烃类化合物利用的组合方法
文献发布时间:2023-06-19 19:30:30
技术领域
本发明涉及石油化工领域,具体涉及一种烃类化合物利用的组合方法。
背景技术
随着来我国新能源技术的应用推广及汽油的升级,未来车用汽油需求量将逐渐呈下降趋势;另一方面,随着汽油质量的升级,对汽油中芳烃、烯烃及苯的含量要求将进一步降低。因此,实现劣质汽油的质量升级及化工转化是拓宽汽油利用的有效途径。
汽油组分主要来自催化裂化及催化重整单元,其中催化裂化单元汽油组分烯烃含量较高,而催化重整单元汽油调和组分含有较高的芳烃组分,同时也是汽油中苯的主要来源。目前实现汽油的质量升级主要通过选择性加氢或芳构化等反应降低烯烃及苯含量,中国专利CN101767035B公开了一种用于催化裂化汽油生产BTX芳烃的催化剂及其制备方法,该催化剂成分的重量百分比为VIII族贵金属0.05-2.0%,Zn含量0.2-5.0%,Sn含量0.2-5.0%,ZSM-5/ZSM-11共结晶分子筛含量5.0-80%,具有很好的芳构化活性和BTX选择性及耐硫抗烯烃性能,可用于催化裂化汽油或和直馏汽油或掺和焦化、裂化等汽油组分制芳烃。中国专利申请CN1923965A公开了一种催化裂化汽油制取乙烯、丙烯和芳烃的方法,将原料与催化剂一次接触转化为乙烯、丙烯和芳烃的混合物。
此外,通过苯与烯烃的烷基化反应也是降低汽油中苯含量的常用方法,如CN103562161B公布了一种通过烯烃烷基化苯降低汽油中苯含量的方法,汽油物料与包括C2-C5烯烃的烷基化及接触,生产低苯含量的流出物。CN103289730A公开了一种含烯烃低碳烃与苯烷基化生产高辛烷值汽油的方法,以裂解干气及苯为原料,通过多级冷激式固定床反应器发生烷基化反应。
发明内容
本发明的目的是为了克服现有技术存在的汽油组成中烯烃和苯含量较高的问题,提供一种烃类化合物利用的组合方法,该方法具有反应物耗低,产品结构优化的特点。
为了实现上述目的,本发明提供一种烃类化合物利用的组合方法,该方法包括以下步骤:
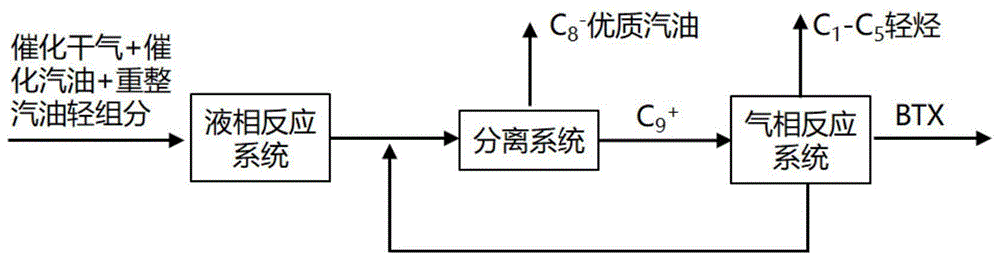
(1)将含碳七及其以下组分的烃类混合物进入液相反应器与第一催化剂接触,在至少部分液相条件下发生包括烯烃与芳烃烷基化反应,生成含低烯烃含量及低苯含量的产物A;
(2)将包含产物A中的C9+组分进入气相反应器与第二催化剂接触,发生包含非芳裂解、脱烷基及烷基转移反应,生成含有苯、甲苯、二甲苯的产物B。
优选地,所述第一催化剂的中强酸量占总酸量比例高于70mol%。
优选地,所述第一催化剂的制备方法包括:
a)将所述分子筛经碱性介质进行处理;
b)将步骤a)所得分子筛经酸性介质进行处理;
c)将步骤b)所得分子筛与粘结剂进行成型、焙烧,得到催化剂半成品;
d)将所述催化剂半成品进行铵交换和焙烧。
通过上述技术方案,能够降低汽油中烯烃和苯含量,并联产高纯度BTX芳烃,同时,该方法具有反应物耗低,产品结构优化的特点。优选情况下,采用优选地第一催化剂,能够更好控制烯烃与苯及芳烃间的烷基化反应程度,进一步降低汽油组分中烯烃和苯含量,同时抑制重组分的生成。
附图说明
图1是本发明实施例1的工艺流程图;
图2是本发明实施例2的工艺流程图。
具体实施方式
在本文中所披露的范围的端点和任何值都不限于该精确的范围或值,这些范围或值应当理解为包含接近这些范围或值的值。对于数值范围来说,各个范围的端点值之间、各个范围的端点值和单独的点值之间,以及单独的点值之间可以彼此组合而得到一个或多个新的数值范围,这些数值范围应被视为在本文中具体公开。
在本发明中,催化剂的中强酸量占总酸量的比例为NH
催化剂孔容根据N
在本发明中,对所述液相反应器和气相反应器的具体选择没有特别的限定,可以为本领域常规使用的各自能够在液相条件下进行反应的反应器或者在气相条件下进行反应的反应器。
本发明提供一种烃类化合物利用的组合方法,该方法包括以下步骤:
(1)将含碳七及其以下组分的烃类混合物进入液相反应器与第一催化剂接触,在至少部分液相条件下发生包括烯烃与芳烃烷基化反应,生成含低烯烃含量及低苯含量的产物A;
(2)将包含产物A中的C9+组分进入气相反应器与第二催化剂接触,发生包含非芳裂解、脱烷基及烷基转移反应,生成含有苯、甲苯、二甲苯的产物B。
本发明对所述碳七及其以下组分的来源选自范围较宽,优选地,所述碳七及其以下组分来自芳构化产物、催化重整产物、催化裂化产物、蒸汽裂解产物、重油加氢裂化产物及其任意组合。
根据本发明的一种具体实施方式,优选地,所述碳七及其以下组分含有烯烃和芳烃;进一步优选地,以碳七及其以下组分的总量为基准,所述烯烃含量为5-40重量%,所述芳烃的含量为10-50重量%。
具体地,所述碳七及其以下组分中还可以含有烷烃,所述烷烃的含量优选为20-60重量%。
本发明步骤(1)所述“在至少部分液相条件下”指的是原料中处于液相的烃类化合物的质量分数至少为1%。
根据本发明,优选地,所述第一催化剂含有具有MWW、FAU或BEA骨架结构的分子筛;进一步优选地,所述分子筛选自MCM-49、MCM-22、Y分子筛和Beta分子筛中的至少一种。
优选地,所述第一催化剂还含有粘结剂,所述第一催化剂中分子筛的含量不低于50重量%,优选为60-95重量%。
本发明对所述粘结剂的种类选择范围较宽,优选为氧化铝和/或氧化硅。
根据本发明,优选地,所述第一催化剂的中强酸量占总酸量比例在70mol%以上,优选在80mol%以上,更优选为80-90mol%。采用该种优选实施方式更有利于抑制强酸性位上发生的过度烷基化及烯烃高度聚合等副反应,减少重组分的生成。
根据本发明,优选地,所述第一催化剂的孔容在0.1cm
只要使用上述结构和组成的第一催化剂即有利于更进一步优化产品结构,对其制备方法选择范围较宽,为了更有利于优化产品结构,优选地,所述第一催化剂的制备方法包括:
a)将所述分子筛经碱性介质进行处理;
b)将步骤a)所得分子筛经酸性介质进行处理;
c)将步骤b)所得分子筛与粘结剂进行成型、焙烧,得到催化剂半成品;
d)将所述催化剂半成品进行铵交换和焙烧。
优选地,所述碱性介质优选自NaOH、KOH、NH
优选地,步骤a)的处理温度为40-100℃,处理时间为1-20小时,步骤a)所述处理过程中,液固质量比为3-10。
根据本发明的一种优选实施方式,该方法还包括对步骤a)所得分子筛进行洗涤,然后进行所述步骤b)。
优选地,所述酸性介质选自有机酸和/或无机酸的溶液(例如为有机酸和/或无机酸的水溶液)。在本发明中,所述酸性介质可以是单独的有机酸溶液或者是无机酸溶液,还可以是有机酸和无机酸的溶液,优选为有机酸以及任选地无机酸的溶液,更优选为有机酸和无机酸的溶液。当所述酸性介质为有机酸和无机酸的溶液时,对其中的有机酸和无机酸摩尔比选自范围较宽,优选为1:(0.1-10),例如为1:0.5。
优选地,所述有机酸选自柠檬酸、草酸、乙酸、酒石酸中的一种或多种。
优选地,所述无机酸选自盐酸、硝酸、硫酸中的一种或多种。
根据本发明的一种优选实施方式,所述酸性介质选自柠檬酸、草酸、乙酸、酒石酸、盐酸、硝酸和硫酸溶液中的一种或多种。进一步优选所述酸性介质的浓度为0.1-5mol/L;
优选地,步骤b)的处理温度为50-90℃,处理时间为1-20小时,步骤b)所述处理过程中,液固质量比为3-10。
根据本发明的一种优选实施方式,该方法还包括对步骤b)所得分子筛进行洗涤,然后进行烘干,再进行所述步骤c)。
本发明对步骤c)所述成型选自范围较宽,例如可以为捏合成型。
步骤c)所述焙烧可以在空气气氛下进行。优选地,步骤c)所述焙烧的条件包括:温度为300-600℃,时间为2-8h。
本发明对步骤d)所述铵交换的选择范围较宽,只要得到铵型催化剂即可。所述铵交换可以采用水溶性铵盐,包括但不限于硝酸铵。所述铵交换可以进行1次或多次,例如2-5次。对所述铵交换的具体条件,本发明不做具体限定,可以按照本领域常规操作进行,在此不再赘述。
本发明对步骤d)所述焙烧的条件选自范围较宽,优选地,步骤d)所述焙烧的条件包括:温度为300-600℃,时间为2-8h。
根据本发明的一种优选实施方式,所述液相反应器的反应条件包括:温度为100-400℃,压力为1-7MPa,进料质量空速为1-8h
优选地,步骤(1)中,产物A中烯烃含量较液相反应器进料中烯烃含量下降10%以上,优选下降20%以上。
优选地,步骤(1)中,产物A中苯含量较液相反应器进料中苯含量下降20%以上,优选下降30%以上。
采用上述优选实施方式,更有利于降低汽油中烯烃和苯含量。
可以知晓的是,在上述公开内容前提下,步骤(1)除了发生本发明所述的烯烃与芳烃烷基化反应,还不可避免的发生其他副反应,例如烯烃叠合反应。
根据本发明的一种优选实施方式,该方法还包括将产物A进行分离,得到C8以下组分以及所述C9+组分。优选地,该方法还包括从所述产物A中分离出的C8以下组分作为改质汽油调和组分。
根据本发明的一种优选实施方式,所述第二催化剂按照物流方向由上至下设置有至少一种加氢保护催化剂和至少一种芳烃转化催化剂。所述加氢保护催化剂和芳烃转化催化剂可以按照物流方向由上至下设置在一个催化剂床层内,也可以按照物流方向由上至下设置在两个催化剂床层内。优选地,所述加氢保护催化剂和芳烃转化催化剂按照物流方向由上至下设置在两个催化剂床层内。
采用上述优选实施方式,采用加氢保护催化剂更有利于延长芳烃转化催化剂的使用寿命。所述加氢保护催化剂的作用包括但不限于饱和烯烃。
根据本发明,优选地,所述加氢保护催化剂包括无酸性氧化物和/或弱酸性氧化物以及金属活性组分。所述无酸性氧化物和/或弱酸性氧化物作为载体。
优选地,无酸性氧化物或弱酸性氧化物选自Al
优选地,所述金属活性组分选自VIII族非贵金属组分、贵金属组分和VIB族金属中的至少一种,进一步优选为Pt、Ni和Pd中的至少一种。
本发明对加氢保护催化剂中无酸性氧化物和/或弱酸性氧化物以及金属活性组分的含量选择范围较宽,优选地,以所述加氢保护催化剂的总量为基准,无酸性氧化物和/或弱酸性氧化物的含量为80-99.9重量%,以氧化物计,金属活性组分的含量为0.1-20重量%。
根据本发明的一种优选实施方式,所述芳烃转化催化剂含有十元环孔道的硅铝分子筛以及十二元环孔道的硅铝分子筛和任选地加氢金属组分。采用该种优选实施方式更有利于获得高纯度苯、甲苯及二甲苯产物。
优选地,所述十元环孔道的硅铝分子筛为ZSM-5。
优选地,所述十二元环孔道的硅铝分子筛选自MOR、Beta和ZSM-12中的至少一种。
优选地,以芳烃转化催化剂中分子筛的总量为基准,十元环孔道的硅铝分子筛的含量为10-90重量%,十二元环孔道的硅铝分子筛的含量为10-90重量%。
优选地,所述芳烃转化催化剂中,以芳烃转化催化剂的总量为基准,分子筛的含量为50-99重量%,加氢金属组分的含量为0.01-10重量%。所述芳烃转化催化剂中还优选含有粘结剂,所述粘结剂的种类可以如上文所述。对粘结剂的含量选自范围较宽,以能够满足100%原则为准。
优选地,所述加氢金属组分选自Mo、Pt、Re和Bi中的至少一种。更优选地,所述加氢金属组分为Mo和Re。对Mo和Re的含量选自范围较宽,优选地,以芳烃转化催化剂的总量为基准,Mo的含量为0.5-5重量%,Re的含量为0.05-1重量%。
根据本发明的一种优选实施方式,所述加氢保护催化剂和芳烃转化催化剂在使用前,还包括分别对二者进行还原的步骤。所述还原的条件优选各自独立地包括:在氢气气氛下进行,温度为300-600℃,时间为1-5h。
本发明对所述加氢保护催化剂和芳烃转化催化剂的制备方法不作特别限定,均可以采用本领域常规手段制得,本发明在此不再赘述。
根据本发明,优选地,所述气相反应器的反应条件包括:温度为300-600℃,压力为1.5-5MPa,进料质量空速为1-20h
根据本发明的一种优选实施方式,该方法还包括:将产物B中含有的苯、甲苯、二甲苯作为产品采出,更优选产物苯、甲苯、二甲苯纯度大于99.9%。采用本发明提供的方法,能够得到高纯度的二甲苯。
优选地,该方法还包括将产物B中的C9+组分循环回气相反应器。该种优选实施方式可以将产物B中的C9+组分循环利用。
以下将通过实施例对本发明进行详细描述。
以下制备实施例1-10用于说明本发明所述第一催化剂的制备。
制备实施例1
取20克Beta沸石(硅铝分子比(Si/Al2)为50),加入100克浓度为0.5mol/L的NaOH溶液中,在搅拌下升温至90℃并恒温4小时,用去离子水洗涤过滤。所得滤饼加入100克浓度为0.3mol/L柠檬酸溶液中,在搅拌下升温至90℃并恒温4小时,用去离子水洗涤过滤、烘干,得到酸改性分子筛。取酸改性分子筛与7.7克氧化铝捏合成型,空气气氛下550℃焙烧3小时制得催化剂半成品;将催化剂半成品经过浓度为1mol/L的硝酸铵溶液于90℃下交换4小时,过滤,重复铵交换3次获得铵型催化剂,再在500℃下焙烧3小时制得第一催化剂,结果如表1所示。
制备实施例2
取20克MCM-22沸石(硅铝分子比(Si/Al2)为30),加入100克浓度为0.3mol/L的NaOH溶液中,在搅拌下升温至60℃并恒温4小时,用去离子水洗涤过滤。所得滤饼加入100克浓度为0.3mol/L柠檬酸溶液中,在搅拌下升温至90℃并恒温4小时,用去离子水洗涤过滤、烘干,得到酸改性分子筛。取酸改性分子筛与7.7克氧化铝捏合成型,空气气氛下550℃焙烧3小时制得催化剂半成品;将催化剂半成品经过浓度为1mol/L的硝酸铵溶液于90℃下交换4小时,过滤,重复铵交换3次获得铵型催化剂,再在500℃下焙烧3小时制得第一催化剂,结果如表1所示。
制备实施例3
取20克USY沸石(硅铝分子比(Si/Al2)为25),加入100克浓度为0.8mol/L的NaOH溶液中,在搅拌下升温至80℃并恒温4小时,用去离子水洗涤过滤。所得滤饼加入100克浓度为0.3mol/L柠檬酸溶液中,在搅拌下升温至90℃并恒温4小时,用去离子水洗涤过滤、烘干,得到酸改性分子筛。取酸改性分子筛与7.7克氧化铝捏合成型,空气气氛下550℃焙烧3小时制得催化剂半成品;将催化剂半成品经过浓度为1mol/L的硝酸铵溶液于90℃下交换4小时,过滤,重复铵交换3次获得铵型催化剂,再在500℃下焙烧3小时制得第一催化剂,结果如表1所示。
制备实施例4
按照实施例2的方法处理,不同的是,将柠檬酸溶液替换为0.3mol/L的草酸溶液,结果如表1所示。
制备实施例5
按照实施例2的方法处理,不同的是,将柠檬酸溶液替换为0.2mol/L柠檬酸+0.1mol/L草酸溶液混合液,结果如表1所示。
制备实施例6
按照实施例2的方法处理,不同的是,将柠檬酸溶液替换为0.2mol/L柠檬酸+0.1mol/L盐酸溶液混合液,结果如表1所示。
制备实施例7
按照实施例2的方法处理,不同的是,将柠檬酸溶液替换为0.2mol/L柠檬酸+0.1mol/L硫酸溶液混合液,结果如表1所示。
制备实施例8
取20克MCM-22沸石,加入100克浓度为0.3mol/L的NaOH溶液中,在搅拌下升温至60℃并恒温4小时,用去离子水洗涤过滤。取改性分子筛与7.7克氧化铝捏合成型,空气气氛下550℃焙烧3小时制得催化剂半成品;将催化剂半成品经过浓度为1mol/L的硝酸铵溶液于90℃下交换4小时,过滤,重复铵交换3次获得铵型催化剂,再在500℃下焙烧3小时制得第一催化剂。
制备实施例9
取20克MCM-22沸石,与7.7克氧化铝捏合成型,空气气氛下550℃焙烧3小时制得催化剂半成品;将催化剂半成品经过浓度为1mol/L的硝酸铵溶液于90℃下交换4小时,过滤,重复铵交换3次获得铵型催化剂,再在500℃下焙烧3小时制得第一催化剂。
制备实施例10
取20克MCM-22沸石,加入100克浓度为0.3mol/L柠檬酸溶液中,在搅拌下升温至90℃并恒温4小时,用去离子水洗涤过滤、烘干,得到酸改性分子筛。取改性分子筛与7.7克氧化铝捏合成型,空气气氛下550℃焙烧3小时制得催化剂半成品;将催化剂半成品经过浓度为1mol/L的硝酸铵溶液于90℃下交换4小时,过滤,重复铵交换3次获得铵型催化剂,再在500℃下焙烧3小时制得第一催化剂。
表1各制备实施例所得催化剂的组成
烃类混合物处理方法实施例
以下实施例用于说明本发明的组合方法,其中:
加氢保护催化剂:在氧化铝载体上负载Pt和Ni的催化剂,其中Pt含量0.1wt%,Ni含量(以金属元素计)8wt%,余量为氧化铝载体,投用前催化剂先经氢气于500℃下还原3小时;
芳烃转化催化剂:Re-Mo改性的MOR+ZSM-5沸石,其中Re含量(以金属元素计)0.3wt%,Mo含量(以金属元素计)2wt%,MOR含量为30重量%,ZSM-5含量为30%,余量为氧化铝,投用前催化剂先经氢气于400℃下还原3小时。
实施例1
现参考图1更全面地说明本发明。100吨/小时的催化裂化干气、催化裂化汽油及催化重整汽油轻组分混合物(馏程:<130℃)经脱硫脱氮预处理(得到的原料组成情况见表2),进入液相烷基化单元,发生包括烯烃与芳烃间的烷基化反应,催化剂为制备实施例1制得的第一催化剂A1,反应产物分离出C8及以下组分作为汽油组分,C9以上组分再进入气相反应单元,发生包括加氢裂解及脱烷基的反应(各反应单元的操作条件见表3),生成的产物进入分离单元进行顺序分离,获得C1-C5轻烃,高纯度BTX及C9+组分。其中C
实施例2
现参考图2更全面地说明本发明。100吨/小时的催化裂化汽油及催化重整汽油轻组分混合物(馏程:40-120℃)经脱硫脱氮预处理(得到的原料组成情况见表2),进入液相烷基化单元,发生包括烯烃与芳烃间的烷基化反应,催化剂为制备实施例1制得的第一催化剂A1,反应产物分离出C8及以下组分作为汽油组分,C9以上组分再进入气相反应单元,发生包括加氢裂解、非芳裂解及脱烷基的反应(各反应单元的操作条件见表3),生成的产物进入分离单元进行顺序分离,获得C1-C5轻烃,高纯度BTX及C9+组分。其中C
实施例3-11
按照实施例1的方法,不同的是,将第一催化剂分别替换为制备实施例2-10得到的第一催化剂,其余操作条件不变,结果见表4。
表2各实施例所用原料的组成
表3实施例1-2的操作条件
表4装置产品收率
/>
通过实施例结果可以看出,采用本发明技术方案具有降低汽油中苯含量及烯烃含量,同时抑制重组分生成的效果。
以上详细描述了本发明的优选实施方式,但是,本发明并不限于此。在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,包括各个技术特征以任何其它的合适方式进行组合,这些简单变型和组合同样应当视为本发明所公开的内容,均属于本发明的保护范围。