一种提高大肠杆菌鲁棒性的重组菌及其构建方法
文献发布时间:2024-04-18 19:52:40
技术领域
本发明属于生物技术领域,涉及微生物合成生物学领域,具体涉及一种提高大肠杆菌鲁棒性的重组菌及其构建方法,尤其涉及一种提高大肠杆菌耐受性和产物合成能力的重组菌及其构建方法。
背景技术
生物质是一种可再生的清洁资源,通过生物制造技术,可以被转化为燃料、大宗化学品和天然产物,从而部分替代石油化工炼制和植物提取。目前,在工业生物技术中,一些重要的微生物,包括细菌和酵母,已经被广泛应用于发酵过程,作为细胞工厂以生产药品、保健品、酶、食品成分、燃料和生物化学品(Marta Tous Mohedano et al.,2022)。为了满足商业需求,微生物细胞通常需要被改造,用于有效地生物合成有价值的化学物质,最终实现特定的指标,如效度、产量和生产力。微生物发酵具有成本低、无化学残留物、产量高等明显优点,被认为是生物制造的核心,而其中的大肠杆菌由于具有遗传背景清晰、生理背景明确、易于培养、迭代时间短、遗传操作手法成熟等一系列优点,被作为最常用的底盘细胞广泛用于微生物细胞工厂的构建。
生物制造的核心技术是构建高效的微生物细胞工厂,将生物质原材料转化为各种终端产品。但是微生物细胞工厂在生产化合物的过程中,往往会暴露在各种不同的胁迫中,这些压力来自原料、新陈代谢或工业生产过程,如上游生物质衍生糖(例如纤维素水解液)制备过程中产生的呋喃以及酚类化合物等杂质,生产过程积累的有毒中间体、不良副产物,下游发酵过程中存在的高温、酸碱、高盐等不利的工业条件,所合成的终产物对细胞自身的反噬,这一系列不利条件会对细胞工厂产生毒害作用,从而极大的限制菌株的生长和产物合成能力。
研究表明,合成产物引起E.coli细胞毒害的机制有多种,包括导致细胞膜损伤、DNA损伤、引起蛋白质变性和诱导活性氧(ROS)的产生等。尽管不同化合物对细胞的毒性机制各有不同,但细胞膜的损伤被认为是多种化合物引起细胞毒害的重要共性机制。E.coli细胞膜细胞膜作为细胞屏障,负责维持细胞内微环境稳定,并参与同外界环境进行物质交换、能量运输和信息传递的过程;另外,细胞膜上含有丰富的酶系,执行许多重要的代谢功能,对维持和调节细胞生理和代谢至关重要。因此,构建鲁棒性的E.coli细胞膜,提高其面对有毒产物和底物时的耐受性,是解决细胞膜损伤这一问题的有效策略。
发明内容
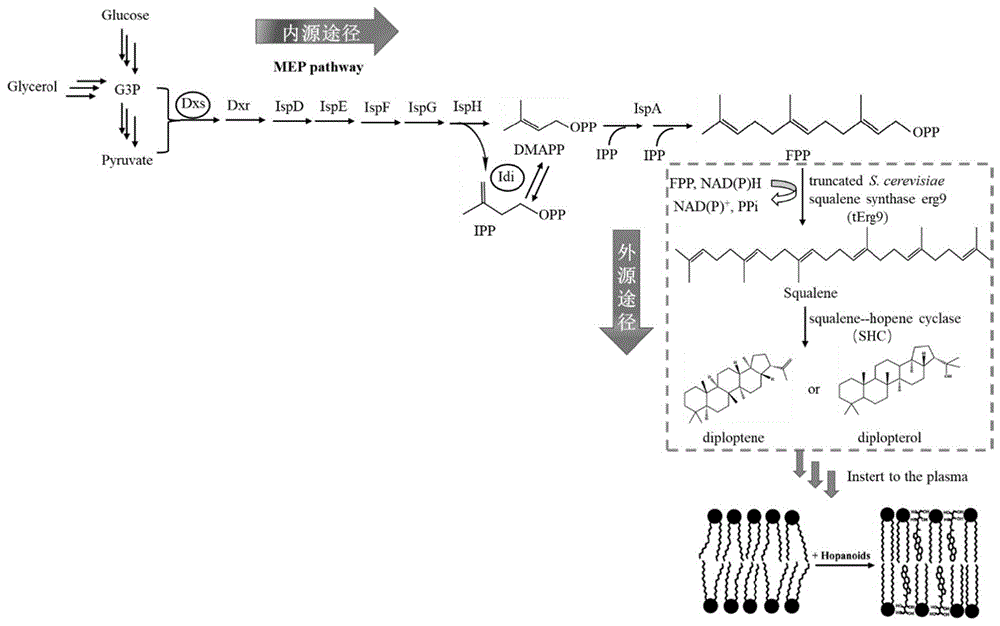
为解决上述技术问题,本发明的目的是提供一种提高大肠杆菌鲁棒性的重组菌及其构建方法。本发明通过人工合成途径将非天然膜组分引入大肠杆菌,通过重塑E.coli细胞膜改造大肠杆菌底盘细胞耐受性,构建出高效的微生物细胞工厂,能够极大地提升目前工业微生物的发酵能力,提高其生理性能。具体来说,本发明采用CRISPR-cas9基因编辑技术,将宿主菌MG1655中编码甲基乙二醛合酶基因mgsA替换为来自酵母的截短的角鲨烯合酶基因tErg9,并运用人工启动子策略上调tErg9的表达;在所得重组菌基础上采用CRISPR-cas9基因编辑技术继续将重组菌磷酸乙酰转移酶基因pta替换为来自酸热脂环酸芽孢杆菌的角鲨烯-藿烯环化酶基因shc,并运用人工启动子策略上调shc的表达,得到重组工程菌,所得到的重组大肠杆菌没有抗性,对多种化合物抑制剂具有广谱耐受性,能够生产C8化合物。其中,所述人工启动子元件为M1-93,其核苷酸序列如SEQ ID NO.5所示。
本发明的目的是通过以下技术方案来实现的:
第一方面,本发明提供一种鲁棒性提高的重组大肠杆菌,该重组菌为引入了非天然膜组分hopanoids的重组大肠杆菌;非天然膜组分hopanoids的合成是通过大肠杆菌內源MEP途径结合引入的外源人工合成途径进行。
进一步的,对內源MEP途径关键限速酶进行人工调控。
內源MEP途径关键限速酶內源MEP途径关键限速酶包括1-脱氧-D-木酮糖-5-磷酸合酶基因Dxs和异戊烯基焦磷酸异构酶基因idi。
大肠杆菌內源MEP途径包括(副产物合成基因)甲基乙二醛合酶(Methylglyoxalsynthase)基因mgsA和磷酸乙酰转移酶(Phosphate acetyltransferase)基因pta。
引入的外源人工合成途径包括(来自酵母的截短的)角鲨烯合酶(truncatedS.cerevisiae squalene synthase gene erg9)基因tErg9和(来自酸热脂环酸芽孢杆菌的)角鲨烯-藿烯环化酶(Squalene--hopene cyclase)基因shc。
具体的,所述重组大肠杆菌依次将两个副产物合成基因敲除,并引入一条人工合成途径,即依次将来自酵母的截短的角鲨烯合酶基因tErg9、来自酸热脂环酸芽孢杆菌的角鲨烯-藿烯环化酶基因shc整合到MgsA和pta两个位点。
本发明中,来自酵母的截短的角鲨烯合酶能够催化大肠杆菌体内前体物质法尼基二磷酸酯FPP合成角鲨烯(squalene),随后来自酸热脂环酸芽孢杆菌的角鲨烯-藿烯环化酶将角鲨烯催化为藿烷,是一种固醇类似物。藿烷插入到磷脂双分子层中,降低了细胞膜的流动性,增强了大肠杆菌耐酸碱与耐渗透压的能力。
作为本发明的一个实施方案,将大肠杆菌中的甲基乙二醛合酶基因mgsA替换为角鲨烯合酶基因tErg9,并运用人工启动子策略上调tErg9的表达;将磷酸乙酰转移酶基因pta替换为角鲨烯-藿烯环化酶基因shc,并运用人工启动子策略上调shc的表达;得到(引入了外源人工合成途径的)重组大肠杆菌。记为重组E.coli菌ΔmgsA::tErg9Δpta::shc。
作为本发明的一个实施方案,编码大肠杆菌中甲基乙二醛合酶基因mgsA的核苷酸的序列包括SEQ ID NO.1所示的序列;编码大肠杆菌中磷酸乙酰转移酶基因pta的核苷酸的序列包括SEQ ID NO.3所示的序列;编码所述(外源合成途径的来自酵母的截短的)角鲨烯合酶基因tErg9、(来自酸热脂环酸芽孢杆菌的)角鲨烯-藿烯环化酶基因shc的核苷酸的序列包括SEQ ID NO.2、SEQ ID NO.4所示的序列。
作为本发明的一个实施方案,所述酵母包括S.cerevisiae BY4742,所述酸热脂环酸芽孢杆菌包括Alicyclobacillus acidocaldarius subsp.acidocaldarius。
作为本发明的一个实施方案,所述外源人工合成途径包括人工调控元件M1-93-tErg9+M1-93-shc;具体是采用M1-93作为tErg9和shc的人工调控元件,组成M1-93-tErg9和M1-93-shc。M1-93的核苷酸序列如SEQ ID NO.5所示。
作为本发明的一个实施方案,还包括对所得重组工程菌內源MEP途径关键限速酶进行人工调控的步骤。所述对內源MEP途径关键限速酶进行人工调控是将1-脱氧-D-木酮糖-5-磷酸合酶基因Dxs和异戊烯基焦磷酸异构酶基因idi的启动子分别替换为人工增强启动子M1-37和M1-46;M1-37的核苷酸序列包括SEQ ID NO.6所示,M1-46的核苷酸序列包括SEQ ID NO.7所示。对內源MEP途径两个关键酶1-脱氧-D-木酮糖-5-磷酸合酶基因Dxs和异戊烯基焦磷酸异构酶基因idi进行人工加强调控,使更多碳代谢流走向IPP/DMAPP的合成,从而进一步提高工程菌生成hopanoids的水平。
本发明通过CRISPR-Cas9技术得到引入了外源人工合成途径的重组大肠杆菌。作为本发明的一个实施方案,重组大肠杆菌的宿主为大肠杆菌E.coli MG1655。
进一步地,同时构建重组大肠杆菌的对照菌株大肠杆菌E.coli MG1655ΔmgsAΔpta。
第二方面,本发明提供一种鲁棒性提高的重组大肠杆菌的构建方法,包括以下步骤:
(1)根据所要替换的基因mgsA,所需整合的M1-93-tErg9人工调控元件,构建相应的pTargetF-N20质粒及DonorDNA;
根据所要替换的基因pta,所需整合的M1-93-shc人工调控元件,构建相应的pTargetF-N20质粒及DonorDNA;
(2)将其中一种pTargetF-N20质粒及DonorDNA采用电转化法转化至含有pCas质粒的大肠杆菌感受态中;
诱导质粒pCas质粒上sgRNA的转录,消除pTargetF-N20质粒并筛选出基因改造成功的菌株;
(3)将另一种未转化的pTargetF-N20质粒及DonorDNA转化至步骤(2)中得到的菌株中,诱导质粒pCas质粒上sgRNA的转录,消除pTargetF-N20质粒并筛选出基因改造成功的菌株;
(4)消除pCas质粒,得到引入了外源人工合成途径的重组大肠杆菌。记为E.coliMG1655ΔmgsA::tErg9Δpta::shc。
本发明中,人工调控元件M1-93-tErg9、M1-93-shc是通过CRISPR-cas9基因编辑技术进行操作。
作为本发明的一个实施方案,在步骤(2)中,含有pCas质粒的大肠杆菌感受态,是将质粒pCas转化到胞内,获得含pCas质粒的受体菌。
进一步地,将所述引入了外源人工合成途径的重组大肠杆菌通过λ-Red一步重组法将內源限速酶基因dxs和idi的启动子分别替换为人工增强启动子M1-37和M1-46,M1-37的核苷酸序列包括SEQ ID NO.6所示,M1-46的核苷酸序列包括SEQ ID NO.7所示。得到的鲁棒性提高的重组大肠杆菌,记为大肠杆菌ΔmgsA::tErg9Δpta::smo
M1-37-dxs M1-46-idi;与此同时,构建工程菌株的对照菌株ΔmgsAΔpta M1-37-dxs
M1-46-idi。
作为本发明的一个实施方案,在步骤(2)中转化的是根据所需整合的人工调控元件M1-93-tErg9的pTargetF-N20质粒及DonorDNA;步骤(3)中转化的是根据所要替换的基因pta,所需整合的M1-93-shc人工调控元件,构建相应的pTargetF-N20质粒及DonorDNA。
作为本发明的一个实施方案,在步骤(1)中,构建pTargetF-N20质粒时,根据所要替换的基因mgsA、pta,在网站(https://www.benchling.com/)上找到合适的N20核苷酸序列,以质粒pTargrtF为模板并进行全质粒PCR。
作为本发明的一个实施方案,在步骤(1)中,构建DonorDNA时,根据所要整合的人工调控元件M1-93-tErg9、M1-93-shc上、下游同源臂,再分别与M1-93和tErg9、M1-93和shc进行一步同源重组整合构建而得。
作为本发明的一个实施方案,在步骤(2)、(3)中,采用IPTG诱导。
作为本发明的一个实施方案,在步骤(2)、(3)中,通过壮观霉素及卡那霉素抗性平板筛选获得工程菌;
作为本发明的一个实施方案,在步骤(4)中,不加任何抗生素37℃过夜培养步骤(3)构建的菌株,以消除pCas质粒。
第三方面,本发明提供了一种鲁棒性提高的重组大肠杆菌,所述重组大肠杆菌为Hop1菌株,保藏编号为CGMCC NO.26270,属名Escherichia coli,中文译名大肠埃希氏菌,保藏日期为2022年12月26日。
本发明将Hop1菌株提交专利认可的机构进行保藏;Hop1菌株的微生物保藏编号为:CGMCC NO.26270;分类命名为:大肠埃希氏菌Escherichia coli;保藏单位:中国微生物菌种保藏管理委员会普通微生物中心;保藏时间是2022年12月26日;保藏地址:北京市朝阳区北辰西路1号院3号中国科学院微生物研究所。
第四方面,本发明提供了一种鲁棒性提高的重组大肠杆菌在作为底盘细胞生产目标化合物中的应用。
作为本发明的一个实施方案,上述应用包括多种目标化合物,如长短链脂肪酸、三羟基丙酸、间苯三酚等不同化合物的产物展示,具体包括以下步骤:
(1)将不同产物的利用质粒分别转入对照菌和工程菌中;
(2)进一步地,在步骤(1)中,产物利用质粒对应为下表:
(3)对于长短链脂肪酸,以MOPS+2%(wt/v)Glucose培养基进行培养;
对于三羟基丙酸,以M9+2%(wt/v)Glycerol培养基进行培养;
对于间苯三酚,以M9+2%(wt/v)Glucose培养基进行培养。
其中:MOPS培养基配方为:
40X“M”
其中micronutrient solution成分为:
ZnCl
M9+2%(wt/v)Glycerol培养基配方为:
M9+2%(wt/v)Glucose培养基配方为:
5X M9 salts
(4)以0.1mM的IPTG为诱导剂,30℃培养72h,期间每24h取样。长短链脂肪酸发酵液上清液经萃取和衍生成酯之后进行GC-MS分析;三羟基丙酸和间苯三酚发酵液经离心过滤后经高效液相色谱分析。
作为本发明的另一个实施方案,所述的应用包括以下步骤:
(1)将产物利用质粒pXZ18Z(长链脂肪酸)、pJMYEE182564(短链脂肪酸)、pTrc99A-phlD(间苯三酚)、pTrc99a-dhaB-aldH分别转入所述重组大肠杆菌中;
(2)以MOPS+2%(wt/v)Glucose培养基进行培养;
(3)以0.1-0.5mM的IPTG为诱导剂,30℃培养24-72h,即得所述目标化合物。
上述步骤(3)中,培养期间每隔24h取样发酵液上清液经萃取和衍生成酯之后进行GC-MS分析。
与现有技术相比,本发明具有如下有益效果:
1)本发明采用CRISPR-cas9基因编辑技术将非天然膜组分hopanoids的合成途径引入大肠杆菌中,通过人工构建,重塑大肠杆菌细胞膜,改善了细胞膜流动性,有效提高了大肠杆菌的耐受性。
2)本发明的工程菌株鲁棒性提高的重组大肠杆菌在耐受性提高的基础上同时提高了对目标化合物的合成能力。
3)本发明的工程菌株不含质粒,不存在质粒丢失的风险,发酵时不加入抗生素,具有应用于工业化生产的潜力。
附图说明
通过阅读参照以下附图对非限制性实施例所作的详细描述,本发明的其它特征、目的和优点将会变得更明显:
图1为非天然膜组分引入大肠杆菌细胞膜构建鲁棒性提高的重组大肠杆菌构建策略及模式图;
图2为消除pCas质粒的步骤图;
图3为实施例2中pTargetF-mgsA重组质粒结构图;
图4为实施例2中pTargetF-pta重组质粒结构图;
图5为实施例3中含shc的载体质粒图谱pCDF-P1-SHC;
图6为构建工程菌株E.coli MG1655ΔmgsAΔpta M1-37-dxs M1-46-idi、E.coliMG1655ΔmgsA::tErg9Δpta::shc M1-37-dxs M1-46-idi对不利工业条件及多种化合物的耐受性测试;
图7为构建工程菌株E.coli MG1655ΔmgsAΔpta M1-37-dxs M1-46-idi、E.coliMG1655ΔmgsA::tErg9Δpta::shcM1-37-dxs M1-46-idi对于纤维素水解液中的典型抑制物的耐受性测试;
图8为构建工程菌株E.coli MG1655ΔmgsAΔpta M1-37-dxs M1-46-idi、E.coliMG1655ΔmgsA::tErg9Δpta::shcM1-37-dxs M1-46-idi对一碳化合物的耐受性测试;
图9为构建工程菌株E.coli MG1655ΔmgsAΔpta M1-37-dxs M1-46-idi、E.coliMG1655ΔmgsA::tErg9Δpta::shcM1-37-dxs M1-46-idi对抑制性产物的耐受测试;
图10为构建工程菌株E.coli MG1655ΔmgsAΔpta M1-37-dxs M1-46-idi、E.coliMG1655ΔmgsA::tErg9Δpta::shcM1-37-dxs M1-46-idi一定浓度的纤维素水解液复配物中生长情况;
图11为pTrc99A-phlD载体及验证图谱,核酸序列上方细箭头所示为测序比对情况;
图12为pTrc99A-dha-aldH载体及验证图谱,核酸序列上方细箭头为测序比对情况;
图13为构建工程菌株E.coli MG1655ΔmgsAΔpta M1-37-dxs M1-46-idi、E.coliMG1655ΔmgsA::tErg9Δpta::shcM1-37-dxs M1-46-idi对于短链脂肪酸的产物合成能力;
图14为构建工程菌株E.coli MG1655ΔmgsAΔpta M1-37-dxs M1-46-idi、E.coliMG1655ΔmgsA::tErg9Δpta::shcM1-37-dxs M1-46-idi对于长链脂肪酸的产物合成能力;
图15为构建工程菌株E.coli MG1655ΔmgsAΔpta M1-37-dxs M1-46-idi、E.coliMG1655ΔmgsA::tErg9Δpta::shcM1-37-dxs M1-46-idi对于三羟基丙酸的产物合成能力;
图16为构建工程菌株E.coli MG1655ΔmgsAΔpta M1-37-dxs M1-46-idi、E.coliMG1655ΔmgsA::tErg9Δpta::shcM1-37-dxs M1-46-idi对于对间苯三酚的产物合成能力。
具体实施方式
下面结合实施例,对本发明的具体实施方式作进一步详细描述。以下实施例用于说明本发明,将有助于本领域的技术人员进一步理解本发明,但不用来限制本发明的范围。应当指出的是,对本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变化和改进。这些都属于本发明的保护范围。
本发明提供了一种提高大肠杆菌鲁棒性的重组菌及其构建方法和应用;该重组菌包含一种非天然膜组分,使其细胞膜得到重塑,从而全面提高了大肠杆菌的鲁棒性和产物合成能力。所述非天然膜组分为来自于酸热脂环酸芽孢杆菌,是一种耐热嗜酸古生菌,能够适应极端环境。本发明的重组菌具有如下特性:A、包含一条外源人工合成途径,该合成途径不存在于E.coli中(图1);B、人工引入的该途径能够合成非天然膜组分hopanoids,并插入到E.coli细胞膜磷脂双分子层中使E.coli细胞膜得到重塑;C、不同于以往的对其天然组分的改造,该重组菌细胞膜中包含全新的非天然膜组分,重组工程菌的耐受性和产物合成能力得到显著提高。
具体的,大肠杆菌CRISPR-Cas9系统由两个基本质粒pCas和pTargetF组成,质粒pCas是大肠杆菌游离型质粒,含有L-阿拉伯糖诱导表达Red重组酶元件、Cas9蛋白的编码基因Cas9、温敏型元件及诱导消除质粒pTargetF所使用的sgRNA、卡那霉素抗性基因KanR等。含有pCas衍生质粒系列均需在30℃环境中培养以保证质粒能够正常复制而不发生丢失。质粒pTargetF是大肠杆菌游离型质粒,含有壮观霉素抗性基因aadA和用于转录sgRNA的启动子pJ23119。
根据目标编辑位点的序列,设计引物扩增得到含有与目标位点匹配的20个碱基序列(N20),然后将序列克隆到pTargetF载体中获得敲除质粒;其次,将质粒pCas转化到宿主细胞中,诱导λ-Red重组系统表达,制备感受态细胞,然后将敲除质粒pTargetF和DonorDNA转化到感受态细胞进行基因编辑和重组,涂布培养获得转化子,测序验证基因组重组情况;然后,诱导菌株中pCas质粒上的质粒修复系统工作进行剪切pTargetF敲除质粒,完成一轮基因改造。依次进行上述工作可以对宿主基因组的多个基因位点进行改造,实现基因的缺失或插入;最后37℃培养,消除pCas质粒。
实施例1E.coli MG1655 CRISPR-Cas9系统的构建
(1)基础质粒pCas通过热激转化的方法转化大肠杆菌
制备大肠杆菌E.coli MG1655的感受态,将质粒pCas转化到胞内获得含pCas质粒的受体菌E.coli MG1655/pCas。
(2)受体菌电转感受态制备
a、将含pCas质粒的大肠杆菌(E.coli MG1655/pCas)接种到液体LB(含卡那霉素终浓度为50μg/mL)培养基中,30℃、220rpm/min培养至对数期。
b、以1%的接种量接种于装有20mL LB(含卡那霉素终浓度为50μg/mL)培养基的50mL三角瓶中,30℃、200rpm/min条件培养至OD600为0.2~0.3时,加入终浓度为10mmol/L的L-阿拉伯糖诱导剂,诱导pCas上的λ-Red重组酶充分表达,OD
c、在超净台内将培养液转移到50mL无菌离心管中,在冰中放置15min。
d、将离心管中的菌液4℃、6000rpm离心10min。
e、弃上清,加入20mL预冷的ddH
f、弃上清,加入20mL预冷的10%甘油温柔重悬菌体,4℃、6000rpm离心10min。
e、弃上清,将菌体重悬浮于5mL预冷的10%甘油中,按每管50-80μL感受态细胞分装于1.5mL离心管中,冰上放置备用。
(3)pTargetF-N20质粒和DonorDNA的转化
a、将待转化的DNA加入感受态细胞中,温柔混合均匀,冰上放置30min;
b、将混合物转入预冷的2mm电转杯中,放置冰上至少2min,待电转;
c、打开电转仪,将参数设定为2.5kV;
d、从冰中取出电转杯,用纸巾吸去表面的水分,放入样品槽中电击。电击后立即加入常温LB培养基悬浮细胞,30℃复苏2h,涂布含50μg/mL卡那霉素和50μg/mL壮观霉素的LB平板,30℃培养过夜;
e、挑取平板长出的克隆子,使用验证引物进行菌落PCR验证[对于MgsA位点的编辑验证引物为(MgsA-L-up1:TCACATGAGGCCTGCCAG SEQ ID NO.12
&MgsA-R-down1:GCCGATTCCGGTAAAGCT SEQ ID NO.13
),对于pta位点的编辑,验证引物为pta-L-up1:TGAGCGTTGACGCAATCA SEQIDNO.14&pta-R-down1:GATCCTGAGGTTAATCCTTCAAA SEQ ID NO.15],获得正确得基因编辑菌株。
(4)pTargetF-N20质粒的消除
a、将验证正确的基因编辑克隆子接种至5mL LB培养基(含卡那霉素终浓度为50μg/mL),加入终浓度为0.5mM IPTG诱导质粒pCas质粒上sgRNA的转录,30℃培养12-16小时,用接种环蘸取适量菌液在终浓度为50μg/mL卡那霉素的LB平板上划线;
b、挑取20-30个分离的单菌落一一对应在含终浓度为50μg/mL卡那霉素平板、50μg/mL卡那霉素平板和50μg/mL壮观霉素的双抗平板上点板,平板于30℃培养。挑取不在双抗平板生长,而在卡那霉素平板生长的单菌落扩培保种,此单菌落为消除pTargetF质粒但保留了pCas质粒的新菌株,如图2。
c、将构建的其他基因整合质粒转化到上述菌株,并筛选获得基因整合菌株而后进行敲除质粒的消除,从而实现循环的基因组编辑。如:先将根据M1-93-tErg9构建的pTargetF-N20质粒及DonorDNA转化到含pCas质粒的大肠杆菌感受态中,在进行pTargetF-N20质粒的消除;再将另一种,根据M1-93-shc构建的pTargetF-N20质粒及DonorDNA转化到前者获得的保留pCas质粒的菌株中,再进行pTargetF-N20质粒的消除;最后再进行基础质粒pCas的消除。本实施例详述了crispr-cas9的操作步骤,实施例2、3即利用crispr具体操作。
(5)基础质粒的消除
基因组编辑完成后,将只含pCas质粒的基因组编辑菌株接种至LB培养基,不加任何抗生素37℃过夜培养,用接种环蘸取适量菌液在LB平板上划线;稀释后涂布于LB平板,37℃培养。挑取20-30个分离的单菌落一一对应在不含任何抗生素的LB平板(37℃培养)、含终浓度为50μg/mL卡那霉素LB平板(30℃培养)上点板。挑取不在卡那霉素LB平板生长而对应在LB平板生长的单菌落,此单菌落为消除基础质粒pCas的菌株,最终获得不含任何质粒的基因编辑菌株。
实施例2:E.coli MG1655ΔmgsA与E.coli MG1655ΔmgsA::tErg9菌株的构建(1)根据大肠杆菌mgsA基因上下游序列设计引物。根据NCBI上公布的大肠杆菌基因组序列,找到甲基乙二醛合酶基因mgsA(Accession IDs:G6497(EcoCyc))序列,在mgsA基因上选取切割位点N 2 0(AACGTCAACGCGATGTTGAG SEQ ID NO.16),设计pTargetF全质粒PCR的双向扩增引物,得到pTargetF-mgsA敲除质粒,如图3所示,其中,引物如下:
pTargetF-mgsA-up:
gtcctaggtataatactagtAACGTCAACGCGATGTTGAGgttttagagctagaaatagc
SEQ ID NO.17
pTargetF-down:
actagtattatacctaggactgag SEQ ID NO.18
(2)采用PCR的方法扩增mgsA敲除的上游同源臂、下游同源臂引物如下:
a)扩增mgsA敲除的上游同源臂引物,模板为Escherichia coli K-12substr.MG1655(Strain)基因组:
MgsA-L up1:TCACATGAGGCCTGCCAG SEQ ID NO.12
MgsA-L-KO down:
CTGATGAGCTGGGTGGAACGATCCAGTCGCCGCATTTCAA SEQ ID NO.19
b)扩增mgsA敲除的下游同源臂引物,模板为MG1655基因组:
MgsA-R up:CGTTCCACCCAGCTCATCAG SEQ ID NO.20
MgsA-R down1:GCCGATTCCGGTAAAGCT SEQ ID NO.21
c)扩增mgsA敲除的DonorDNA1片段,模板为上述a+b的重组片段(所述序列为SEQID NO.8):
MgsA-L up2:CAACACGCTGGCCGAAGT SEQ ID NO.22
MgsA-R down2:CTCGCCATTACCTCAACGG SEQ ID NO.23
(3)采用PCR的方法扩增M1-93-tErg9替换的上游同源臂、下游同源臂以及人工调控元件M1-93、tErg9,引物如下:
d)扩增tErg9替换上游同源臂引物,模板为MG1655基因组:
MgsA-L up1:TCACATGAGGCCTGCCAG SEQ ID NO.12
MgsA-L down:ATCCAGTCGCCGCATTTCAA SEQ ID NO.24
e)扩增人工调控元件M1-93,模板为M1-93基因组
M1-tErg9-up:TTGAAATGCGGCGACTGGATTTATCTCTGGCGGTGTTG SEQ IDNO.25;
M1-tErg9-down:AATTGTAATAGCTTTCCCATAGCTGTTTCCTGGTTTAAAC SEQ ID NO.26;
f)扩增tErg9上下游引物,模板为酵母BY4742基因组:
tErg9-up:ATGGGAAAGCTATTACAATTGGC SEQ ID NO.27
tErg9-down:CTGATGAGCTGGGTGGAACGTCAGTACTCTTCTTCTTGTTGGG SEQ ID NO.28
g)扩增tErg9替换下游同源臂引物,模板为MG1655基因组:
MgsA-R up:CGTTCCACCCAGCTCATCAG SEQ ID NO.20
MgsA-R down1:GCCGATTCCGGTAAAGCT SEQ ID NO.21
h)扩增M1-93-tErg9的DonorDNA2片段引物,模板为上述d+e+f+g四个片段的同源重组产物(所述序列为SEQ ID NO.9):
MgsA-L up2:CAACACGCTGGCCGAAGT SEQ ID NO.22
MgsA-R down2:CTCGCCATTACCTCAACGG SEQ ID NO.23;
运用一步同源重组技术将a+b(所述序列为SEQ ID NO.8)进行重组以及将d+e+f+g(所述序列为SEQ ID NO.9)进行重组后,接着,进行一轮PCR扩增回收目的片段,分别得到DonorDNA1和DonorDNA2,随后将pTargetF-mgsA敲除质粒与DonorDNA1片段或将pTargetF-mgsA敲除质粒与DonorDNA2片段共转化入含pCas9质粒的MG1655菌株中,利用CRISPR-Cas9技术完成基因敲除或替换,筛选得到成功敲除了mgsA的对照菌E.coli MG1655ΔmgsA或替换了人工元件M1-93-tErg9的工程菌E.coli MG1655ΔmgsA::tErg9。具体转化步骤同实施例1。
实施例3:E.coli MG1655ΔmgsAΔpta与E.coli MG1655ΔmgsA::tErg9Δpta::shc菌株的构建
(1)根据大肠杆菌pta基因上下游序列设计引物。根据NCBI上公布的大肠杆菌基因组序列,找到磷酸乙酰转移酶基因pta(Accession IDs:EG20173(EcoCyc))序列,在pta基因上选取切割位点N 20(GCTGATTCCGCTGCGGCCTT SEQ ID NO.29),设计pTargetF全质粒PCR的双向扩增引物,得到pTargetF-pta敲除质粒,如图4所示,其中,引物如下:
pTargetF-pta-up:
gtcctaggtataatactagtGCTGATTCCGCTGCGGCCTTgttttagagctagaaatagc SEQ IDNO.30;
pTargetF-down:
actagtattatacctaggactgag SEQ ID NO.18;
(2)采用PCR的方法扩增pta敲除的上游同源臂、下游同源臂,引物如下:
a)扩增pta敲除的上游同源臂引物,模板MG1655基因组:
pta-L up1:TGAGCGTTGACGCAATCA SEQ ID NO.14;
pta-L-KO down:AGCTGCGGATGATGACGAGAGGTTTATCCTCTTTCGTTACCG SEQ IDNO.31;
b)扩增pta敲除的下游同源臂引物,模板MG1655基因组:
pta-R up:TCTCGTCATCATCCGCAG SEQ ID NO.32;
pta-R down1:GATCCTGAGGTTAATCCTTCAAA SEQ ID NO.15;
c)扩增pta敲除的DonorDNA3片段,模板为上述a+b的重组片段(所述序列为SEQ IDNO.10):
pta-L up2:TGACCAAAGAGTCTGGCCT SEQ ID NO.33;
pta-R down2:GTCGTGAACAGCTGTACGC SEQ ID NO.34;
(3)采用人工合成的方法,按照E.coli密码子偏好性合成来自酸热脂环酸芽孢杆菌的shc,采用PCR的方法扩增M1-93-shc替换的上游同源臂、下游同源臂以及人工调控元件M1-93、shc,引物如下:
d)扩增shc上游同源臂引物,模板为MG1655基因组:
pta-L up1:TGAGCGTTGACGCAATCA SEQ ID NO.14;
pta-L down:GGTTTATCCTCTTTCGTTACCG SEQ ID NO.35;
e)扩增人工调控元件M1-93,模板为M1-93基因组:
M1-SMO-up:GTAACGAAAGAGGATAAACCTTATCTCTGGCGGTGTTG SEQ ID NO.36;
M1-SMO-down:CCCAGTTCGATGCTGCTCATAGCTGTTTCCTGGTTTAAAC SEQ ID NO.37;
f)扩增shc上下游引物,模板为合成的含shc的载体质粒(图5):
shc-up:ATGGCTGAACAGCTGGTTGA SEQ ID NO.38;
shc-down:CTGCGGATGATGACGAGATTAACGACGTTCAATTGCCTGT SEQ ID NO.39;
g)扩增shc替换下游同源臂引物,模板MG1655基因组:
pta-R up:TCTCGTCATCATCCGCAG SEQ ID NO.32;
pta-R down1:GATCCTGAGGTTAATCCTTCAAA SEQ ID NO.15;
h)扩增DonorDNA4片段引物,模板为上述四个片段的同源重组产物(所述序列为SEQ ID NO.11):
pta-L up2:TGACCAAAGAGTCTGGCCT SEQ ID NO.33;
pta-R down2:GTCGTGAACAGCTGTACGC SEQ ID NO.34;
运用一步同源重组技术将a+b(所述序列为SEQ ID NO.10)进行重组以及将d+e+f+g(所述序列为SEQ ID NO.11)进行重组后,接着进行一轮PCR扩增回收目的片段,分别得到DonorDNA3和DonorDNA4,随后将pTargetF-pta敲除质粒与DonorDNA3片段或将pTargetF-pta敲除质粒与DonorDNA4片段分别共转化入含pCas质粒的E.coli MG1655ΔmgsA或E.coliMG1655ΔmgsA::tErg9菌株中,利用CRISPR-Cas9技术完成基因敲除或替换,筛选得到成功敲除了pta的对照菌株E.coli MG1655ΔmgsAΔpta或替换了人工元件M1-93-shc的工程菌E.coli MG1655ΔmgsA::tErg9Δpta::shc。具体转化步骤同实施例1。
实施例4:重组菌株E.coli MG1655ΔmgsA::tErg9Δpta::shc的内源途径限速酶Dxs的调控
通过λ-Red一步重组法对实施例3构建的E.coli MG1655ΔmgsA::tErg9Δpta::shc重组菌株及其对照菌株E.coli MG1655ΔmgsAΔpta进行内源途径限速酶Dxs及Idi的表达调控。
λ-Red一步重组法对dxs基因的表达进行原位启动子替换:
首先将pKD46质粒分别化学转化至E.coli MG1655ΔmgsA::tErg9Δpta::shc菌株中或对照菌株E.coli MG1655ΔmgsAΔpta,挑取单菌落至液体培养基中,30℃,220rpm培养至OD
将构建好的重组片段FKF::M1-37-dxs电转入感受态细胞中,30℃培养得到转化子;
通过一步重组在染色体上实现M1-37启动子替代dxs自身启动子,达到精确调控dxs转录水平的目的。
重组片段FKF::M1-37-dxs通过PCR扩增回收目的片段获得,模板为M1-37基因组,引物如下:
Dxs-M1-up:
ACTACATCATCCAGCGTAATAAATAAACAATAAGTATTAATAGGCCCCTGAGGAACACTTAACGGCTGAC SEQ ID NO.40;
Dxs-M1-down:
GTGGAGTCGACCAGTGCCAGGGTCGGGTATTTGGCAATATCAAAACTCATAGCTGTTTCCTGGTTTAAAC SEQ ID NO.41;
通过37℃高温诱导消除温度敏感质粒pKD46,获得dxs改造后的工程菌株E.coliMG1655ΔmgsA::tErg9Δpta::shc M1-37-dxs及相应对照菌株E.coli MG1655ΔmgsAΔptaM1-37-dxs。
实施例5:重组菌株E.coli MG1655ΔmgsA::tErg9Δpta::shc M1-37-dxs及其对照菌株E.coli MG1655ΔmgsAΔpta M1-37-dxs的内源途径限速酶Idi的调控
具体操作方法同实施例4,其中有区别的为重组片段FKF::M1-46-idi的引物序列如下,模板为M1-46基因组:
Idi-M1-up:
TCACTTGGTTAATCATTTCACTCTTCAATTATCTATAATGATGAGTGATCAGGAACACTTAACGGCTGAC SEQ ID NO.42;
Idi-M1-down:
CCCGTGGGAACTCCCTGTGCATTCAATAAAATGACGTGTTCCGTTTGCATAGCTGTTTCCTGGTTTAAAC SEQ ID NO.43。
实施例6:重组菌株E.coli MG1655ΔmgsA::tErg9Δpta::shc M1-37-dxs M1-46-idi的耐受性分析
挑取实施例5构建的E.coli MG1655ΔmgsA::tErg9Δpta::shc M1-37-dxsM1-46-idi重组菌株进行多种化合物的耐受性测试。
另外为了作为对照,将实施例5构建的对照菌株E.coli MG1655ΔmgsAΔpta M1-37-dxs M1-46-idi按照同样方法进行测试。
具体操作步骤如下:
挑取经划线活化的单克隆,接种至15mL MOPS培养基中,37℃,200rpm,过夜培养至对数期(OD550~1.5-2.0);测定OD
广谱耐受性测试结果显示改造之后工程菌株的耐受性得到了显著提升。首先,工程菌株提高了对不利工业化条件的耐受性(图6),如在42℃高温条件下,比生长速率提升312%;在35g/L NaCl高渗透压条件下,比生长速率提升167%;在5Mm H2O2强氧化条件下,比生长速率提升60%。另外,提升了对纤维素水解液(图7)、一碳化合物(图8)、抑制性产物如有机酸和有机醇的耐受性(图9);纤维素水解液复配物(20g/L glucose+2g/L HMF+5g/LLevulinic acid+0.5g/L Vanilic acid)测定葡萄糖消耗情况发现,对照菌株未生长,而工程菌株最高OD值将近3.0,葡萄糖消耗达到10g/L(图10)。
实施例7:重组菌株E.coli MG1655ΔmgsA::tErg9Δpta::shc M1-37-dxs M1-46-idi的产物合成能力分析
通过本发明构建的一种鲁棒性提高的重组大肠杆菌,可以作为底盘细胞生产多种目标化合物,如长短链脂肪酸、三羟基丙酸、间苯三酚等不同化合物的产物展示,具体包括以下步骤:
(1)将不同产物的利用质粒分别转入对照菌和工程菌中;
(2)进一步地,在步骤(1)中,产物利用质粒对应为下表:
其中,对于pTrc99A-phlD载体上的phlD基因,对于pTrc99A-dha-aldH的dha-aldH基因,均委托GENEWIZ(苏州金唯智生物科技有限公司)优化并常规合成,二者分别为来自Pseudomonas protegens的phloroglucinol synthase基因,以及来自Klebsiellapneumoniae 21的glycerol dehydratase gene dhaB和来自Ralstonia eutropha的aldehyde dehydrogenase gene aldH基因,将基因克隆至载体pCDFDuet-1(Spectinomycin),制备mini-scale重组质粒DNA和含有该重组质粒的穿刺菌。
本实验室在得到重组质粒后,通过gilbson同源重组的方法将合成的基因构建到pTrc99A载体中。具体的:
通过设计含有与pTrc99A载体插入位点左右侧同源序列的引物,引物序列如下:
载体骨架引物:
99A-F:TACGTGATTGATAAATCCGC SEQ ID NO.44;
99A-R:GGTCTGTTTCCTGTGTGAAA SEQ ID NO.45;
PhlD基因:
phlD-up:TTTCACACAGGAAACAGACCATGAGCACCCTGTGCCTG SEQ ID NO.46;
PhlD-down:GCGGATTTATCAATCACGTATTACGCGGTCCATTCGCC SEQ ID NO.47;
dha-aldH基因:
3-HP-up:TTTCACACAGGAAACAGACCATGTATCAGGATCTGGCGCT SEQ ID NO.48;
3-HP-down:GCGGATTTATCAATCACGTAttaattcgcctgaccggc SEQ ID NO.49;经PCR分别克隆载体骨架及基因片段,纯化回收PCR产物后进行载体骨架与基因片段的无缝克隆,组装产物用于热激转化筛选单克隆进行测序验证得到目标质粒pTrc99A-phlD(载体及验证图谱见图11)与pTrc99A-dha-aldH(载体及验证图谱见图12)。
(3)对于长短链脂肪酸,以MOPS+2%(wt/v)Glucose培养基进行培养;
对于三羟基丙酸,以M9+2%(wt/v)Glycerol培养基进行培养;
对于间苯三酚,以M9+2%(wt/v)Glucose培养基进行培养;
其中:MOPS培养基配方为:
40X“M”
其中micronutrient solution成分为:
ZnCl
M9+2%(wt/v)Glycerol培养基配方为:
M9+2%(wt/v)Glucose培养基配方为:
5X M9 salts
(5)以0.1mM的IPTG为诱导剂,30℃培养72h,期间每24h取样。长短链脂肪酸发酵液上清液经萃取和衍生成酯之后进行GC-MS分析;三羟基丙酸和间苯三酚发酵液经离心过滤后经高效液相色谱分析。
最终工程菌株显著提高了对常见目标产物如短链脂肪酸(图13)、长链脂肪酸(图14)、三羟基丙酸(图15)、间苯三酚(图16)的合成能力。可见,本发明采用基因工程技术,将人工合成途径引入大肠杆菌中,结合内源途径的优化,能够有效提高重组工程菌的耐受性。
以上对本发明的具体实施例进行了描述。需要理解的是,本发明并不局限于上述特定实施方式,本领域技术人员可以在权利要求的范围内做出各种变化或修改,这并不影响本发明的实质内容。在不冲突的情况下,本申请的实施例和实施例中的特征可以任意相互组合。
- 一种谷氨酸棒杆菌重组菌及其构建方法
- 一种高产L-亮氨酸的谷氨酸棒杆菌重组菌及其构建方法
- 一种生物鲁棒性提高的大肠杆菌底盘细胞及其构建方法与应用
- 一种生物鲁棒性提高的大肠杆菌底盘细胞及其构建方法与应用