如下步骤:将烯酸II、缩合剂与活化剂,溶于有机溶剂,加入四[(4-羟基)芳基]卟啉V,四[(4-羟基)芳基]卟啉V、烯酸II、缩合剂与活化剂的摩尔比为1∶4~8∶4~8∶0.1~16,反应物先在-10℃~5℃搅拌0.5h~2h,再在5℃~40℃搅拌反应0.5h~96h,经柱层析提纯制备含乙二醇单元的烯基封端的四芳基卟啉类化合物I。
其中:缩合剂选自N,N’-二环己基碳二亚胺(DCC)、1-乙基-(3-二甲基氨基丙基)碳二亚胺盐酸盐(EDCI·HCl)、O-(7-氮杂苯并三唑-1-基)-N,N,N’,N’-四甲基脲六氟磷酸酯(HATU)、苯并三氮唑-N,N,N”N’-四甲基脲六氟磷酸酯(HBTU)、6-氯苯并三氮唑-1,1,3,3-四甲基脲六氟磷酸酯(HCTU)、O-苯并三氮唑-N,N,N’,N’-四甲基脲四氟硼酸酯(TBTU)、O-(N-琥珀酰亚胺基)-N N N’,N′-四甲基四氟硼酸脲(TSTU)或O-(5-降冰片烯基-2,3-二羰亚胺)-N,N,N’,N’-四甲基脲四氟硼酸(TNTU);活化剂选自DMAP、二乙胺、三乙胺、吡啶、N,N-二甲基苯胺或DIEA;有机溶剂选自二氯甲烷、三氯甲烷、四氯化碳、乙腈、四氢呋喃、二氧六环、N,N-二甲基甲酰胺(DMF)或N,N-二甲基乙酰胺(DMAC)。
通式III所示的丙烯酸羟基酯由二醇与丙烯酰氯脱除一分子氯化氢制备,可参照文献Bioconjugate Chemistry,2018,29(9),3203-3212,其反应式为:
通式V所示的四[(4-羟基)芳基]卟啉可按如下方法制备:将1摩尔的吡咯和1摩尔的甲氧基芳醛VII加入丙酸溶液,回流反应0.5h~3h,经柱层析分离提纯得到通式VI的四[(4-甲氧基)芳基]卟啉,再用三氯化硼或三溴化硼在室温下脱除甲基,制备通式V的化合物,其反应式为:
通式IV所示的酸酐,通式VI所示的甲氧基芳醛,可直接从阿拉丁、毕得医药等试剂公司购买。
本发明中,含乙二醇单元的烯基封端的四芳基金属卟啉类化合物I-M的制备方法如下:以化合物I与二价金属醋酸盐反应,制备化合物I-M。其中,二价金属醋酸盐选自醋酸锌、醋酸镁、醋酸铜等,所述的化合物I与二价金属醋酸盐的摩尔比为1∶1~10,溶剂选自二氯甲烷、三氯甲烷或DMF,搅拌反应温度为25℃~150℃,产物需柱层析分离提纯。其反应式为:
本发明内容还包括所述的含乙二醇单元的烯基封端的四芳基(金属)卟啉类化合物在制备防有害蓝光材料方面的应用。
其中,所述的防有害蓝光材料采用热引发聚合固化制备,添加的含乙二醇单元的烯基封端的四芳基(金属)卟啉类化合物占反应体系总重量的0.01%~1%。
其中,所述的防有害蓝光材料的制备过程包括反应单体和引发剂,所述反应单体包括甲基丙烯酸羟乙酯(HEMA)、N-乙烯基吡咯烷酮(NVP)、甲基丙烯酸(MAA)、甲基丙烯酸甘油酯、甲基丙烯酸缩水甘油酯(GMA)、N,N二甲基丙烯酰胺、N-甲基-N-乙烯基乙酰胺、甲基丙烯酸甲酯(MMA)、醋酸丁酸纤维素、有机硅等单体中的一种或多种;所述引发剂包括偶氮二异丁腈(AIBN)、过氧化苯甲酰(BPO)、过氧化二碳酸二异丙酯、过氧化二碳酸二环己酯、偶氮二异庚氰和过氧化二碳酸双(2-苯基乙氧基)酯中的一种或多种。
本发明内容还包括所述的含乙二醇单元的烯基封端的四芳基(金属)卟啉类化合物在制备抗蓝光角膜接触镜方面的应用。
其中,所述的抗蓝光角膜接触镜由至少一种反应单体,一种交联剂,一种引发剂以及一种抗蓝光剂通过热引发聚合而成,抗蓝光剂为含乙二醇单元的烯基封端的四芳基(金属)卟啉类化合物。
其中,所述的抗蓝光角膜接触镜,按照质量份数计,反应单体为100份,抗蓝光剂占反应单体质量的0.01%~0.5%;引发剂占反应单体质量的0.01~2%;交联剂占反应单体质量的0.01~2%。
其中,所述的反应单体包括具有亲水性基团的单体和无亲水性基团的单体,亲水性基团单体的份数为40~100份,无亲水性基团单体的份数为0~60份。
其中,所述的具有亲水性基团的单体包括3-(3-甲基丙烯酰氧基-2-羟丙基)丙基双(三甲基硅氧烷)甲基硅烷(SIGMA)、3-丙烯酰胺丙基三(三甲基甲硅烷氧基)硅烷、单甲基丙烯酰胺氧基丙基封端的单正丁基封端的聚二甲基硅氧烷、甲基丙烯酸羟乙酯、NVP、甲基丙烯酸缩水甘油酯、甲基丙烯酸甘油酯、N,N二甲基丙烯酰胺、N-甲基-N-乙烯基乙酰胺中的一种或多种;所述的具有无亲水性基团的单体包括甲基丙烯酸甲酯、甲基丙烯酸乙酯、甲基丙烯酸异冰片酯、甲基丙烯酰氧丙基三(三甲基硅氧烷基)硅烷、3-[二乙氧基(甲基)甲硅烷基]甲基丙烯酸丙酯、单甲基丙烯酰氧基丙基封端的单正丁基封端的聚二甲基硅氧烷、氟化硅氧烷甲基丙烯酸酯中的一种或多种。
其中,所述的引发剂包括AIBN、BPO、偶氮二异庚腈中的一种或多种;所述的交联剂包括二乙烯基聚乙二醇酯、二甲基丙烯酸乙二醇酯、二甲基丙烯酸三乙二醇酯、异氰脲酸三烯丙酯、二乙二醇二乙烯基醚、聚乙二醇二甲基丙烯酸酯中的一种或多种。
本发明内容还包括所述的抗蓝光角膜接触镜的制备方法,包括以下步骤:所述抗蓝光角膜接触镜是按比例将抗蓝光剂添加到角膜接触镜镜片原料中,搅拌,充填入角膜接触镜模具中,热引发聚合固化,通过纯水或醇的水溶液浸泡后制得抗蓝光角膜接触镜。
其中,所述的抗蓝光角膜接触镜由抗蓝光剂和其他反应活性单体聚合制备而成;该镜片在纯水和乙醇溶液中无析出,镜片无色或淡粉色,且能有效吸收有害蓝光,透过有益蓝光。抗蓝光剂与HEMA、SIGMA等单体有较好的相溶性,添加较少的抗蓝光剂即可起到较好的抗蓝光效果,调节抗蓝光剂的含量可以有效的改变角膜接触镜在385~445nm的蓝光透过率,而对镜片整体的性能影响较小。
其中,所述的抗蓝光角膜接触镜在385~445nm的光透过率低于80%,在446~505nm的光透过率高于95%,可见光透过率大于95%。
其中,有实施例的抗蓝光角膜接触镜在385~445nm的光透过率低于60%,在446~505nm的光透过率高于95%,可见光透过率大于95%。
其中,有实施例的抗蓝光角膜接触镜在385~445nm的光透过率低于40%,在446~505nm的光透过率高于95%,可见光透过率大于90%。
有益效果:与现有技术相比,本发明具有以下优点:
1、本发明提供的含乙二醇单元的烯基封端的四芳基(金属)卟啉为新化合物,因结构中引入了多酯基以及可增加亲水性的乙二醇单元,更容易与HEMA、有机硅等单体互溶,进而发生聚合反应,与已有类似结构(Dyes and Pigments,2019,161,155-161)相比,具有更优异的溶解性能和更好的生物相容性。
2、与已有蓝光吸收剂相比,添加量更少,仅添加0.05%,即能有效阻隔40%有害蓝光(385~445nm)而几乎不吸收有益蓝光(446~505nm),视网膜蓝光辐射危害降低率H
3、苯环侧链乙二醇单元的增长,有助于减小聚合固化反应的空间位阻、提高材料的透氧率和含水量。因结构含有四个末端双键,可作为交联剂使用,无需再添加其他小分子交联剂,具有有效交联聚合物单体形成空间网状结构、增强材料稳定性和均匀性的作用。
4、添加含乙二醇单元的烯基封端的四芳基(金属)卟啉类化合物作为抗蓝光剂制备的角膜接触镜,其力学性能未发生明显变化,且随着抗蓝光剂添加量的增加,抗蓝光镜片在385~445nm的平均透光率低于40%时,镜片的可见光透过率依旧可以保持在90%以上,满足角膜接触镜正常配戴的光学需求。
附图说明
图1实施例2中化合物V-1的氢谱图。
图2实施例3中化合物I-1的氢谱图。
图3实施例8中化合物I-6的氢谱图。
图4实施例23中化合物I-1-Cu的氢谱图。
图5实施例45中制备的阻隔有害蓝光的高分子材料图。
图6实施例46中制备的阻隔有害蓝光的高分子材料图。
图7对比例1中制备的高分子材料图。
图8实施例47添加0.05%化合物I-1的抗蓝光水凝胶角膜接触镜。
图9实施例52添加0.05%化合物I-1的抗蓝光硅水凝胶角膜接触镜。
图10实施例3中化合物I-1的DSC分析图。
图11实施例24中化合物I-1-Zn的DSC分析图。
图12实施例47、48、49和51与市售抗蓝光镜片的光谱透过率对比图。
图13实施例49的体外细胞增殖率图。
图14实施例54的体外细胞增殖率图。
图15对比例4的体外细胞增殖率图。
图16对比例9的体外细胞增殖率图。
具体实施方式:
以下通过具体实施例对本发明作进一步详细说明。
实施例1丁二酸单[2-[(2-甲基-丙烯酰基)氧]乙基]酯(化合物II-1)的合成
丁二酸单[2-[(2-甲基丙烯酰基)氧]乙基]酯(化合物II-1)的合成路线:
N
实施例2四[(4-羟基)苯基]卟啉(化合物V-1)的合成
四[(4-羟基)苯基]卟啉(化合物V-1)的合成路线:
在三口圆底烧瓶中,依次加入500mL丙酸溶液、20.8mL吡咯和40.92g4-甲氧基苯甲醛,回流反应0.5h。冷却至室温,过滤,粗产品用乙酸乙酯/石油醚重结晶,得紫色固体VI-1。将VI-1与三溴化硼(BBr
实施例3四[(4-甲基丙烯酰氧乙基丁二酰氧基)苯基]卟啉(化合物I-1)的合成
四[(4-甲基丙烯酰氧乙基丁二酰氧基)苯基]卟啉(化合物I-1)的合成路线:
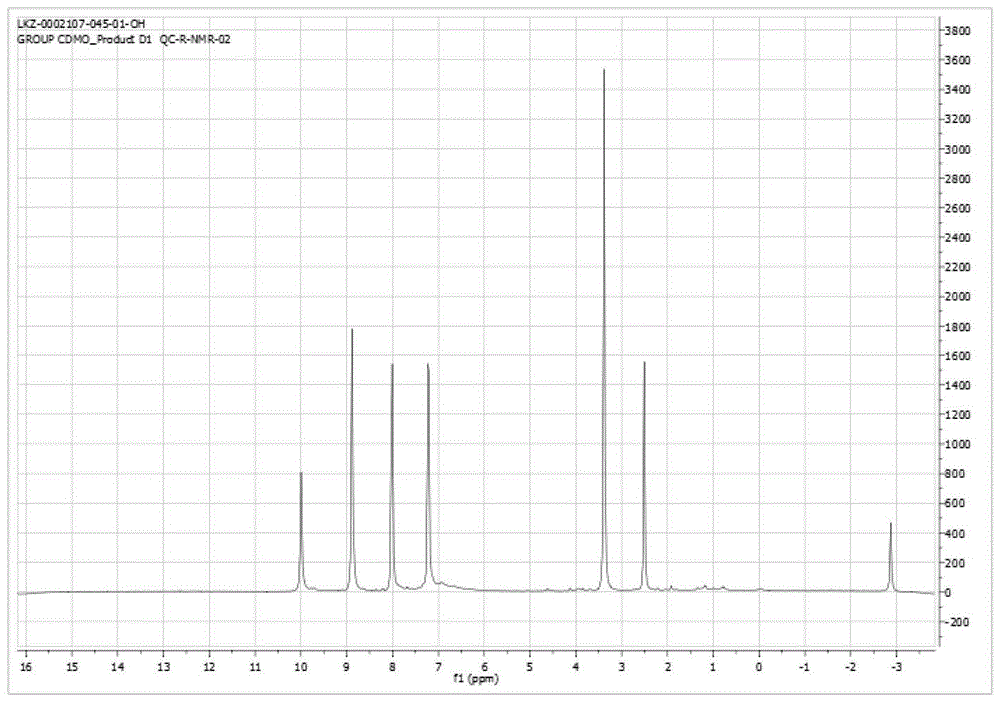
将1.012g化合物II-1溶于50mL二氯甲烷,冷却至-5℃,加入0.91gN,N’-二环己基碳二亚胺(DCC)和0.07g 4-二甲氨基吡啶(DMAP),搅拌1h,加入0.679g化合物V-1,继续搅拌0.5h,然后升温至35℃,搅拌24h,浓缩,经柱层析分离提纯,得紫色固体I-1。核磁数据(
实施例4四[(2-甲基-4-甲基丙烯酰氧乙基丁二酰氧基)苯基]卟啉(化合物I-2)的合成
四[(2-甲基-4-甲基丙烯酰氧乙基丁二酰氧基)苯基]卟啉(化合物I-2)的合成路线:
先参照实施例2化合物V-1的合成方法制备四[(2-甲基-4-羟基)苯基]卟啉(化合物V-2)。然后将1.012g化合物II-1溶于50mL三氯甲烷,冷却至-10℃,加入0.92gDCC和0.08gDMAP,搅拌0.5h,再加入0.735g化合物V-2,继续搅拌0.5h,然后升至室温,搅拌72h,浓缩,经柱层析分离提纯,得紫色固体I-2。元素分析数据如下:计算值w(C)=66.74%,w(H)=5.47%,w(N)=3.54%;实验值w(C)=66.52%,w(H)=5.45%,w(N)=3.56%。
实施例5四[(3-甲基-4-甲基丙烯酰氧乙基丁二酰氧基)苯基]卟啉(化合物I-3)的合成
四[(3-甲基-4-甲基丙烯酰氧乙基丁二酰氧基)苯基]卟啉(化合物I-3)的合成路线:
先参照实施例2化合物V-1的合成方法制备四[(3-甲基-4-羟基)苯基]卟啉(化合物V-3)。然后将1.012g化合物II-1溶于50mL三氯甲烷,冷却至0℃,加入0.87g EDCI·HCl和0.22g DMAP,搅拌0.5h,再加入0.735g化合物V-3,继续搅拌1.5h,然后升温至40℃,搅拌12h,浓缩,经柱层析分离提纯,得紫色固体I-3。元素分析数据如下:计算值w(C)=66.74%,w(H)=5.47%,w(N)=3.54%;实验值w(C)=66.47%,w(H)=5.50%,w(N)=3.56%。
实施例6四[(3-氟-4-甲基丙烯酰氧乙基丁二酰氧基)苯基]卟啉(化合物I-4)的合成
四[(3-氟-4-甲基丙烯酰氧乙基丁二酰氧基)苯基]卟啉(化合物I-4)的合成路线:
先参照实施例2化合物V-1的合成方法制备四[(3-氟-4-羟基)苯基]卟啉(化合物V-4)。然后将1.012g化合物II-1溶于50mL三氯甲烷,冷却至-5℃,加入1.05g DCC和0.15g三乙胺,搅拌0.5h,再加入0.751g化合物V-4,继续搅拌1h,然后在5℃搅拌反应96h,浓缩,经柱层析分离提纯,得紫色固体I-4。元素分析数据如下:计算值w(C)=63.08%,w(H)=4.66%,w(N)=3.50%;实验值w(C)=63.35%,w(H)=4.64%,w(N)=3.48%。
实施例7四[(3-氯-4-甲基丙烯酰氧乙基丁二酰氧基)苯基]卟啉(化合物I-5)的合成
四[(3-氯-4-甲基丙烯酰氧乙基丁二酰氧基)苯基]卟啉(化合物I-5)的合成路线:
先参照实施例2化合物V-1的合成方法制备四[(3-氯-4-羟基)苯基]卟啉(化合物V-5)。然后将1.012g化合物II-1溶于20mL DMF,冷却至5℃,加入1.80g HATU和0.87g DIEA,搅拌0.5h,再加入0.816g化合物V-5,继续搅拌0.5h,然后升温至30℃,搅拌6h,浓缩,经柱层析分离提纯,得紫色固体I-5。元素分析数据如下:计算值w(C)=60.58%,w(H)=4.48%,w(N)=3.36%;实验值w(C)=60.32%,w(H)=4.50%,w(N)=3.38%。
实施例8四[(4-甲基丙烯酰二氧乙基丁二酰氧基)苯基]卟啉(化合物I-6)的合成
四[(4-甲基丙烯酰二氧乙基丁二酰氧基)苯基]卟啉(化合物I-6)的合成路线:
先参照实施例1化合物II-1的合成方法制备丁二酸单甲基丙烯酰氧乙基氧乙酯(化合物II-2)。然后将1.372g化合物II-2溶于20mL DMAC,冷却至0℃,加入2.0g HATU和1.0g DIEA,搅拌0.5h,再加入0.678g化合物V-1,继续搅拌1.5h,然后升至室温,搅拌40h,浓缩,经柱层析分离提纯,得紫色固体I-6。核磁数据(
实施例9四[(3-甲基-4-甲基丙烯酰二氧乙基丁二酰氧基)苯基]卟啉(化合物I-7)的合成
四[(3-甲基-4-甲基丙烯酰二氧乙基丁二酰氧基)苯基]卟啉(化合物I-7)的合成路线:
将1.372g化合物II-2溶于50mL二氧六环,冷却至5℃,加入1.93g HBTU和0.925gN,N-二甲基苯胺,搅拌1h,再加入0.735g化合物V-3,继续搅拌1h,然后升温至40℃,搅拌0.5h,浓缩,经柱层析分离提纯,得紫色固体I-7。元素分析数据如下:计算值w(C)=65.52%,w(H)=5.84%,w(N)=3.18%;实验值w(C)=65.82%,w(H)=5.87%,w(N)=3.16%。
实施例10四[(4-甲基丙烯酰三氧乙基丁二酰氧基)苯基]卟啉(化合物I-8)的合成
四[(4-甲基丙烯酰三氧乙基丁二酰氧基)苯基]卟啉(化合物I-8)的合成路线:
先参照实施例1化合物II-1的合成方法制备丁二酸单甲基丙烯酰二氧乙基氧乙酯(化合物II-3)。然后将1.592g化合物II-3溶于50mL四氢呋喃,冷却至5℃,加入2.31g HCTU和0.53g吡啶,搅拌0.5h,再加入0.679g化合物V-1,继续搅拌1h,然后升至室温,搅拌72h,浓缩,经柱层析分离提纯,得紫色固体I-8。元素分析数据如下:计算值w(C)=63.89%,w(H)=5.90%,w(N)=2.98%;实验值w(C)=63.62%,w(H)=5.88%,w(N)=2.99%。
实施例11四[(4-甲基丙烯酰三氧乙基丁二酰氧基)苯基]卟啉(化合物I-9)的合成
四[(4-甲基丙烯酰三氧乙基丁二酰氧基)苯基]卟啉(化合物I-9)的合成路线:
将1.592g化合物II-3溶于50mL二氯甲烷,冷却至0℃,加入1.82g TBTU和0.92g三乙胺,搅拌0.5h,再加入0.751g化合物V-4,继续搅拌0.5h,然后升至室温,搅拌72h,浓缩,经柱层析分离提纯,得紫色固体I-9。元素分析数据如下:计算值w(C)=61.53%,w(H)=5.47%,w(N)=2.87%;实验值w(C)=61.79%,w(H)=5.45%,w(N)=2.86%。
实施例12四[(4-甲基丙烯酰四氧乙基丁二酰氧基)苯基]卟啉(化合物I-10)的合成
四[(4-甲基丙烯酰四氧乙基丁二酰氧基)苯基]卟啉(化合物I-10)的合成路线:
先参照实施例1化合物II-1的合成方法制备丁二酸单甲基丙烯酰三氧乙基氧乙酯(化合物II-4)。然后将1.812g化合物II-4溶于20mL DMF,冷却至5℃,加入1.75g TSTU和0.64g二乙胺,搅拌1h,再加入0.679g化合物V-1,继续搅拌1h,然后升温至35℃,搅拌6h,浓缩,经柱层析分离提纯,得紫色固体I-10。元素分析数据如下:计算值w(C)=63.09%,w(H)=6.18%,w(N)=2.72%;实验值w(C)=63.37%,w(H)=6.16%,w(N)=2.73%。
实施例13四[(3-甲基-4-甲基丙烯酰四氧乙基丁二酰氧基)苯基]卟啉(化合物I-11)的合成
四[(3-甲基-4-甲基丙烯酰四氧乙基丁二酰氧基)苯基]卟啉(化合物I-11)的合成路线:
将1.812g化合物II-4溶于20mL DMAC,冷却至5℃,加入2.15g TNTU和1.15g DIEA,搅拌1h,再加入0.735g化合物V-3,继续搅拌1h,然后升温至35℃,搅拌8h,浓缩,经柱层析分离提纯,得紫色固体I-11。元素分析数据如下:计算值w(C)=63.69%,w(H)=6.39%,w(N)=2.65%;实验值w(C)=63.37%,w(H)=6.37%,w(N)=2.66%。
实施例14四[(4-丙烯酰氧乙基丁二酰氧基)苯基]卟啉(化合物I-12)的合成
四[(4-丙烯酰氧乙基丁二酰氧基)苯基]卟啉(化合物I-12)的合成路线:
先参照实施例1化合物II-1的合成方法制备丁二酸单丙烯酰氧乙基氧乙酯(化合物II-5)。然后将0.951g化合物II-5溶于50mL二氯甲烷,冷却至-10℃,加入1.5g DCC和0.12g吡啶,搅拌0.5h,再加入0.678g化合物V-1,继续搅拌1h,然后升温至30℃,搅拌12h,浓缩,柱层析分离提纯,得紫色固体I-12。元素分析数据如下:计算值w(C)=65.30%,w(H)=4.80%,w(N)=3.81%;实验值w(C)=65.62%,w(H)=4.78%,w(N)=3.83%。
实施例15四[(3-甲基-4-丙烯酰氧乙基丁二酰氧基)苯基]卟啉(化合物I-13)的合成
四[(3-甲基-4-丙烯酰氧乙基丁二酰氧基)苯基]卟啉(化合物I-13)的合成路线:
将0.951g化合物II-5溶于50mL四氯化碳,冷却至0℃,加入1.15g DCC和0.15g三乙胺,搅拌1h,再加入0.735g化合物V-1,继续搅拌1h,然后升至室温,搅拌24h,浓缩,柱层析分离提纯,得紫色固体I-13。元素分析数据如下:计算值w(C)=66.05%,w(H)=5.15%,w(N)=3.67%;实验值w(C)=65.36%,w(H)=5.13%,w(N)=3.69%。
实施例16四[(4-丙烯酰二氧乙基丁二酰氧基)苯基]卟啉(化合物I-14)的合成
四[(4-丙烯酰二氧乙基丁二酰氧基)苯基]卟啉(化合物I-14)的合成路线:
先参照实施例1化合物II-1的合成方法制备丁二酸单丙烯酰氧乙基氧乙酯(化合物II-6)。然后将1.145g化合物II-6溶于20mL DMF,冷却至0℃,加入1.08g EDCI·HCl和1.10g N,N-二甲基苯胺,搅拌0.5h,再加入0.678g化合物V-1,继续搅拌1.5h,然后升至室温,搅拌72h,浓缩,柱层析分离提纯,得紫色固体I-14。元素分析数据如下:计算值w(C)=64.15%,w(H)=5.26%,w(N)=3.40%;实验值w(C)=64.44%,w(H)=5.23%,w(N)=3.38%。
实施例17四[(3-甲基-4-丙烯酰二氧乙基丁二酰氧基)苯基]卟啉(化合物I-15)的合成
四[(3-甲基-4-丙烯酰二氧乙基丁二酰氧基)苯基]卟啉(化合物I-15)的合成路线:
将1.145g化合物II-6溶于50mL三氯甲烷,冷却至5℃,加入2.12g HATU和0.95gDIEA,搅拌0.5h,再加入0.735g化合物V-3,继续搅拌0.5h,然后升温至40℃,搅拌0.5h,浓缩,经柱层析分离提纯,得紫色固体I-15。元素分析数据如下:计算值w(C)=64.86%,w(H)=5.56%,w(N)=3.29%;实验值w(C)=64.58%,w(H)=5.58%,w(N)=3.31%。
实施例18四[(3-甲基-4-丙烯酰二氧乙基丁二酰氧基)苯基]卟啉(化合物I-16)的合成
四[(3-甲基-4-丙烯酰二氧乙基丁二酰氧基)苯基]卟啉(化合物I-16)的合成路线:
先参照实施例1化合物II-1的合成方法制备丁二酸单甲基丙烯酰二氧乙基氧乙酯(化合物II-7)。然后将1.340g化合物II-7溶于30mL乙腈,冷却至0℃,加入1.18g DCC和0.12g DMAP,搅拌0.5h,再加入0.679g化合物V-1,继续搅拌0.5h,然后升温至40℃,搅拌8h,浓缩,经柱层析分离提纯,得紫色固体I-16。元素分析数据如下:计算值w(C)=63.22%,w(H)=5.64%,w(N)=3.07%;实验值w(C)=63.50%,w(H)=5.62%,w(N)=3.05%。
实施例19四[(4-甲基丙烯酰氧乙基戊二酰氧基)苯基]卟啉(化合物I-17)的合成
四[(4-甲基丙烯酰氧乙基戊二酰氧基)苯基]卟啉(化合物I-17)的合成路线:
先参照实施例1化合物II-1的合成方法制备戊二酸单[2-[(2-甲基丙烯酰基)氧]乙基]酯(化合物II-8)。然后将1.22g化合物II-8溶于20mL DMF,冷却至0℃,加入2.14gHBTU和0.18g三乙胺,搅拌0.5h,加入0.68g化合物V-1,继续搅拌0.5h,然后升至室温,搅拌36h,浓缩,柱层析分离提纯,得紫色固体I-17。元素分析数据如下:计算值w(C)=66.74%,w(H)=5.47%,w(N)=3.54%;实验值w(C)=66.45%,w(H)=5.49%,w(N)=3.56%。
实施例20四[(4-甲基丙烯酰氧乙基己二酰氧基)苯基]卟啉(化合物I-18)的合成
四[(4-甲基丙烯酰氧乙基己二酰氧基)苯基]卟啉(化合物I-18)的合成路线:
先参照实施例1化合物II-1的合成方法制备己二酸单[2-[(2-甲基丙烯酰基)氧]乙基]酯(化合物II-9)。然后将1.29g化合物II-9溶于50mL三氯甲烷,冷却至0℃,加入1.25gDCC和0.21g吡啶,搅拌0.5h,加入0.68g化合物V-1,继续搅拌1h,然后升至室温,搅拌24h,浓缩,柱层析分离提纯,得紫色固体I-18。元素分析数据如下:计算值w(C)=67.39%,w(H)=5.78%,w(N)=3.42%;实验值w(C)=67.69%,w(H)=5.75%,w(N)=3.40%。
实施例21四[(4-甲基丙烯酰四氧乙基戊二酰氧基)苯基]卟啉(化合物I-19)的合成
四[(4-甲基丙烯酰四氧乙基戊二酰氧基)苯基]卟啉(化合物I-19)的合成路线:
先参照实施例1化合物II-1的合成方法制备戊二酸单甲基丙烯酰三氧乙基氧乙酯(化合物II-10)。然后将1.88g化合物II-10溶于50mL四氢呋喃,冷却至5℃,加入2.23g HATU和1.21g DIEA,搅拌1h,加入0.68g化合物V-1,继续搅拌0.5h,然后升温至40℃,搅拌4h,浓缩,柱层析分离提纯,得紫色固体I-19。元素分析数据如下:计算值w(C)=63.69%,w(H)=6.39%,w(N)=2.65%;实验值w(C)=63.35%,w(H)=6.42%,w(N)=2.66%。
实施例22四[(4-甲基丙烯酰四氧乙基己二酰氧基)苯基]卟啉(化合物I-20)的合成
四[(4-甲基丙烯酰四氧乙基己二酰氧基)苯基]卟啉(化合物I-20)的合成路线:
先参照实施例1化合物II-1的合成方法制备己二酸单甲基丙烯酰三氧乙基氧乙酯(化合物II-11)。然后将1.95g化合物II-11溶于50mL二氯甲烷,冷却至-5℃,加入1.24g DCC和0.16g DMAP,搅拌0.5h,加入0.65g化合物V-1,继续搅拌1h,然后升温至35℃,搅拌8h,浓缩,柱层析分离提纯,得紫色固体1-20。元素分析数据如下:计算值w(C)=64.25%,w(H)=6.60%,w(N)=2.58%;实验值w(C)=64.55%,w(H)=6.58%,w(N)=2.57%。
实施例23四[(4-甲基丙烯酰氧乙基丁二酰氧基)苯基]卟啉铜(化合物I-1-Cu)的合成
四[(4-甲基丙烯酰氧乙基丁二酰氧基)苯基]卟啉铜(化合物I-1-Cu)的合成路线:
将0.76g化合物I-1溶于20mLCHCl
实施例24四[(4-甲基丙烯酰氧乙基丁二酰氧基)苯基]卟啉锌(化合物I-1-Zn)的合成
四[(4-甲基丙烯酰氧乙基丁二酰氧基)苯基]卟啉锌(化合物I-1-Zn)的合成路线:
将0.76g化合物I-1溶于10mL二氯甲烷,加入0.2g醋酸锌,室温搅拌过夜。旋蒸除去溶剂,柱层析分离提纯得产品I-1-Zn。元素分析数据如下:计算值w(C)=63.42%,w(H)=4.82%,w(N)=3.52%;实验值w(C)=63.15%,w(H)=4.84%,w(N)=3.50%。
实施例25四[(2-甲基-4-甲基丙烯酰氧乙基丁二酰氧基)苯基]卟啉锌(化合物I-2-Zn)的合成
四[(2-甲基-4-甲基丙烯酰氧乙基丁二酰氧基)苯基]卟啉锌(化合物I-2-Zn)的合成路线:
将0.79g化合物I-2溶于10mL二氯甲烷,加入0.2g醋酸锌,室温搅拌过夜。旋蒸除去溶剂,柱层析分离提纯得产品I-2-Zn。元素分析数据如下:计算值w(C)=64.17%,w(H)=5.14%,w(N)=3.40%;实验值w(C)=64.47%,w(H)=5.12%,w(N)=3.38%。
实施例26四[(3-甲基-4-甲基丙烯酰氧乙基丁二酰氧基)苯基]卟啉镁(化合物I-3-Mg)的合成
四[(3-甲基-4-甲基丙烯酰氧乙基丁二酰氧基)苯基]卟啉镁(化合物I-3-Mg)的合成路线:
将0.79g化合物I-3溶于10mL DMF,加入0.54g醋酸镁,回流搅拌过夜。旋蒸除去溶剂,柱层析分离提纯得产品I-3-Mg。元素分析数据如下:计算值w(C)=66.74%,w(H)=5.47%,w(N)=3.54%;实验值w(C)=66.45%,w(H)=5.49%,w(N)=3.55%。
实施例27四[(3-氟-4-甲基丙烯酰氧乙基丁二酰氧基)苯基]卟啉镁(化合物I-4-Mg)的合成
四[(3-氟-4-甲基丙烯酰氧乙基丁二酰氧基)苯基]卟啉镁(化合物I-4-Mg)的合成路线:
将0.80g化合物I-4溶于10mL DMF,加入0.54g醋酸镁,回流搅拌过夜。旋蒸除去溶剂,柱层析分离提纯得产品I-4-Mg。元素分析数据如下:计算值w(C)=62.21%,w(H)=4.48%,w(N)=3.45%;实验值w(C)=62.49%,w(H)=4.50%,w(N)=3.43%。
实施例28四[(3-氯-4-甲基丙烯酰氧乙基丁二酰氧基)苯基]卟啉铜(化合物I-5-Cu)的合成
四[(3-氯-4-甲基丙烯酰氧乙基丁二酰氧基)苯基]卟啉铜(化合物I-5-Cu)的合成路线:
将0.83g化合物I-5溶于20mLCHCl
实施例29四[(4-甲基丙烯酰二氧乙基丁二酰氧基)苯基]卟啉铜(化合物I-6-Cu)的合成
四[(4-甲基丙烯酰二氧乙基丁二酰氧基)苯基]卟啉铜(化合物I-6-Cu)的合成路线:
将0.85g化合物I-6溶于20mLCHCl
实施例30四[(3-甲基-4-甲基丙烯酰二氧乙基丁二酰氧基)苯基]卟啉锌(化合物I-7-Zn)的合成
四[(3-甲基-4-甲基丙烯酰二氧乙基丁二酰氧基)苯基]卟啉锌(化合物I-7-Zn)的合成路线:
将0.88g化合物I-7溶于10mL二氯甲烷,加入0.2g醋酸锌,室温搅拌过夜。旋蒸除去溶剂,柱层析分离提纯得产品I-7-Zn。元素分析数据如下:计算值w(C)=63.24%,w(H)=5.53%,w(N)=3.07%;实验值w(C)=63.52%,w(H)=5.51%,w(N)=3.05%。
实施例31四[(4-甲基丙烯酰三氧乙基丁二酰氧基)苯基]卟啉铜(化合物I-8-Cu)的合成
四[(4-甲基丙烯酰三氧乙基丁二酰氧基)苯基]卟啉铜(化合物I-8-Cu)的合成路线:
将0.85g化合物I-8溶于20mLCHCl
实施例32四[(4-甲基丙烯酰三氧乙基丁二酰氧基)苯基]卟啉锌(化合物I-9-Zn)的合成
四[(4-甲基丙烯酰三氧乙基丁二酰氧基)苯基]卟啉锌(化合物I-9-Zn)的合成路线:
将0.98g化合物I-9溶于10mL二氯甲烷,加入0.2g醋酸锌,室温搅拌过夜。旋蒸除去溶剂,柱层析分离提纯得产品I-9-Zn。元素分析数据如下:计算值w(C)=59.60%,w(H)=5.20%,w(N)=2.78%;实验值w(C)=59.87%,w(H)=5.18%,w(N)=2.76%。
实施例33四[(4-甲基丙烯酰四氧乙基丁二酰氧基)苯基]卟啉铜(化合物I-10-Cu)的合成
四[(4-甲基丙烯酰四氧乙基丁二酰氧基)苯基]卟啉铜(化合物I-10-Cu)的合成路线:
将1.03g化合物I-10溶于20mLCHCl
实施例34四[(3-甲基-4-甲基丙烯酰四氧乙基丁二酰氧基)苯基]卟啉镁(化合物I-11-Mg)的合成
四[(3-甲基-4-甲基丙烯酰四氧乙基丁二酰氧基)苯基]卟啉镁(化合物I-11-Mg)的合成路线:
将0.98g化合物I-11溶于10mL DMF,加入0.54g醋酸镁,回流搅拌过夜。旋蒸除去溶剂,柱层析分离提纯得产品I-4-Mg。元素分析数据如下:计算值w(C)=61.88%,w(H)=6.12%,w(N)=2.58%;实验值w(C)=61.59%,w(H)=6.15%,w(N)=2.60%。
实施例35四[(4-丙烯酰氧乙基丁二酰氧基)苯基]卟啉铜(化合物I-12-Cu)的合成
四[(4-丙烯酰氧乙基丁二酰氧基)苯基]卟啉铜(化合物I-12-Cu)的合成路线:
将0.72g化合物I-12溶于20mLCHCl
实施例36四[(3-甲基-4-丙烯酰氧乙基丁二酰氧基)苯基]卟啉镁(化合物I-13-Mg)的合成
四[(3-甲基-4-丙烯酰氧乙基丁二酰氧基)苯基]卟啉镁(化合物I-13-Mg)的合成路线:
将0.76g化合物I-13溶于10mL DMF,加入0.54g醋酸镁,回流搅拌过夜。旋蒸除去溶剂,柱层析分离提纯得产品I-13-Mg。元素分析数据如下:计算值w(C)=65.10%,w(H)=4.94%,w(N)=3.62%;实验值w(C)=64.82%,w(H)=4.96%,w(N)=3.64%。
实施例37四[(4-丙烯酰二氧乙基丁二酰氧基)苯基]卟啉锌(化合物I-14-Zn)的合成
四[(4-丙烯酰二氧乙基丁二酰氧基)苯基]卟啉锌(化合物I-14-Zn)的合成路线:
将0.83g化合物I-14溶于10mL二氯甲烷,加入0.2g醋酸锌,室温搅拌过夜。旋蒸除去溶剂,柱层析分离提纯得产品I-14-Zn。元素分析数据如下:计算值w(C)=61.77%,w(H)=4.95%,w(N)=3.27%;实验值w(C)=61.50%,w(H)=4.97%,w(N)=3.28%。
实施例38四[(4-丙烯酰二氧乙基丁二酰氧基)苯基]卟啉铜(化合物I-14-Cu)的合成
四[(4-丙烯酰二氧乙基丁二酰氧基)苯基]卟啉铜(化合物I-14-Cu)的合成路线:
将0.83g化合物I-14溶于20mLCHCl
实施例39四[(3-甲基-4-丙烯酰二氧乙基丁二酰氧基)苯基]卟啉锌(化合物I-15-Zn)的合成
四[(3-甲基-4-丙烯酰二氧乙基丁二酰氧基)苯基]卟啉锌(化合物I-15-Zn)的合成路线:
将0.85g化合物I-15溶于10mL二氯甲烷,加入0.2g醋酸锌,室温搅拌过夜。旋蒸除去溶剂,柱层析分离提纯得产品I-15-Zn。元素分析数据如下:计算值w(C)=62.53%,w(H)=5.25%,w(N)=3.17%;实验值w(C)=62.25%,w(H)=5.28%,w(N)=3.19%。
实施例40四[(3-甲基-4-丙烯酰二氧乙基丁二酰氧基)苯基]卟啉铜(化合物I-16-Cu)的合成
四[(3-甲基-4-丙烯酰二氧乙基丁二酰氧基)苯基]卟啉铜(化合物I-16-Cu)的合成路线:
将0.92g化合物I-16溶于20mLCHCl
实施例41四[(4-甲基丙烯酰氧乙基戊二酰氧基)苯基]卟啉(化合物I-17-Cu)的合成
四[(4-甲基丙烯酰氧乙基戊二酰氧基)苯基]卟啉(化合物I-17-Cu)的合成路线:
将0.85g化合物I-17溶于20mLCHCl
实施例42四[(4-甲基丙烯酰氧乙基己二酰氧基)苯基]卟啉锌(化合物I-18-Zn)的合成
四[(4-甲基丙烯酰氧乙基己二酰氧基)苯基]卟啉锌(化合物I-18-Zn)的合成路线:
将0.88g化合物I-18溶于10mL二氯甲烷,加入0.2g醋酸锌,室温搅拌过夜。旋蒸除去溶剂,柱层析分离提纯得产品I-18-Zn。元素分析数据如下:计算值w(C)=64.88%,w(H)=5.45%,w(N)=3.29%;实验值w(C)=64.59%,w(H)=5.48%,w(N)=3.31%。
实施例43四[(4-甲基丙烯酰四氧乙基戊二酰氧基)苯基]卟啉镁(化合物I-19-Mg)的合成
四[(4-甲基丙烯酰四氧乙基戊二酰氧基)苯基]卟啉镁(化合物I-19-Mg)的合成路线:
将0.95g化合物I-19溶于10mL DMF,加入0.54g醋酸镁,回流搅拌过夜。旋蒸除去溶剂,柱层析分离提纯得产品I-19-Mg。元素分析数据如下:计算值w(C)=63.02%,w(H)=6.23%,w(N)=2.62%;实验值w(C)=63.30%,w(H)=6.21%,w(N)=2.61%。
实施例44四[(4-甲基丙烯酰四氧乙基己二酰氧基)苯基]卟啉铜(化合物I-20-Cu)的合成
四[(4-甲基丙烯酰四氧乙基己二酰氧基)苯基]卟啉铜(化合物I-20-Cu)的合成路线:
将1.05g化合物I-20溶于20mLCHCl
实施例45防有害蓝光高分子材料的制备
防有害蓝光高分子材料的制备路线:
将16.38g甲基丙烯酸羟乙酯(HEMA)、3.38g N-乙烯基吡咯烷酮(NVP)、2.25g甲基丙烯酰氧基甲基三(三甲基硅氧基)硅烷、0.0113g化合物I-1(0.05%)和0.158g引发剂偶氮二异丁腈(AIBN),超声10min,过滤,滴注到模具中,在90℃烘箱中反应4h,脱模,制得防有害蓝光材料。合成产品见图5所示。图5所示的防有害蓝光材料为粉色,打破了传统防有害蓝光材料偏黄的特性,为防蓝光材料的选择提供了更多的选择性,且该材料在385-445nm的有害蓝光透过率为58.5%,在446nm-505nm的有益蓝光透过率为98.9%,主要针对有害蓝光进行阻隔,具有颜色特别、蓝光阻隔选择性好的特点。
实施例46防有害蓝光高分子材料的制备
防有害蓝光高分子材料的制备路线:
将16.38g HEMA、3.38g NVP、2.25g甲基丙烯酰氧基甲基三(三甲基硅氧基)硅烷、0.0113g化合物I-6(0.05%)和0.158g AIBN,超声10min,过滤,滴注到模具中,在90℃烘箱中反应3h,脱模,制得防有害蓝光材料。合成产品见见图6。图6所示的防有害蓝光材料为粉色,打破了传统防有害蓝光材料偏黄的特性,为防蓝光材料的选择提供了更多的选择性,且该材料在385-445nm的有害蓝光透过率为59.4%,在446nm-505nm的有益蓝光透过率为98.7%主要针对有害蓝光进行阻隔,具有颜色特别、蓝光阻隔选择性好的特点。
对比例1高分子材料的制备
高分子材料的合成路线:
将16.38g HEMA、3.38g NVP、2.25g甲基丙烯酰氧基甲基三(三甲基硅氧基)硅烷、0.158gAIBN和0.338gN,N’-亚甲基双丙烯酰胺,超声10min,过滤,滴注到模具中,在90℃烘箱中反应8h,脱模,制得对比例高分子材料。合成产品见图7。
实施例47抗蓝光水凝胶镜片的制备
将12.6g HEMA、7.2g甲基丙烯酸缩水甘油酯、0.01g抗蓝光剂(化合物I-1)、0.06gAIBN和0.05g二甲基丙烯酸乙二醇酯,室温混合搅拌5h,过滤,逐滴滴入到模具中,在75℃烘箱中反应12h,反应完后脱模,放入纯水中水合3h,后移入到标准盐水中,制得抗蓝光水凝胶角膜接触镜。合成产品见图8。图8所示的抗蓝光水凝胶角膜接触镜为粉色,在保存液或护理液中更易被找到,无需添加其他染料如蓝染,且该镜片在385-445nm的有害蓝光透过率为61.0%,在446nm-505nm的有益蓝光透过率为99.0%,380-800nm光透过率为98.0%。说明该抗蓝光水凝胶角膜接触镜主要具有颜色特别、蓝光阻隔选择性好、光透过率高的特点。
对比例2
将12.6g HEMA、7.2g甲基丙烯酸缩水甘油酯、0.06gAIBN和0.05g二甲基丙烯酸乙二醇酯室温混合搅拌5h,过滤,逐滴滴入到模具中,在75℃烘箱中反应12h,反应完后脱模,放入纯水中水合3h,后移入到标准盐水中,制得水凝胶角膜接触镜。
实施例48抗蓝光水凝胶镜片的制备
将12.6g HEMA、3.6g甲基丙烯酸缩水甘油酯、3.6g NVP、0.015g的抗蓝光剂(化合物I-6)、0.03g AIBN、0.02g二甲基丙烯酸乙二醇酯、0.02g二乙二醇二乙烯基醚,室温混合搅拌5h,过滤,逐滴滴入到模具中,在85℃烘箱中反应16h,反应完后脱模,放入纯水中水合4h,后移入到标准盐水中,制得抗蓝光水凝胶角膜接触镜。
对比例3
将12.6g HEMA、3.6g甲基丙烯酸缩水甘油酯、3.6g NVP、0.03g AIBN、0.02g二甲基丙烯酸乙二醇酯、0.02g二乙二醇二乙烯基醚,室温混合搅拌5h,过滤,逐滴滴入到模具中,85℃烘箱中反应16h,反应完后脱模,放入纯水中水合4h,后移入到标准盐水中,制得水凝胶角膜接触镜。
实施例49抗蓝光水凝胶镜片的制备
将18g甲基丙烯酸甘油酯、2gN,N二甲基丙烯酰胺、0.02g的抗蓝光剂(化合物I-16)、0.2g AIBN和0.16g聚乙二醇二乙烯基醚,室温混合搅拌6h,过滤,逐滴滴入到模具中,在110℃烘箱中反应12h,反应完后脱模,放入20%乙醇溶液中水合3h,后移入纯水中水合12h,最终移到标准盐水中,制得抗蓝光水凝胶角膜接触镜。
对比例4
将18g甲基丙烯酸甘油酯、2g N,N二甲基丙烯酰胺、0.2g AIBN和0.16g聚乙二醇二乙烯基醚,室温混合搅拌6h,过滤,逐滴滴入到模具中,在110℃烘箱中反应12h,反应完后脱模,放入20%乙醇溶液中水合3h,后移入纯水中水合12h,最终移到标准盐水中,制得水凝胶角膜接触镜。
实施例50抗蓝光水凝胶镜片的制备
将16g甲基丙烯酸甘油酯、4g N-甲基-N-乙烯基乙酰胺、0.002g的抗蓝光剂(化合物I-1-Cu)、0.3gAIBN、0.2g聚乙二醇二甲基丙烯酸酯、0.1g二乙二醇二乙烯基醚室温混合搅拌5h,过滤,逐滴滴入到模具中,在70℃烘箱中反应24h,反应完后脱模,放入30%乙醇溶液中水合5h,后移入纯水中水合12h,最终移到标准盐水中,制得抗蓝光水凝胶角膜接触镜。
对比例5
将16g甲基丙烯酸甘油酯、4g N-甲基-N-乙烯基乙酰胺、0.3g AIBN、0.2g聚乙二醇二甲基丙烯酸酯和0.16g二乙二醇二乙烯基醚,室温混合搅拌5h,过滤,逐滴滴入到模具中,在70℃烘箱中反应24h,反应完后脱模,放入30%乙醇溶液中水合5h,后移入纯水中水合12h,最终移到标准盐水中,制得水凝胶角膜接触镜。
实施例51抗蓝光硅水凝胶镜片的制备
将11.88g SIGMA、7.92g NVP、0.005g的抗蓝光剂(化合物I-1-Zn)、0.1g偶氮二异庚腈和0.1g二甲基丙烯酸三乙二醇酯,室温混合搅拌12h,过滤,逐滴滴入到模具中,在80℃烘箱中反应18h,反应完后脱模,20%的异丙醇溶液中水合5h,后移入到纯水中水合12h,最后放入标准盐水中,制得抗蓝光硅水凝胶角膜接触镜。
对比例6
将11.88g SIGMA、7.92g NVP、0.1g偶氮二异庚腈和0.1g二甲基丙烯酸三乙二醇酯,室温混合搅拌12h,过滤,逐滴滴入到模具中,在80℃烘箱中反应18h,反应完后脱模,20%的异丙醇溶液中水合5h,后移入到纯水中水合12h,最后放入标准盐水中,制得硅水凝胶角膜接触镜。
实施例52抗蓝光硅水凝胶镜片的制备
6.8g SIGMA、3.8g甲基丙烯酰氧丙基三(三甲基硅氧烷基)硅烷、7.92g HEMA、0.01g的抗蓝光剂(化合物I-1)、0.06g BPO,0.14g聚乙二醇二甲基丙烯酸酯,室温混合搅拌24h,过滤,逐滴滴入到模具中,在60℃烘箱中反应22h,反应完后脱模,5%的聚乙二醇200的溶液中水合3h,后移入纯水中水合5h,最后放入标准盐水中,制得抗蓝光硅水凝胶角膜接触镜。合成产品见图9。图9所示的抗蓝光硅水凝胶角膜接触镜颜色呈粉色,避免了传统的抗蓝光镜片偏黄的情况,且该镜片在385-445nm的有害蓝光透过率为57.7%,在446nm-505nm的有益蓝光透过率为98.7%,380-800nm光透过率为97.5%。说明该抗蓝光水凝胶角膜接触镜主要具有颜色特别、蓝光阻隔选择性好、光透过率高的特点。
对比例7
6.8g SIGMA、3.8g甲基丙烯酰氧丙基三(三甲基硅氧烷基)硅烷、7.92g HEMA、0.06gBPO、0.14g聚乙二醇二甲基丙烯酸酯,室温混合搅拌24h,过滤,逐滴滴入到模具中,在60℃烘箱中反应22h,反应完后脱模,5%的聚乙二醇200的溶液中水合3h,后移入纯水中水合5h,最后放入标准盐水中,制得硅水凝胶角膜接触镜。
实施例53抗蓝光硅水凝胶镜片的制备
6.8g SIGMA、3.8g甲基丙烯酰氧丙基三(三甲基硅氧烷基)硅烷、7.92g N,N二甲基丙烯酰胺、0.02g的抗蓝光剂(化合物I-6)、0.08g BPO,0.4g聚乙二醇二甲基丙烯酸酯,室温混合搅拌24h,过滤,逐滴滴入到模具中,在65℃烘箱中反应18h,反应完后脱模,3%的聚乙二醇200的溶液中水合3h,后移入到纯水中水合5h,最后放入标准盐水中,制得抗蓝光硅水凝胶角膜接触镜。
对比例8
6.8g SIGMA、3.8甲基丙烯酰氧丙基三(三甲基硅氧烷基)硅烷、7.92g N,N二甲基丙烯酰胺、0.08g BPO、0.4g聚乙二醇二甲基丙烯酸酯,室温混合搅拌24h,过滤,逐滴滴入到模具中,在65℃烘箱中反应18h,反应完后脱模,3%的聚乙二醇200的溶液中水合3h,后移入到纯水中水合5h,最后放入标准盐水中,制得硅水凝胶角膜接触镜。
实施例54抗蓝光硅水凝胶镜片的制备
6.8g SIGMA、3.8g甲基丙烯酸异冰片酯、7.92g N,N二甲基丙烯酰胺、0.1g的抗蓝光剂(化合物I-16)、0.12g偶氮二异庚腈,0.16g异氰脲酸三烯丙酯,室温混合搅拌24h,过滤,逐滴滴入到模具中,在70℃烘箱中反应20h,反应完后脱模,5%的甘油水溶液中水合3h,后移入到纯水中水合5h,最后放入标准盐水中,制得抗蓝光硅水凝胶角膜接触镜。
对比例9
6.8g SIGMA、3.8g甲基丙烯酸异冰片酯、7.92g N,N二甲基丙烯酰胺、0.12g AIBN,0.16g异氰脲酸三烯丙酯,室温混合搅拌24h,过滤,逐滴滴入到模具中,在70℃烘箱中反应20h,反应完后脱模,5%的甘油水溶液中水合3h,后移入到纯水中水合5h,最后放入标准盐水中,制得硅水凝胶角膜接触镜。
实施例55抗蓝光水凝胶镜片的制备
将18g HEMA、2g MMA、0.015g的抗蓝光剂(化合物I-1-Cu)、0.05g BPO,0.08g二甲基丙烯酸三乙二醇酯,室温混合搅拌4h,过滤,逐滴滴入到模具中,在60℃烘箱中反应26h,反应完后脱模,5%的聚乙二醇400的水溶液中水合2h,后移入到纯水中水合5h,最后放入标准盐水中,制得抗蓝光水凝胶角膜接触镜。
对比例10
将18g HEMA、2g MMA、0.05g BPO,0.08g二甲基丙烯酸三乙二醇酯,室温混合搅拌4h,过滤,逐滴滴入到模具中,在60℃烘箱中反应26h,反应完后脱模,5%的聚乙二醇400的水溶液中水合2h,后移入到纯水中水合5h,最后放入标准盐水中,制得水凝胶角膜接触镜。
实施例56抗蓝光硅水凝胶镜片的制备
将9.28g甲基丙烯酰氧丙基三(三甲基硅氧烷基)硅烷、10.72g HEMA,0.005g的抗蓝光剂(化合物I-1-Zn)、0.085g偶氮二异庚腈,0.075g二乙二醇二乙烯基醚,室温混合搅拌4h,过滤,逐滴滴入到模具中,在60℃烘箱中反应24h,反应完后脱模,1%的聚乙烯醇溶液中水合2h,后移入到纯水中水合12h,最后放入标准盐水中,制得抗蓝光硅水凝胶角膜接触镜。
对比例11
将12.20g甲基丙烯酰氧丙基三(三甲基硅氧烷基)硅烷、7.8g HEMA、0.085g偶氮二异庚腈,0.075g二乙二醇二乙烯基醚,室温混合搅拌4h,过滤,逐滴滴入到模具中,在60℃烘箱中反应24h,反应完后脱模,1%的聚乙烯醇溶液中水合2h,后移入到纯水中水合12h,最后放入标准盐水中,制得硅水凝胶角膜接触镜。
性能测试实施例:
实施例57热稳定性
称取3mg含乙二醇单元的烯基封端的四芳基(金属)卟啉类化合物,采用差示扫描量热法(DSC),测定其分解温度,测试结果如表1所示,部分结果见图10和11。
表1 DSC测试结果
/>
实施例58防有害蓝光性能
针对实施例3~44,采用DMF为溶剂,配制10μmol/L的含乙二醇单元的烯基封端的四芳基(金属)卟啉类化合物溶液,分别在220~800nm波长下测定其透过率。针对实施例45~46、对比例1,将制备的高分子材料紧贴装有标准盐水(标准盐水配方参照GB11417.4-2012)的比色皿光学玻璃透光面,分别在220~800nm波长下测定其透过率。测试结果如表2所示。
表2透过率测试结果
/>
实施例59防有害蓝光性能
将实施例47~56和对比例2~11制备的角膜接触镜以及市售抗蓝光镜片(北京自然美光学有限公司的优瞳小黄芯),紧贴装有生理盐水(标准盐水参照GB11417.4-2012)的比色皿光学玻璃透光面,在380~800nm波长下测定其透过率;将镜片切成长条状,宽为7mm,初始夹具长度5mm,运行速率为10mm/min,测试得到镜片的断裂伸长率,镜片含水量按照GB/T11417.7中称重法的测试方法进场测试。测试结果如表3所示,部分结果见附图12。
表3抗蓝光角膜接触镜的透过率数据
结果表明:本发明中抗蓝光角膜接触镜片在385~445nm的平均透光率低于80%,更优的配方在385~445nm的平均透光率低于40%,在446~505nm的平均透光率高于95%,同时其可见光透过率可以达到90%以上,视网膜蓝光辐射危害降低率H
实施例60镜片细胞毒性测试
优选镜片实施例49和54及对比例4和9,按照GB/T16886.5-2017体外细胞毒性实验的方法测试抗蓝光镜片的细胞毒性,加入抗蓝光剂的镜片和未加抗蓝光剂镜片均为无毒,细胞增殖率见图13~16。GB/T16886.5-2017医疗器械生物学评价第5部分:体外细胞毒性试验中提出,定量评价细胞增殖率下降大于30%则被认为具有细胞毒性。实施例49和54及对比例4和9的镜片在不同浓度浸提液中的细胞增殖率均在70%以上,添加抗蓝光剂前后的镜片均无毒,则说明加入抗蓝光剂对镜片的细胞毒性无影响,符合镜片的安全性评价要求。