一种多糖中单糖组成的分析方法
文献发布时间:2023-06-19 18:27:32
技术领域
本发明属于多糖分析技术领域,特别涉及一种多糖中单糖组成的分析方法。
背景技术
糖是广泛存在于自然界的重要有机化合物,因其具有抗肿瘤、抗氧化、增强免疫等生物活性而受到广泛关注。但由于其结构特点、极性大、水溶性强,难以定性和定量分析。单糖组成是多糖初级结构研究的重要组成部分。研究单糖的组成对多糖的结构特征、理化性质及构效关系具有重要意义。
传统分析黄精多糖中单糖组成的方法主要采用PMP柱前衍生化试剂与多糖水解后的单糖作用,继而使用HPLC(高效液相色谱法)分离后测定,该方法简便快速、高效灵敏等特点,灵敏度优于液相色谱常用的示差检测器(RID)和蒸发光散射(ELSD)检测器。此方法具有简便实用、重复性好、灵敏度高、通量大等优点,其原理如下:
但该方法存在以下不足:1.仅适用于醛糖分析,对酮糖不适用;2.分析的灵敏度不高,可实现100μg左右必须含醛基基团的单糖的定性、定量分析。
还有一种做法是外利用离子色谱-脉冲安培检测法测定样品中的糖,该方法无需衍生即可测定样品中的糖,此方法灵敏度高,操作简单,分离度好,但离子色谱测定时对流动性有所限定,流动性需要是碱性溶液,呈电化学惰性,工作电压下背景电流小,但是像果糖与葡萄糖是差向异构体,在碱性条件下会发生酮式-烯醇式互变,导致对单糖进行柱前衍生化时,果糖会转变为葡萄糖,从而干扰这两种糖的定量与定性测定。
还有利用气相色谱(GC)法测定,该方法利用GC对糖腈衍生物的乙酰化反应十分敏感的特性,将乙酸酐生成糖腈衍生物应用于多糖分析,糖腈衍生物反应原理如下:
有学者利用该方法建立了气相色谱法测定脂肪含量小于5%食品中可溶性、不溶性部分单糖和总非淀粉多糖含量的方法。在宽口径毛细管柱上用火焰电离法检测乙腈糖醇衍生物。该方法准确,但只适用于醛糖的分析,像果糖类似的酮糖不行。
基于上述问题,提出一种应用广泛的多糖中单糖组分的分析方法是亟需解决的问题。
发明内容
针对上述问题,一方面本发明公开了一种多糖中单糖组成的分析方法,所述分析方法包括以下步骤:
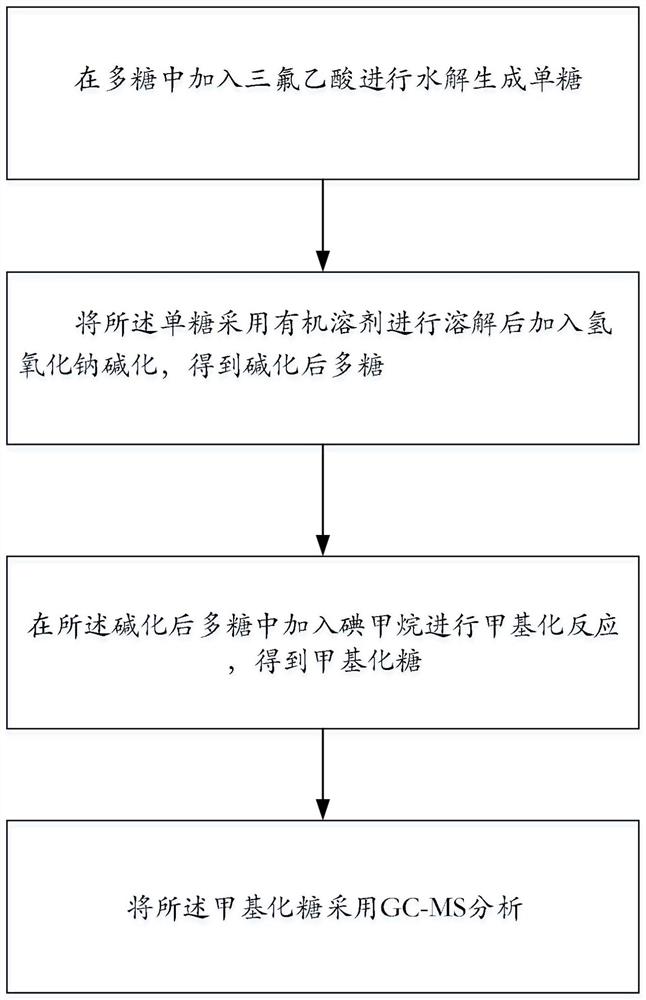
在多糖中加入三氟乙酸进行水解生成单糖;
将所述单糖采用有机溶剂进行溶解后加入氢氧化钠碱化,得到碱化后单糖;
在所述碱化后单糖中加入碘甲烷进行甲基化反应,得到甲基化糖;
将所述甲基化糖采用GC-MS分析。
进一步地,将所述甲基化糖采用GC-MS分析包括以下步骤:
采用GC-MS对多种单糖标准品进行分析,并绘制每种单糖的色谱峰面积相对于浓度的标准曲线;
根据单糖标准品的GC-MS结果对比所述甲基化糖的GC-MS结果确定多糖中的单糖组成和构型;
根据每种单糖的标准曲线计算多糖样品中的峰面积,确定多糖中各单糖组分的含量。
进一步地,所述多糖和三氟乙酸的投料比为每20mg多糖中加入0.1-5mL 2mol/L三氟乙酸。
进一步地,水解条件为:在密封条件下反应6小时,温度为100℃。
进一步地,所述有机溶剂为丙酮、N,N-二甲基甲酰胺、N-甲基吡咯烷酮、二甲基亚砜等或二甲基亚砜。
进一步地,所述氢氧化钠和多糖的质量比为1:1-6。
进一步地,所述碱化条件为:35℃碱化30分钟。
进一步地,所述碘甲烷的加入量为每20mg多糖中加入1mLCH
进一步地,甲基化反应条件为在密封条件下避光反应12小时,温度为35℃。
进一步地,所述GC-MS的测试条件为:
BR-17型弹性毛细管色谱柱(0.25μm×250μm×30m);载气:高纯氦气(纯度≥99.999%)、高纯氮气(纯度≥99.999%);柱流量:1.0mL/min;采用不分流进样,进样口温度为250℃;程序升温条件:初始温度为50℃,以25℃/min的速度升温至100℃,保持3min,再以4℃/min的速度升温至184℃;
EI离子源,电子能量70eV;离子源温度为220℃;传输线温度为280℃;四级杆温度40℃;全扫描模式,扫描范围m/z 40~600;质谱检索标准库:NIST 11.L。
本发明的有益效果:
本发明将多糖水解为单糖,二甲亚砜溶解,碱化,经完全甲基化经GC-MS分析,为多糖类组分中单糖组成测定提供了新方法,整个多糖处理过程条件温和、操作简单对环境友好;
本发明提出的分析方法可以分析多糖中十种单糖组成,该方法不仅可用于醛糖的分析,更能够对酮糖(果糖)的分析,具有明确的化学反应方程式;分析的灵敏度较高,可完成10μg以下单糖的定性、定量分析。
本发明的其它特征和优点将在随后的说明书中阐述,并且,部分地从说明书中变得显而易见,或者通过实施本发明而了解。本发明的目的和其他优点可通过在说明书、权利要求书以及附图中所指出的结构来实现和获得。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作一简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
图1示出了本发明实施例中一种多糖中单糖组成的分析方法流程图;
图2示出了本发明实施例中各单糖标准品的GC-MS图谱;
图3示出了本发明实施例中各单糖标准品甲基化后结构式;
图4示出了本发明实施例中黄精提取物中多糖单糖组成GC-MS图谱。
具体实施方式
为使本发明实施例的目的、技术方案和优点更加清楚,下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地说明,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
本发明通过将多糖水解,利用二甲基亚砜溶解后再碱化,后经CH
其反应原理如下:
即二甲基亚砜在碱性环境中形成二甲基亚砜磺酰阴离子,二甲基亚砜磺酰阴离子和糖反应生成糖羟基-醇钠,糖羟基-醇钠再经碘甲烷完全甲基化后生成甲醚甲基化糖。
如图1所示,本发明提出的分析方法按照以下步骤进行:
在多糖中加入三氟乙酸进行水解生成单糖;其中多糖和三氟乙酸的投料比为每20mg黄精多糖中加入0.1-5mL 2mol/L三氟乙酸;水解条件为:在密封条件下反应6小时,温度为100℃;
将所述单糖采用有机溶剂进行溶解后加入氢氧化钠碱化,得到碱化后单糖;有机溶剂为丙酮、N,N-二甲基甲酰胺、N-甲基吡咯烷酮或二甲基亚砜,优选为二甲基亚砜,其中,氢氧化钠和多糖的质量比为1:1-6,碱化条件为:35℃碱化30分钟;
在所述碱化后单糖中加入碘甲烷进行甲基化反应,得到甲基化糖;碘甲烷的加入量为每20mg黄精多糖加入1mLCH
将所述甲基化糖采用GC-MS分析。具体的是先采用GC-MS对多种单糖标准品进行分析,并绘制每种单糖的色谱峰面积相对于浓度的标准曲线;
根据单糖标准品的GC-MS结果对比所述甲基化糖的GC-MS结果确定多糖中的单糖组成和构型;
根据每种单糖的标准曲线计算多糖样品中的峰面积,确定多糖中各单糖组分的含量。
以下结合具体实施例对上述分析方法进行示例性说明,本发明实施例以黄精多糖为例进行说明。
实施例1测量各单糖标准品的GC-MS。
取葡萄糖(Glc)、甘露糖(Man)、果糖(Fru)、半乳糖(Gal)、鼠李糖(Rha)、核糖(Rib)、木糖(Xyl)、阿拉伯糖(Ara)、葡萄糖酸(GlcA)、半乳糖醛酸(GalA)10种不同的单糖标准品进行GC-MS测定,其具体过程如下:
1.1称取各单糖标准品各5mg,置于10mL Schlenk管内,加入1mL二甲基亚砜超声溶解10分钟,置于10mL Schlenk管内,加入30mgNaOH碱化30分钟,在搅拌下加入0.5mL CH
1.2GC-MS色谱条件:
BR-17型弹性毛细管色谱柱(0.25μm×250μm×30m);
载气:高纯氦气(纯度≥99.999%)、高纯氮气(纯度≥99.999%);柱流量:1.0mL/min;采用不分流进样,进样口温度为250℃;程序升温条件:初始温度为50℃,以25℃/min的速度升温至100℃,保持3min,再以4℃/min的速度升温至184℃。
质谱条件:EI离子源,电子能量70eV;离子源温度为220℃;传输线温度为280℃;四级杆温度40℃。全扫描模式,扫描范围m/z 40~600;质谱检索标准库:NIST 11.L。
各单糖标准品甲基化后的GC-MS图谱如图2所示,图2中1-12对应的图谱分别为10种单糖的混合标样、二苯甲酮、葡萄糖、甘露糖、核糖、半乳糖、鼠李糖、阿拉伯糖、果糖、葡萄糖醛酸、半乳糖醛酸和木糖的图谱;表1为各单糖标准品甲基化后出峰时间、化学式、分子量和离子碎片信息。
表1 10种单糖的甲基化质谱数据
各单糖标准品甲基化后的结构式如图3所示,其中葡萄糖(Glc)、甘露糖(Man)、半乳糖(Gal)、核糖(Rib)、木糖(Xyl)、阿拉伯糖(Ara)、葡萄糖酸(GlcA)均有α-D和β-D两种构型,鼠李糖(Rha)有α-L和β-L两种构型,葡萄糖酸(GlcA)有α-D-GlcA一种构型,果糖(Fru)为单一构型。
实施例2以黄精多糖为例对本发明提出的分析方法进行说明。
2.1先对黄精多糖进行处理:加入2mL 2mol/L三氟乙酸,密封100℃反应6小时,反应结束后,5000rpm离心10分钟,甲醇3次旋干除去残余三氟乙酸,酸水解产物加入二甲基亚砜超声溶解10分钟,置于10mL Schlenk管内,加入60mgNaOH碱化30分钟,在搅拌下加入1mLCH
2.2 GC-MS色谱条件:BR-17型弹性毛细管色谱柱(0.25μm×250μm×30m);载气:高纯氦气(纯度≥99.999%)、高纯氮气(纯度≥99.999%);柱流量:1.0mL/min;采用不分流进样,进样口温度为250℃;程序升温条件:初始温度为50℃,以25℃/min的速度升温至100℃,保持3min,再以4℃/min的速度升温至184℃。质谱条件:EI离子源,电子能量70eV;离子源温度为220℃;传输线温度为280℃;四级杆温度40℃。全扫描模式,扫描范围m/z 40~600;质谱检索标准库:NIST 11.L。
2.3对上述分析方法进行方法学考察
2.3.1线性关系考察:称取5.00mg 10个单糖标准品,按照实施例1中1.1的方法配制10个标准溶液。将样品加到GC-MS检测器中进行检测,测定8个不同浓度单糖的色谱峰面积,绘制10个单糖的色谱峰面积(Y)相对于质量浓度(X)的标准曲线。以质量浓度S/N=3和S/N=10为检出限和定量限,在相应范围内对10个单糖化合物进行测定。得到10种单糖化合物的线性范围、回归方程、相关系数、检出限和定量限,结果如表2所示。
表2 10种单糖标准曲线线性关系
2.3.2精密度测定:将甲基化单糖混合标准品连续进样8次测试,在此基础上计算10种单糖混合物的相对标准偏差(RSD)。
2.3.3稳定性测定:将甲基化单糖混合标准品于0,2,12h和24h各进样1次,计算10个单糖的RSD。
2.3.4重复性测定:精确称取20.00mg黄精多糖,按2.1的方法配制溶液。配制6种待测溶液,注射后计算预实验测定的10种单糖的RSD。
2.3.5加样回收率:对6个样品的多糖进行了准确的测定和测试。将10种单糖的3种不同质量浓度与标准溶液在线性范围内混合,进行加样回收率试验。每个样品重复注射3次,计算预实验测定的10种单糖的RSD。
精密度、稳定性、重复性的测定结果以及加样回收率的计算结果如表3所示。
表3 10种单糖精密度、重复性和稳定性
2.4采用GC-MS测定黄精提取物中多糖的含量,结果如图4所示,图4中1为葡萄糖;2为甘露糖;3为半乳糖;4为葡萄糖醛酸;5为半乳糖醛酸;6为二苯甲酮。采用表3中10种单糖的标准曲线计算黄精多糖中各单糖的峰面积,以确定多糖组分的准确含量,各多糖组分的含量及摩尔百分数见表4。黄精中多糖的主要成分为葡萄糖,葡萄糖的含量为12.19μg/mg,摩尔百分数为58.54mol%,其次为半乳糖,半乳糖的含量为3.82μg/mg,摩尔百分数为17.37mol%。
表4黄精多糖单糖组成测定
尽管参照前述实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的精神和范围。
- 一种酸性多糖中单糖组成的测定方法
- 一种分析肉桂多糖中单糖组成的测定方法
- 一种利用微波酸水解和阴离子交换色谱-脉冲安培法分析多糖中单糖组成的检测方法