一种检测弥漫大B细胞淋巴瘤的试剂盒及其应用
文献发布时间:2024-04-18 19:44:28
技术领域
本发明涉及生物检测技术领域,尤其涉及一种检测弥漫大B细胞淋巴瘤的试剂盒及其应用。
背景技术
弥漫性大B细胞淋巴瘤(DLBCL)是最常见的非霍奇金淋巴瘤(NHL)类型,包括一组具有不同的生物学特性、临床表现和对治疗反应的异质性疾病。R-CHOP(利妥昔单抗、环磷酰胺、多柔比星、长春新碱和泼尼松)仍然是治疗的主流,在近90%的局限期患者和高达60%的晚期患者中可以实现长期疾病控制。随着研究的不断推进,学术界对DLBCL的遗传状况和分子特征的理解取得了进展,确定了对化疗-免疫疗法效果不佳的高风险亚群,正在进行临床试验研究,新的治疗方法有可能改善高风险疾病患者的预后。2021年DLBCL的标准治疗方法仍然是R-CHOP的化疗-免疫疗法,尽管这种方式安全有效,但高达45%-50%的患者会复发。目前在了解DLBCL的基因组和转录组方面的努力已经确定了化疗-免疫疗法预后不佳的患者亚群,这些进展指导了在前期和复发情况下评估新型组合疗法的新型试验的设计,并为患者的选择提供信息。
DLBCL分子分型具有独特的预后特征、基因突变、信号通路异常及肿瘤微环境改变。2020年,Staudt团队创建了DLBCL分子分型的概率分类工具(LymphGen算法),通过对574例DLBCL活检样本的研究,将DLBCL分为7种遗传学亚型:MCD、N1、BN2、ST2、A53、EZB(MYC+)和EZB(MYC-)。MCD(以MYD88L265P和CD79B突变为典型特征)和BN2(以BCL6易位和NOTCH2突变为主要特征)依赖于BCR通路,EZB(以EZB突变和BCL2易位为主要特征)常发生表观遗传失调,ST2(以SGK1和TET2突变为主要特征)PI3K和JAK2通路上调,A53的特征是TP53突变和缺失,N1(NOTCH1突变)具有多种免疫细胞抑制表型。这些分子异质性表现决定了各分子分型对于R-CHOP的反应不同,为临床提供了潜在治疗靶点的依据,有望指导靶向治疗策略的优化。
近年来越来越多的靶向药物可应用于淋巴瘤的治疗,并且已有许多临床研究将这些靶向药物与标准R-CHOP联合,主要包括BTK抑制剂(伊布替尼、泽布替尼、奥布替尼)、免疫调节剂(来那度胺)、DNA去甲基化药物(地西他滨)、组蛋白去乙酰化酶抑制剂(西达本胺)等。PHOENIX研究作为Ⅲ期随机对照试验,比较了靶向BCR信号通路的BTK抑制剂伊布替尼联合R-CHOP及R-CHOP治疗初治non-GCB亚型DLBCL的疗效,未发现伊布替尼联合R-CHOP相比R-CHOP在整体患者中的生存获益。在年龄小于60岁的患者中,伊布替尼联合R-CHOP相较于安慰剂联合R-CHOP可增加疗效,由于BTK抑制剂的脱靶作用增大了不良反应的发生率,较多老年患者不能耐受目标剂量,影响疗效。REMARC研究显示在60~80岁经一线治疗有效的初治DLBCL患者中lenalidomide维持治疗可提高患者的无进展生存,且获益与细胞起源无关。然而首个在初治ABC亚型DLBCL患者中比较来那度胺联合R-CHOP和R-CHOP一线治疗疗效的全球多中心Ⅲ期随机对照临床研究(ROBUST研究),未发现来那度胺联合R-CHOP相比R-CHOP在整体患者中的生存获益。这些结果说明初诊时仅根据细胞起源进行分型来选择同一种靶向治疗,是远远不够的。然而亚组分析获得一些有意义的结果,伊布替尼联合R-CHOP对MCD和N1亚型的患者疗效显著。且体内及体外研究均显示,伊布替尼可显著抑制BN2亚型的DLBCL肿瘤生长。有研究表明,来那度胺能够降低Treg细胞,激活CD8+的T淋巴细胞,影响T辅助细胞的亚型。如REMARC研究中,来那度胺组观察到的疗效,是由于上调干扰素基因直接发挥抗肿瘤作用。
DNA测序的出现为DLBCL的基因组和转录组特征提供了新的见解,高通量测序(NGS)已成为完整描述非霍奇金淋巴瘤(NHL)基因变异谱的有用工具。近年来,对NHL分子谱系的研究有了很大进展。各个小组都试图根据基因特征或通过将特征与临床和分析数据相结合的方式来建立预后评分和基因风险群。然而,在很多研究中心应用这些技术的手段仍然具有局限性,从研究层面落实到临床实践中还需要更多验证。因此,虽然NGS淋巴板在临床实践中有所实施,但至今还没有标准的方法,而且各实验室的基因选择、测序平台、读数深度和变体分析等特征也会有所不同。
现有技术中已经发现一些信号通路和基因参与了DLBCL的预后分型,例如中国专利CN114277134A公开了一种弥漫大B细胞淋巴瘤的分型模型、分型方法和试剂盒,通过检测18个基因的突变信息可以完成弥漫大B细胞淋巴瘤的分型,通过一致性的比较发现,和应用NEJM50基因模型分出的结果高度一致,然而18基因在实际应用中对高危型的概括性很强,以致于高危型中的相对低危型无法精准识别,从而出现过度治疗。因此,分型更为细致、从而用药治疗性更有针对性的基因分型方法更具有临床应用价值。
发明内容
本发明的第一个方面提供了一种检测弥漫大B细胞淋巴瘤的试剂盒,所述试剂盒中含有用于捕获目的基因的探针,所述目的基因包括:BCL2基因、BCL6基因、MYC基因、ARID1A基因、B2M基因、BTG1基因、BTG2基因、CCND3基因、CD70基因、CD79B基因、CIITA基因、CREBBP基因、DDX3X基因、DTX1基因、DUSP2基因、EP300基因、EZH2基因、FAS基因、GNA13基因、IRF4基因、IRF8基因、KMT2D基因、MPEG1基因、MYD88基因、NOTCH1基因、NOTCH2基因、PIM1基因、PRDM1基因、SGK1基因、SOCS1基因、STAT3基因、STAT6基因、TBL1XR1基因、TET2基因、TNFAIP3基因、TNFRSF14基因、TP53基因、ZFP36L1基因。
现有技术中对于弥漫大B细胞淋巴瘤可以做到18基因的分型,然而在实际应用中,18基因对高危型的概括性很强,以致于高危型中的相对低危型无法精准识别,从而出现过度治疗。本发明中对弥漫大B细胞淋巴瘤进行了38基因分型,相对于现有技术划分更为细致,从生物曲线图中可知,ST-2型处于曲线图的最上端,表明愈后效果更好,经临床证明BN2型无需过度治疗。
在一些实施方式中,所述探针采用以下方法设计得到:
S1.根据数据库提取目的基因的外显子坐标,获得外显子区域的集合;
S2.截取一个外显子区域的1~2Unit作为第一个探针单元,截取外显子区域的(n-1)Unit~(n+1)Unit bp作为第n个探针单元,依次截取,Unit=60bp,获得该外显子区域的探针;
S3.对探针的核苷酸序列进行评估,评估的参数包括:Tm值、GC含量、Hairpin自由能、序列复杂度和基因组内同源性;
S4.对于评分低于阈值的探针进行调整,调整为调整评分低于阈值的探针的序列截取时的Unit值为60±10bp,重复S2、S3步骤优化探针的序列;
S5.对外显子区域的集合按照S1~S5方法获取探针,获得探针的集合。
在一些实施方式中,所述n的取值范围为1~50。
在一些实施方式中,S4中所述的调整包括调整探针长度、平移探针位置等本领域常用方式中的任意一种。
在一些实施方式中,所述探针为双链DNA探针。
现有技术中的探针一般是双链RNA探针,本发明中采用的是双链DNA探针,经试验分析和临床验证发现,双链DNA探针针对GC含量高的区域其捕获效率更高。
进一步地,所述双链DNA探针的信息如下表:
在一些实施方式中,所述检测试剂盒包括建库试剂盒和杂交捕获试剂盒。
在一些实施方式中,所述建库试剂盒包括片段化酶和末端修复酶、片段化和末端修复缓冲液、连接酶、连接缓冲液、接头混合液、前PCR扩增反应液、I5端接头引物液、I7端接头引物液、杂交缓冲液、封闭液、探针保护液、探针、后PCR扩增反应液、后PCR扩增引物混合液。
本发明的第二个方面提供了一种弥漫大B细胞淋巴瘤相关基因的建库方法,包括以下步骤:
S1.基因组DNA片段化,末端修复,5’端磷酸化,3‘端加“A”尾;
S2.接头连接;
S3.连接后纯化;
S4.前PCR扩增预文库;
S5.前PCR扩增后磁珠纯化;
S6.混合预文库、杂交液、封闭液、探针保护液和探针,进行杂交;
S7.杂交后洗脱纯化;
S8.后PCR扩增终文库;
S9.后PCR扩增后磁珠纯化;
S10.上机测序。
本发明的第三个方面提供了一种所述试剂盒在制备弥漫大B细胞淋巴瘤检测产品中的应用。
本发明的第四个方面提供了一种建立弥漫大B细胞淋巴瘤的分型模型的方法,包括以下步骤:
S1、获取最低样本量的弥漫大B细胞淋巴瘤样本;
S2、检测所述弥漫大B细胞淋巴瘤样本中目的基因的突变信息;
S3、将所述突变信息采用PAM算法聚类,建立分型模型,获得分出包含MCD型、N1型、EZB MYC+型、EZB MYC-型、BN2型、ST2型、TP53突变型、Others型的分型模型;所述PAM算法聚类中初始中心点的参数如下:
针对MCD型:BCL2基因的权重为0.05±0.005、BCL6基因的权重为0.2±0.02、ARID1A基因的权重为0.05±0.005、B2M基因的权重为0.15±0.015、BTG1基因的权重为0.5±0.05、BTG2基因的权重为0.2±0.02、CCND3基因的权重为0.05±0.005、CD70基因的权重为0、CD79B基因的权重为0.2±0.02、CIITA基因的权重为0.05±0.005、CREBBP基因的权重为0.1±0.01、DDX3X基因的权重为0、DTX1基因的权重为0.3±0.03、DUSP2基因的权重为0.3±0.03、EP300基因的权重为0.05±0.005、EZH2基因的权重为0.05±0.005、FAS基因的权重为0.05±0.005、GNA13基因的权重为0、IRF4基因的权重为0.15±0.015、IRF8基因的权重为0、KMT2D基因的权重为0.35±0.035、MPEG1基因的权重为0.5±0.05、MYD88基因的权重为0.8±0.08、NOTCH1基因的权重为0、NOTCH2基因的权重为0.05±0.005、PIM1基因的权重为0.85±0.085、PRDM1基因的权重为0.2±0.02、SGK1基因的权重为0.05±0.005、SOCS1基因的权重为0、STAT3基因的权重为0.05±0.005、STAT6基因的权重为0、TBL1XR1基因的权重为0.35±0.035、TET2基因的权重为0、TNFAIP3基因的权重为0.05±0.005、TNFRSF14基因的权重为0、ZFP36L1基因的权重为0.1;
针对BN2型:BCL2基因的权重为0.024±0.0024、BCL6基因的权重为0.643±0.0643、ARID1A基因的权重为0.071±0.0071、B2M基因的权重为0.143±0.0143、BTG1基因的权重为0.143±0.0143、BTG2基因的权重为0.405±0.0405、CCND3基因的权重为0.119±0.0119、CD70基因的权重为0.548±0.0548、CD79B基因的权重为0.048±0.0048、CIITA基因的权重为0.024±0.0024、CREBBP基因的权重为0.071±0.0071、DDX3X基因的权重为0.095±0.0095、DTX1基因的权重为0.405±0.0405、DUSP2基因的权重为0.095±0.0095、EP300基因的权重为0.095±0.0095、EZH2基因的权重为0.024±0.0024、FAS基因的权重为0.143±0.0143、GNA13基因的权重为0.048±0.0048、IRF4基因的权重为0.048±0.0048、IRF8基因的权重为0.024±0.0024、KMT2D基因的权重为0.167±0.0167、MPEG1基因的权重为0.167±0.0167、MYD88基因的权重为0.143±0.0143、NOTCH1基因的权重为0、NOTCH2基因的权重为0.31±0.031、PIM1基因的权重为0.143±0.0143、PRDM1基因的权重为0.143±0.0143、SGK1基因的权重为0.048±0.0048、SOCS1基因的权重为0.071±0.0071、STAT3基因的权重为0.095±0.0095、STAT6基因的权重为0.024±0.0024、TBL1XR1基因的权重为0、TET2基因的权重为0.119±0.0119、TNFAIP3基因的权重为0.286±0.0286、TNFRSF14基因的权重为0.119±0.0119、ZFP36L1基因的权重为0.095;
针对N1型:BCL2基因的权重为0、BCL6基因的权重为0、ARID1A基因的权重为0.083±0.0083、B2M基因的权重为0.083±0.0083、BTG1基因的权重为0.25±0.025、BTG2基因的权重为0、CCND3基因的权重为0.083±0.0083、CD70基因的权重为0、CD79B基因的权重为0、CIITA基因的权重为0.167±0.0167、CREBBP基因的权重为0、DDX3X基因的权重为0、DTX1基因的权重为0、DUSP2基因的权重为0、EP300基因的权重为0.083±0.0083、EZH2基因的权重为0、FAS基因的权重为0、GNA13基因的权重为0.083±0.0083、IRF4基因的权重为0.083±0.0083、IRF8基因的权重为0.083±0.0083、KMT2D基因的权重为0.417±0.0417、MPEG1基因的权重为0.083±0.0083、MYD88基因的权重为0.417±0.0417、NOTCH1基因的权重为1±0.1、NOTCH2基因的权重为0、PIM1基因的权重为0.417±0.0417、PRDM1基因的权重为0.083±0.0083、SGK1基因的权重为0.083±0.0083、SOCS1基因的权重为0.083±0.0083、STAT3基因的权重为0.167±0.0167、STAT6基因的权重为0、TBL1XR1基因的权重为0.167±0.0167、TET2基因的权重为0.25±0.025、TNFAIP3基因的权重为0、TNFRSF14基因的权重为0、ZFP36L1基因的权重为0;
针对EZB型:BCL2基因的权重为0.737±0.0737、BCL6基因的权重为0.105±0.0105、ARID1A基因的权重为0.211±0.0211、B2M基因的权重为0.211±0.0211、BTG1基因的权重为0.105±0.0105、BTG2基因的权重为0.105±0.0105、CCND3基因的权重为0、CD70基因的权重为0.105±0.0105、CD79B基因的权重为0、CIITA基因的权重为0.158±0.0158、CREBBP基因的权重为0.211±0.0211、DDX3X基因的权重为0、DTX1基因的权重为0.053±0.0053、DUSP2基因的权重为0.053±0.0053、EP300基因的权重为0.158±0.0158、EZH2基因的权重为0.211±0.0211、FAS基因的权重为0.211±0.0211、GNA13基因的权重为0.053±0.0053、IRF4基因的权重为0.053±0.0053、IRF8基因的权重为0、KMT2D基因的权重为0.368±0.0368、MPEG1基因的权重为0.053±0.0053、MYD88基因的权重为0、NOTCH1基因的权重为0、NOTCH2基因的权重为0、PIM1基因的权重为0.105±0.0105、PRDM1基因的权重为0、SGK1基因的权重为0.105±0.0105、SOCS1基因的权重为0.421±0.0421、STAT3基因的权重为0.158±0.0158、STAT6基因的权重为0.105±0.0105、TBL1XR1基因的权重为0.105±0.0105、TET2基因的权重为0.053±0.0053、TNFAIP3基因的权重为0.211±0.0211、TNFRSF14基因的权重为0.684±0.0684、ZFP36L1基因的权重为0.053;
针对ST2型:BCL2基因的权重为0.071±0.0071、BCL6基因的权重为0.143±0.0143、ARID1A基因的权重为0、B2M基因的权重为0.071±0.0071、BTG1基因的权重为0.214±0.0214、BTG2基因的权重为0.286±0.0286、CCND3基因的权重为0.071±0.0071、CD70基因的权重为0、CD79B基因的权重为0.071±0.0071、CIITA基因的权重为0.071±0.0071、CREBBP基因的权重为0、DDX3X基因的权重为0.429±0.0429、DTX1基因的权重为0.214±0.0214、DUSP2基因的权重为0.214±0.0214、EP300基因的权重为0.143±0.0143、EZH2基因的权重为0、FAS基因的权重为0.143±0.0143、GNA13基因的权重为0.071±0.0071、IRF4基因的权重为0.286±0.0286、IRF8基因的权重为0.071±0.0071、KMT2D基因的权重为0.071±0.0071、MPEG1基因的权重为0.143±0.0143、MYD88基因的权重为0、NOTCH1基因的权重为0、NOTCH2基因的权重为0、PIM1基因的权重为0.429±0.0429、PRDM1基因的权重为0、SGK1基因的权重为0.5±0.05、SOCS1基因的权重为0.429±0.0429、STAT3基因的权重为0.143±0.0143、STAT6基因的权重为0、TBL1XR1基因的权重为0、TET2基因的权重为0.5±0.05、TNFAIP3基因的权重为0、TNFRSF14基因的权重为0.214±0.0214、ZFP36L1基因的权重为0.286;
针对Others型:BCL2基因的权重为0.048±0.0048、BCL6基因的权重为0.11±0.011、ARID1A基因的权重为0.057±0.0057、B2M基因的权重为0.062±0.0062、BTG1基因的权重为0.048±0.0048、BTG2基因的权重为0.071±0.0071、CCND3基因的权重为0.048±0.0048、CD70基因的权重为0.024±0.0024、CD79B基因的权重为0.067±0.0067、CIITA基因的权重为0.048±0.0048、CREBBP基因的权重为0.057±0.0057、DDX3X基因的权重为0.124±0.0124、DTX1基因的权重为0.057±0.0057、DUSP2基因的权重为0.038±0.0038、EP300基因的权重为0.057±0.0057、EZH2基因的权重为0.014±0.0014、FAS基因的权重为0.038±0.0038、GNA13基因的权重为0.048±0.0048、IRF4基因的权重为0.052±0.0052、IRF8基因的权重为0.01±0.001、KMT2D基因的权重为0.157±0.0157、MPEG1基因的权重为0.043±0.0043、MYD88基因的权重为0.129±0.0129、NOTCH1基因的权重为0、NOTCH2基因的权重为0.029±0.0029、PIM1基因的权重为0.157±0.0157、PRDM1基因的权重为0.067±0.0067、SGK1基因的权重为0.033±0.0033、SOCS1基因的权重为0.052±0.0052、STAT3基因的权重为0.024±0.0024、STAT6基因的权重为0.029±0.0029、TBL1XR1基因的权重为0.038±0.0038、TET2基因的权重为0.1±0.01、TNFAIP3基因的权重为0.057±0.0057、TNFRSF14基因的权重为0.019、ZFP36L1基因的权重为0.038。
进一步地,所述PAM算法聚类中初始中心点的参数如下:针对MCD型:BCL2基因的权重为0.05、BCL6基因的权重为0.2、ARID1A基因的权重为0.05、B2M基因的权重为0.15、BTG1基因的权重为0.5、BTG2基因的权重为0.2、CCND3基因的权重为0.05、CD70基因的权重为0、CD79B基因的权重为0.2、CIITA基因的权重为0.05、CREBBP基因的权重为0.1、DDX3X基因的权重为0、DTX1基因的权重为0.3、DUSP2基因的权重为0.3、EP300基因的权重为0.05、EZH2基因的权重为0.05、FAS基因的权重为0.05、GNA13基因的权重为0、IRF4基因的权重为0.15、IRF8基因的权重为0、KMT2D基因的权重为0.35、MPEG1基因的权重为0.5、MYD88基因的权重为0.8、NOTCH1基因的权重为0、NOTCH2基因的权重为0.05、PIM1基因的权重为0.85、PRDM1基因的权重为0.2、SGK1基因的权重为0.05、SOCS1基因的权重为0、STAT3基因的权重为0.05、STAT6基因的权重为0、TBL1XR1基因的权重为0.35、TET2基因的权重为0、TNFAIP3基因的权重为0.05、TNFRSF14基因的权重为0、ZFP36L1基因的权重为0.1;
针对BN2型:BCL2基因的权重为0.024、BCL6基因的权重为0.643、ARID1A基因的权重为0.071、B2M基因的权重为0.143、BTG1基因的权重为0.143、BTG2基因的权重为0.405、CCND3基因的权重为0.119、CD70基因的权重为0.548、CD79B基因的权重为0.048、CIITA基因的权重为0.024、CREBBP基因的权重为0.071、DDX3X基因的权重为0.095、DTX1基因的权重为0.405、DUSP2基因的权重为0.095、EP300基因的权重为0.095、EZH2基因的权重为0.024、FAS基因的权重为0.143、GNA13基因的权重为0.048、IRF4基因的权重为0.048、IRF8基因的权重为0.024、KMT2D基因的权重为0.167、MPEG1基因的权重为0.167、MYD88基因的权重为0.143、NOTCH1基因的权重为0、NOTCH2基因的权重为0.31、PIM1基因的权重为0.143、PRDM1基因的权重为0.143、SGK1基因的权重为0.048、SOCS1基因的权重为0.071、STAT3基因的权重为0.095、STAT6基因的权重为0.024、TBL1XR1基因的权重为0、TET2基因的权重为0.119、TNFAIP3基因的权重为0.286、TNFRSF14基因的权重为0.119、ZFP36L1基因的权重为0.095;
针对N1型:BCL2基因的权重为0、BCL6基因的权重为0、ARID1A基因的权重为0.083、B2M基因的权重为0.083、BTG1基因的权重为0.25、BTG2基因的权重为0、CCND3基因的权重为0.083、CD70基因的权重为0、CD79B基因的权重为0、CIITA基因的权重为0.167、CREBBP基因的权重为0、DDX3X基因的权重为0、DTX1基因的权重为0、DUSP2基因的权重为0、EP300基因的权重为0.083、EZH2基因的权重为0、FAS基因的权重为0、GNA13基因的权重为0.083、IRF4基因的权重为0.083、IRF8基因的权重为0.083、KMT2D基因的权重为0.417、MPEG1基因的权重为0.083、MYD88基因的权重为0.417、NOTCH1基因的权重为1、NOTCH2基因的权重为0、PIM1基因的权重为0.417、PRDM1基因的权重为0.083、SGK1基因的权重为0.083、SOCS1基因的权重为0.083、STAT3基因的权重为0.167、STAT6基因的权重为0、TBL1XR1基因的权重为0.167、TET2基因的权重为0.25、TNFAIP3基因的权重为0、TNFRSF14基因的权重为0、ZFP36L1基因的权重为0;
针对EZB型:BCL2基因的权重为0.737、BCL6基因的权重为0.105、ARID1A基因的权重为0.211、B2M基因的权重为0.211、BTG1基因的权重为0.105、BTG2基因的权重为0.105、CCND3基因的权重为0、CD70基因的权重为0.105、CD79B基因的权重为0、CIITA基因的权重为0.158、CREBBP基因的权重为0.211、DDX3X基因的权重为0、DTX1基因的权重为0.053、DUSP2基因的权重为0.053、EP300基因的权重为0.158、EZH2基因的权重为0.211、FAS基因的权重为0.211、GNA13基因的权重为0.053、IRF4基因的权重为0.053、IRF8基因的权重为0、KMT2D基因的权重为0.368、MPEG1基因的权重为0.053、MYD88基因的权重为0、NOTCH1基因的权重为0、NOTCH2基因的权重为0、PIM1基因的权重为0.105、PRDM1基因的权重为0、SGK1基因的权重为0.105、SOCS1基因的权重为0.421、STAT3基因的权重为0.158、STAT6基因的权重为0.105、TBL1XR1基因的权重为0.105、TET2基因的权重为0.053、TNFAIP3基因的权重为0.211、TNFRSF14基因的权重为0.684、ZFP36L1基因的权重为0.053;
针对ST2型:BCL2基因的权重为0.071、BCL6基因的权重为0.143、ARID1A基因的权重为0、B2M基因的权重为0.071、BTG1基因的权重为0.214、BTG2基因的权重为0.286、CCND3基因的权重为0.071、CD70基因的权重为0、CD79B基因的权重为0.071、CIITA基因的权重为0.071、CREBBP基因的权重为0、DDX3X基因的权重为0.429、DTX1基因的权重为0.214、DUSP2基因的权重为0.214、EP300基因的权重为0.143、EZH2基因的权重为0、FAS基因的权重为0.143、GNA13基因的权重为0.071、IRF4基因的权重为0.286、IRF8基因的权重为0.071、KMT2D基因的权重为0.071、MPEG1基因的权重为0.143、MYD88基因的权重为0、NOTCH1基因的权重为0、NOTCH2基因的权重为0、PIM1基因的权重为0.429、PRDM1基因的权重为0、SGK1基因的权重为0.5、SOCS1基因的权重为0.429、STAT3基因的权重为0.143、STAT6基因的权重为0、TBL1XR1基因的权重为0、TET2基因的权重为0.5、TNFAIP3基因的权重为0、TNFRSF14基因的权重为0.214、ZFP36L1基因的权重为0.286;
针对Others型:BCL2基因的权重为0.048、BCL6基因的权重为0.11、ARID1A基因的权重为0.057、B2M基因的权重为0.062、BTG1基因的权重为0.048、BTG2基因的权重为0.071、CCND3基因的权重为0.048、CD70基因的权重为0.024、CD79B基因的权重为0.067、CIITA基因的权重为0.048、CREBBP基因的权重为0.057、DDX3X基因的权重为0.124、DTX1基因的权重为0.057、DUSP2基因的权重为0.038、EP300基因的权重为0.057、EZH2基因的权重为0.014、FAS基因的权重为0.038、GNA13基因的权重为0.048、IRF4基因的权重为0.052、IRF8基因的权重为0.01、KMT2D基因的权重为0.157、MPEG1基因的权重为0.043、MYD88基因的权重为0.129、NOTCH1基因的权重为0、NOTCH2基因的权重为0.029、PIM1基因的权重为0.157、PRDM1基因的权重为0.067、SGK1基因的权重为0.033、SOCS1基因的权重为0.052、STAT3基因的权重为0.024、STAT6基因的权重为0.029、TBL1XR1基因的权重为0.038、TET2基因的权重为0.1、TNFAIP3基因的权重为0.057、TNFRSF14基因的权重为0.019、ZFP36L1基因的权重为0.038。
进一步地,所述弥漫大B细胞淋巴瘤样本包括但不限于新鲜组织、FFPE、骨髓。
本发明的第五个方面提供了一种弥漫大B细胞淋巴瘤的分型模型。
本发明的第六个方面提供了一种用于弥漫大B细胞淋巴瘤分型的装置,包括:
样本获取模块,用于获得最低样本量的弥漫大B细胞淋巴瘤样本;
突变信息获取模块,用于获取所述弥漫大B细胞淋巴瘤样本中目的基因的突变信息;
分型模型建立模块,用于采用PAM算法聚类。
与现有技术相比,本发明具有以下有益效果:
1.本发明中对弥漫大B细胞淋巴瘤进行了38基因分型,相对于现有技术划分更为细致,从生物曲线图中可知,ST-2型处于曲线图的最上端,表明愈后效果更好,经临床证明BN2型无需过度治疗。
2.本发明的临床试验对比了R-CHOP-X和RCHOP方案治疗初治、非低危(IPI≥2分或IPI=1分伴大包块)弥漫大B细胞淋巴瘤的临床疗效和安全性,可以为淋巴瘤个体化治疗奠定循证医学的基础。
3.利用本发明的试剂盒检测弥漫大B细胞淋巴瘤的临床治疗结果可以提供预后判断和靶向药物预测作用,即实现一次检测同时进行分型诊断、预后判断和靶向药物预测,具有更全面的临床指导意义。
4.本发明将所述38基因的分型和检测方法制备成试剂盒,具有文库构建周期短、覆盖率高、比对率高、均一性好、操作简单等优点,更适用于临床应用。
5.本发明从使用效果和经济效益的角度均具有优异的推广前景。
附图说明
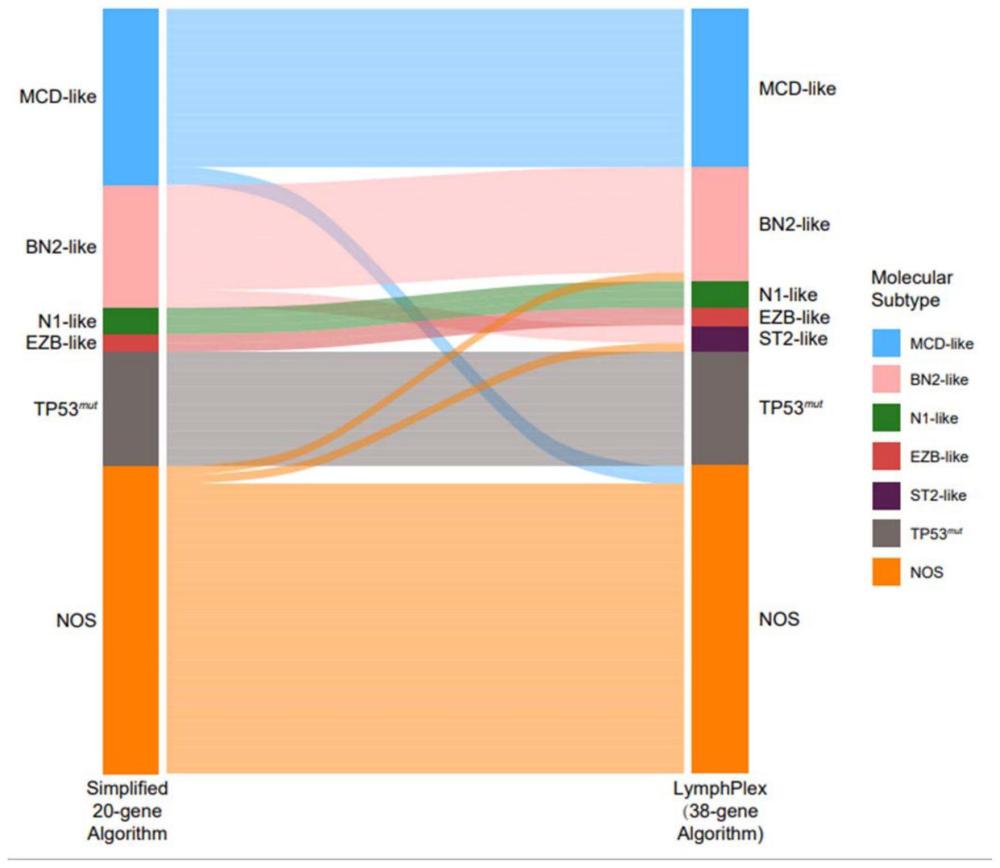
图1为本发明的分型与其他分型的比较;图中左侧为基于20基因的分型结果,右侧为本发明中的38基因分型结果。其中部分在左侧MCD型的患者在本发明中被分到了NOS型,部分在左侧NOS型的患者在本发明中被分到了ST2型和BN2型。
图2为基于现有技术划分的不同亚型患者PFS无进展生存期图。
图3为基于本发明的38基因分型的不同亚型患者PFS无进展生存期图。
图4为基于现有技术划分的不同亚型患者OS生存曲线图。
图5为基于本发明的38基因分型的不同亚型患者OS生存曲线图;
图6为临床试验的技术路线图。
具体实施方式
实施例1
一种检测弥漫大B细胞淋巴瘤的试剂盒,所述试剂盒中含有用于捕获目的基因的探针,所述目的基因包括:BCL2基因、BCL6基因、MYC基因、ARID1A基因、B2M基因、BTG1基因、BTG2基因、CCND3基因、CD70基因、CD79B基因、CIITA基因、CREBBP基因、DDX3X基因、DTX1基因、DUSP2基因、EP300基因、EZH2基因、FAS基因、GNA13基因、IRF4基因、IRF8基因、KMT2D基因、MPEG1基因、MYD88基因、NOTCH1基因、NOTCH2基因、PIM1基因、PRDM1基因、SGK1基因、SOCS1基因、STAT3基因、STAT6基因、TBL1XR1基因、TET2基因、TNFAIP3基因、TNFRSF14基因、TP53基因、ZFP36L1基因。
所述探针为双链DNA探针,采用以下方法设计得到:
S1.根据数据库提取目的基因的外显子坐标,获得外显子区域的集合;
S2.截取一个外显子区域的1~2Unit bp作为第一个探针单元,截取外显子区域的(n-1)Unit~(n+1)Unit bp作为第n个探针单元,依次截取,Unit=60bp,获得该外显子区域的探针;
S3.对探针的核苷酸序列进行评估,评估的参数包括:Tm值、GC含量、Hairpin自由能、序列复杂度和基因组内同源性;
S4.对于评分低于阈值的探针进行调整,调整为调整评分低于阈值的探针的序列截取时的Unit值为60±10bp,重复S2、S3步骤优化探针的序列;
S5.对外显子区域的集合按照S1~S5方法获取探针,获得探针的集合。
测试例1
本测试例提供了实施例1所制备的试剂盒在临床对弥漫大B细胞淋巴瘤的治疗初治、非低危的有效性及安全性的验证。
R-CHOP-X组实施方案如下:
研究组(R-CHOP-X组)
RCHOP方案组实施方案如下:
对照组(R-CHOP组)
继续治疗的必要条件如果符合以下情况,下一个疗程将按计划进行。
实验数据分析方法
本发明的分型结果与现有技术的分型结果的流向关系采用桑基图进行可视化分析,参见图1。主要终点PFS和OS采用Kaplan-Meier(KM)法,绘制生存曲线,采用Log-rank法对组间差异进行统计检验,并通过Cox比例风险回归模型估计组间风险比及其95%置信区间。参见图2-5,从图3可以看出,相比于现有技术分型划分到NOS的患者,通过本发明分型可以划分到ST2型,该亚型患者的PFS得到显著提升,标明从以前疗效不好的亚型中鉴别到了疗效更好的亚型。从图5可以看出,相比于现有技术分型划分到NOS的患者,通过本发明分型可以划分到ST2型,该亚型患者的OS生存曲线得到显著提升,标明从以前疗效不好的亚型中鉴别到了疗效更好的亚型。
临床实验
将实施例1所得到的检测弥漫大B细胞淋巴瘤的试剂盒进行临床试验,具体试验流程如下,流程图如图6所示。
(1)总体设计
本试验采用前瞻性、多中心、随机、对照III期临床研究设计,旨在对比R-CHOP联合新型靶向药物(R-CHOP-X)对比R-CHOP方案治疗初治、非低危(IPI≥2分或IPI=1分伴大包块)弥漫大B细胞淋巴瘤的有效性及安全性。本试验计划纳入约1100例患者,以DLBCL分子分型(CancerCell分型)分层,1:1随机分为R-CHOP-X组(试验组)或R-CHOP组(对照组),每组约550例患者。整个试验包括筛选期(第-28天至-1天)、治疗期及随访期(末次结束后2年)。第1周期仅接受RCHOP方案治疗,第2-6周期接受R-CHOP-X或R-CHOP治疗,21天为一周期。治疗期间每个疗程结束后进行常规评价,3个疗程治疗结束采用PET-CT进行疗效评估,疗效评价为疾病稳定(stabledisease,SD)或疾病进展(progressivedisease,PD)的患者出组,由研究者决定挽救治疗方案,疗效评价为完全缓解(completeresponse,CR)或部分缓解(partialresponse,PR)的患者继续第4-6疗程原方案治疗,完成6疗程治疗后采用PET-CT进行疗效评估,CR患者结束治疗,进入随访期,第一年每3个月一次、后续每6个月一次全身增强CT评估,直至疾病进展,死亡,撤回知情同意或研究结束,以先发生者为准;PR、SD或PD的患者由研究者决定挽救治疗方案。对于CNS-IPI(centralnervoussystem-internationalprognosticindex)高危组(CNS-IPI4-6分)患者、结外累及肾或肾上腺或睾丸或乳腺的患者、“双打击”淋巴瘤的患者,建议常规行至少4次腰穿+鞘注。根据研究者判断,可在其余必要情况下加做腰穿+鞘注进行中枢预防。基线存在大包块(≥7.5cm)或者在治疗结束后存在残余病变的患者,根据研究者判断,可行局部受累野放射治疗。
(2)定义研究终点
本试验以下终点中有效性终点根据2014年Lugano恶性淋巴瘤疗效评价的修订标准进行评估。
1)主要研究终点:
无进展生存时间:研究者评估的无进展生存时间(Progressionfreesurvival,PFS),PFS的定义是随机至疾病进展或死亡(以先发生者为准)的时间。
2)次要研究终点:
完全缓解率,定义为治疗结束达到完全缓解(CR)的患者比例。
总生存:随机至死亡的时间,总生存事件为任何原因的死亡。
安全性:治疗中出现的不良事件,严重不良事件的发生率和严重程度。不良事件按照美国国家癌症研究所的通用不良事件术语标准版本5.0(NCI-CTCAEv5.0)评估。
(3)确定样本量大小
BN2亚型:设对照组2年PFS率为65%,并预期试验组2年PFS率约为80%,在一类错误α=0.05(双侧)、组间比例为1:1、不预设期中分析的情况下,入组1年,随访3年,考虑8%脱落,至少198例样本可提供80%的把握度发现组间差异。该亚组在入组人群中占比约18%,预计整个研究入组需要1100例。
MCD亚型:该亚组在入组人群中占比约15%,约165例,设对照组2年PFS率为60%,并预期试验组2年PFS率约为80%,在一类错误α=0.05(双侧)、组间比例为1:1、考虑8%脱落,不预设期中分析的情况下,165例样本可提供92%的把握度发现组间差异。
P53亚型,该亚组在入组人群中占比约15%,约165例,设对照组2年PFS率为40%,并预期试验组2年PFS率约为70%,在一类错误α=0.05(双侧)、组间比例为1:1、考虑8%脱落,不预设期中分析的情况下,165例样本可提供99%的把握度发现组间差异。
Others亚型:该亚组在入组人群中占比约40%,约440例,设对照组2年PFS率为70%,并预期试验组2年PFS率约为85%,在一类错误α=0.05(双侧)、组间比例为1:1、不预设期中分析的情况下,440例样本可提供99%的把握度发现组间差异。N1亚型:该亚组在入组人群中占比约4%,约44例病例,试验组和对照组各22例。EZB/ST2亚型:该亚组在入组人群中占比约8%,约88例病例,试验组和对照组各44例。
综上,各队列合计总共需要1100例。
(4)研究对象
1)入组标准
1.经肿瘤组织病理确诊的初治弥漫性大B细胞淋巴瘤(DLBCL),至少有一个病灶任一轴线超过1.5cm,有足够标本进行二代测序检查;
2.年龄≥18岁,≤80岁,性别不限;
3.ECOG体力状态评分为0、1或2分;
4.研究者判断预期寿命至少有6个月的患者;
5.患者或其法定代理人必须在进行任何研究特殊检查或程序前提供书面知情同意;
6.国际预后指数(IPI)≥2分或IPI=1分伴大包块;
7.试验筛选前自愿签署书面知情同意书。
2)排除标准符合任何下列标准的受试者不得入选研究
1.既往接受过化疗在内的全身性或局部性治疗;
2.既往接受过自体干细胞移植;
3.既往有其他恶性肿瘤病史,除外皮肤基底细胞癌及原位宫颈癌;
4.伴有未控制的心脑血管疾病,凝血障碍性疾病,结缔组织疾病,严重感染性疾病等疾病;
5.淋巴瘤累及中枢神经系统;
6.原发性纵隔大B细胞淋巴瘤;
7.左室射血分数<50%;
8.筛查时实验室检查值:(除非是因为淋巴瘤引起);a)中性粒细胞<1.5*10
9.精神病患者或其他已知或怀疑不能完全依从研究方案的患者;
10.妊娠或哺乳期妇女;
11.已知患有人类免疫缺陷病毒(HIV)感染,或者活动性乙型或丙型肝炎病毒感染(聚合酶链反应[PCR]显示阳性结果)。如果HbsAg检查结果阳性患者,需行HBVDNA检查,若HBVDNA<10
12.需要用强效和中效CYP3A抑制剂或CYP3A诱导剂进行持续治疗;
13.无法吞咽胶囊或存在显著影响胃肠功能的疾病,如吸收不良综合征、减肥手术、炎症性肠病或部分或完全肠梗阻;
14.其他并发且不受控制的被研究者认为将影响患者对研究参与的医学状况。
(5)研究对象招募
1.研究对象来源为初治、非低危DLBCL患者。
2.研究对象入组后,与研究对象通过微信、电话等方式进行随访提醒。
(6)研究对象分配的方法筛选时将按照先后顺序给予受试者筛选号,采用1:1区块化随机。
(7)研究干预
1)给予研究干预
研究干预描述受试者先接受1个疗程R-CHOP方案,后根据分子分型结果(CancerCell.2020;37(4):551-568.e514.),将淋巴瘤患者分为MCD、BN2、P53(若存在确认的TP53突变,则优先分入该型)、EZB、ST2、N1及Others型,从第2疗程开始,以分子分型分层,按1:1随机接受R-CHOPX或者R-CHOP治疗,MCD、BN2和N1型加用BTK抑制剂奥布替尼,P53型加用DNA去甲基化药物地西他滨,ST2型、EZB型和Others型加用免疫调节剂来那度胺,总计接受5个疗程R-CHOP方案联合新型靶向药物治疗,每21天一次。受试者将在治疗完成后继续接受评估(患者将进入时间和事件表中描述的随访期),直至规定的随访期结束(总研究持续时间为4年)或直至患者符合退出标准。
2)剂量与给药方法
研究组(R-CHOP-X组)
对照组(R-CHOP组)
继续治疗的必要条件如果符合以下情况,下一个疗程将按计划进行:
1.经历骨髓相抑制后,中性粒和血小板计数处于上升阶段;
2.下一疗程的第1天外周血中性粒细胞计数≥1.0×109/L;
3.下一疗程的第1天外周血血小板计数≥75×109/L;
4.非血液学毒性降至1级或基线水平如果未达到(排除淋巴瘤疾病本身所引起),下一疗程应延迟3-4天,同时重复血细胞检查如果仍未达到上述指标,治疗将再延迟3-4天,直至达到上述可化疗标准。
如果延迟治疗超过14天仍不达到标准,患者将退出治疗并记录为不良事件。根据方案要求将继续随访该患者。
5)R-CHOP剂量调整利妥昔单抗不进行剂量调整。其他药物的剂量调整如下:
1.出现2级以上神经毒性,VCR(长春新碱)减为1mg。
2.在中性粒细胞或血小板计数不足时,考虑化疗药物的减量。可以参照以下标准执行:
(1)下一疗程延迟时间在0-7天内:维持原有剂量;
(2)下一疗程延迟时间在8-14天内,剂量调整如下,利妥昔单抗100%,环磷酰胺75%,阿霉素75%,长春新碱100%,泼尼松100%。若患者经过一次剂量调整后,下一疗程仍然延迟超过14天,研究者考虑仍需再次化疗减量,则:利妥昔单抗100%,环磷酰胺50%,阿霉素50%,长春新碱50%,泼尼松50%。
(3)若患者经过两次剂量调整后,研究者考虑仍需再次化疗减量,退出试验。
6)靶向药物剂量调整总体原则
当出现≥3级治疗相关非血液学毒性、≥3级伴感染或发热的中性粒细胞减少,4级中性粒细胞减少超过7天,或其他4级血液学毒性时,可减量或暂停用药并进行对症治疗,待毒性症状消退至1级或基线水平(恢复)时,进行剂量维持或调整原剂量。若患者存在肾功能不全(eGFR<60mL/min/1.73m
7)来那度胺剂量调整
当出现≥3级治疗相关非血液学毒性、≥3级伴感染或发热的中性粒细胞减少,4级中性粒细胞减少超过7天,或其他4级血液学毒性时,若判断与来那度胺相关,中断相关药物治疗。待毒性症状消退至1级或基线水平(恢复)时或根据研究判断,可以原剂量重新开始治疗。如果该毒性第二次发生,应再次判断与来那度胺是否相关,并调整相应药物剂量,将剂量减少一级(来那度胺减为10mgqd)。如果该毒性第三次发生,应再次判断与来那度胺是否相关,可将剂量再减少一级(来那度胺停用)。
8)奥布替尼剂量调整
当出现≥3级治疗相关非血液学毒性、≥3级伴感染或发热的中性粒细胞减少,4级中性粒细胞减少超过7天,或其他4级血液学毒性时,若判断与奥布替尼相关,中断相关药物治疗。待毒性症状消退至1级或基线水平(恢复)时或根据研究判断,可以原剂量重新开始治疗。如果该毒性第二次发生,应再次判断与奥布替尼是否相关,并调整相应药物剂量,将剂量减少一级(奥布替尼减为100mgqd)。如果该毒性第三次发生,应再次判断与奥布替尼是否相关,可将剂量再减少一级(奥布替尼减为50mgqd)。若再次发生则停药。
9)地西他滨剂量调整
当出现因中性粒细胞或血小板计数不足导致的化疗延迟,若判断与治疗相关,延迟时间在0-7天内,维持原剂量;延迟时间在8-14天,地西他滨减量为10mg/m
(8)合并用药及治疗
首程化疗肿瘤负荷较大(巨大肿块或乳酸脱氢酶大于等于500u/L)或PS等于2或胃肠道淋巴瘤(预防胃肠道穿孔)的患者,给予别嘌呤醇、小苏打,先口服强的松,必要时化疗分2天执行,以预防肿瘤溶解综合征。
给与长效粒细胞击落刺激因子(G-CSF)次级预防粒细胞减少。
由于不良事件而给予的伴随性治疗,如果符合报告标准,也需要予以报告,该内容应填写在CRF表的不良事件页。必要时可给予患者足够的支持性治疗,包括输注全血和血制品,抗生素治疗,止吐治疗等治疗的原因,剂量和治疗的日期也应该记录在CRF表中。
对于大包块的患者,由研究者决定是否需要放疗。
(9)研究干预中止及研究对象中止/撤出
1)研究干预中止
如果本试验由于安全性原因而被暂停或中止,研究者将通知监管机构暂停或中止本试验并说明采取这一行动的原因。如果适用的法规要求,研究者必须立即通知IEC/IRB,并提供暂停或中止研究的原因。
研究干预中止后研究对象应尽量继续随访。若患者撤回知情同意,终止治疗且拒绝提供任何疾病相关信息,则无需进行进一步评估或收集信息;否则应尽力收集关于患者疾病的信息,包括安全性随访以获取不良事件、严重不良事件及非预期问题。
研究干预中止并不意味着研究中止,剩余的研究程序仍应按照研究申报书完成。
2)研究对象中止/撤出
受试者可因为下列原因,随时退出研究。
1.患者医学状态(包括妊娠)发生改变,根据研究者判断,出于对受试者的最佳利益考虑。
2.疾病进展或者开始其他抗淋巴瘤治疗。
3.因任何原因不能继续使用治疗,如不可接受的毒性反应。
4.研究受试者随时因任何原因要求退出。(若患者撤回知情同意,终止治疗且拒绝提供任何疾病相关信息,则无需进行进一步评估或收集信息;否则应尽力收集关于患者疾病的信息。)
5.失访。(若患者未进行计划访视,应尽力重新建立联系,尽力记录下患者结局。)
6.死亡。
7.发生严重化疗毒性反应的或患者因不良反应延期化疗超过2周。
8.心脏毒性:左心射血分数(LVEF)≦50%或与基线或上一次心血池扫描相比下降超过15%。
9.不依从研究方案。
3)失访
本试验的随访期为2年,当研究对象停止预定的研究随访、不能完成研究规定的程序或研究者联系不到研究对象时可视为失访。研究者将通过电话/微信保持联系的方式减少失访和资料缺失。
(10)研究结局评价
1)主要及次要结局评价
主要疗效指标:
2年无进展生存(PFS)率,定义为进入研究2年时疾病仍未进展和死亡的患者占所有患者的百分率。即2年PFS率=2年时仍未进展和死亡的患者数/总患者数×100%。
次要疗效指标:
(1)完全缓解率,按2014年Lugano恶性淋巴瘤疗效评价的修订标准。
(2)2年总生存(OS)率,定义为进入研究2年时仍存活的患者占所有患者的百分率。即2年OS率=2年时仍存活的患者数/总患者数×100%。
(3)客观缓解(ORR)率:缓解(CR+PR)的患者占所有患者的百分率,即(CR+PR)患者数/总患者数×100%。
根据2014年Lugano恶性淋巴瘤疗效评价修订标准,客观疗效评估分为完全缓解(CR)、部分缓解(PR)、疾病稳定(SD)、疾病进展(PD)。所有受试者将在第3和第6疗程结束后进行PET-CT疗效评估。如果影像学检查结果显示基线时淋巴瘤骨髓受累患者出现CR,则需要重复进行骨髓活组织检查和穿刺。进入随访期后第1年每3月一次,第2年及后续每6月一次进行抗肿瘤活性评估,直至疾病进展,死亡,撤回知情同意或研究结束,以先发生者为准。评估包括疾病相关症状、体格检查、实验室结果和全身(颈部、胸部、上腹部、盆腔)CT增强检查,和研究者判断需要进行的其他评估手段。对于怀疑PD的受试者建议行PET-CT,并且尽可能获取组织病理结果以确认或排除PD状态。
2)安全性及其他评价
治疗期间不良事件,严重不良事件的发生率和严重程度将根据CTCAEV5.0版进行统计和分析。治疗期间出现的不良事件(TEAE)定义为发生日期或严重程度与基线(治疗前)相比恶化的日期在首次研究药物给药当天或之后至研究药物终止后30天(安全性随访)或启用新的抗癌治疗(以先发生者为准)这段期间的AE。安全性评价指标包括临床试验期间观察到的不良反应。
(11)不良事件与严重不良事件
1)不良事件(AE)定义
临床试验中的受试者在使用药物后出现的任何非期望的和治疗有关或无关的医学事件都是不良事件。它可以是任何不利的和非期望的体征、症状、实验室检查等异常结果,而无论其是否和药物有关。
不良事件实例包括:
·研究过程中身体状况/原有疾病出现显著或非预期恶化或加重。
·慢性或间歇性既存病情恶化,包括严重程度、频率、持续时间的上升和/或伴随明显更坏的结局。
·在试验药物给药后检出或确诊的新疾病,即使该疾病在试验开始前就可能存在。
·疑似药物相互作用的体征、症状或临床后遗症。
·研究药物或伴随药物可疑用药过量的体征、症状或临床后遗症(用药过量本身不作为不良事件或严重不良事件报告)。
·预期药理学或生物学作用显著失效。
不良事件不包括:
·内科或外科操作(例如,内镜检查和阑尾切除术);导致内科或外科操作的疾病是不良事件。
·未发生不良医疗事件的情况(社会原因和/或方便原因住院)。
在试验开始时已存在或检测到的原有疾病或状况的预测每日波动,而未出现恶化。
2)严重不良事件(SAE)定义
4级的血液学毒性反应和3-4级的非血液学毒性反应作为不良事件或严重不良事件进行报告。
严重不良事件的定义为在任何药物剂量下发生的不良事件,并符合以下标准中的任意一项:1)导致死亡;2)危及生命;3)导致住院或延长住院时间;4)永久或显著丧失功能/残疾;5)致畸、致出生缺陷或致癌;6)其他重要医学事件:必须运用医学和科学的判断决定是否对其他的情况加速报告,如重要医学事件可能不会立即危及生命、死亡或住院,但如需要采取医学措施来预防如上情形之一的发生,也通常被视为是严重的。
有些需要住院或延长住院时间的事件可不作为严重不良事件报告,包括:1)因社会原因而非不良事件住院;2)在进入研究前已经预约的择期手术,检查或其它治疗而住院;3)淋巴瘤进展以及相关事件。
3)不良事件分类
事件严重性
CTCAE分级系统中未列出的不良事件将根据下述标准进行分级:
1级,轻度;无症状或症状较轻;仅临床或诊断发现;无需治疗。
2级,中度;最小的,局部的或非侵入性治疗指征;年龄相关工具性日常生活受限。
3级,重度或重要医学意义,但不会立即危及生命;住院治疗或延长住院指征;致残;自理性日常生活受限。
4级,危及生命,需紧急治疗。
5级,死亡。
4)与研究干预的相关性
研究者应对不良事件和研究药物以及合并药之间可能存在的关联做出评估,参照以下2级分类标准评定:
①有关:考虑到包括事件时间,生物学合理性,临床判断和其他潜在原因,怀疑不良事件与研究药物可能相关。且其他干预性治疗或原发病等不能解释观察到的事件。
②无关:考虑到包括事件时间,生物学合理性,临床判断和其他潜在原因,怀疑不良事件与研究药物可能不相关。且其他干预性治疗或原发病等不能解释观察到的事件。
以上①记为治疗相关不良反应。
不良事件发生率=不良事件例数/总例数×100%。
5)预期性
预期的不良反应应采用标准的格式收集,其预期性可根据以往观察到的不良事件评估。应确认不良事件是预期的或是非预期的,如果不良事件的性质、严重性或频率与既往研究干预描述的风险信息不符,可认为是非预期的。
6)收集不良事件和严重不良事件的时间、频率、方法
在签署知情同意书后但在试验药物给药前,只需要报告严重不良事件。
在开始研究药物治疗至最后一次研究药物给药后30天,评估患者并报告所有不良事件和严重不良事件,不考虑与研究药物的因果关系。研究者或其指定代表通过以下问题询问不良事件:
·您感觉如何?
·自您上次访视之后,是否有发生任何医学问题?
·自您上次访视之后,是否服用过任何新的药物?
将在原始文件中记录所有不良事件和严重不良事件。在eCRF中报告所有不良事件和严重不良事件。
7)不良事件和严重不良事件的记录
出现不良事件或严重不良事件后,由研究者负责评审所有事件相关文件(例如,医院病程记录、化验和诊断报告)。之后研究者在严重不良事件报告表上记录有关不良事件或严重不良事件的所有相关信息。
研究中发生的所有不良事件都应该记录在eCRF表格上。研究者将基于体征、症状和/或其他临床信息,尝试诊断事件。在此情况下,应将诊断结果记录为不良事件或严重不良事件,而不是个体的体征/症状。不良事件是研究的独立部分。
8)不良事件的随访
初次报告不良事件或严重不良事件后,要求研究者积极随访每一例受试者,并提供有关受试者状况的进一步信息。
在前次访视/联系时确认且指定为持续事件的所有不良事件和严重不良事件,都将在后续访视/联系时继续随访。
随访所有不良事件和严重不良事件直至消退、状况稳定、事件另有解释说明或患者失访、死亡或开始新的抗肿瘤治疗。一旦事件消退,将相应更新不良事件或严重不良事件报告表。研究者将确保随访时进行有可能阐明不良事件或严重不良事件性质和/或因果关联的任何补充检查。补充检查包括其他实验室检查或研究、组织病理学检查或咨询其他医疗保健专业人员。新信息或更新信息需要重新填写在SAE报告上,所有变更需由研究者签字并注明日期。
9)疾病进展
在本试验的人群中,预期发生并作为有效性终点测量的疾病进展不应作为AE术语进行报告。然而,由疾病进展引起的症状、体征或临床后遗症应作为AE术语进行报告。
例如,患者出现由肺转移的疾病进展引起的胸腔积液。事件术语应报告为“胸腔积液”而不是疾病进展。如果患者由于疾病进展而出现致死性多器官功能衰竭,则术语“多器官功能衰竭”应被报告为死亡结局的SAE,而不是报告为“致死性疾病进展”或“疾病进展导致的死亡”。
10)死亡
死亡是一种结局,通常不视为一起事件。如果唯一已有的信息是死亡和死因不明,则将死亡报告为一起事件,如“死亡”、“不明原因死亡”或“无法解释的死亡”。
11)严重不良事件报告
严重不良事件的快速报告
一旦研究者确定AE达到了方案定义的SAE标准,则必须按下表的描述立即(24小时之内)向研究的发起单位或指定人员报告。
表:向研究的发起单位或指定人员报告严重不良事件(SAE)的时限和记录方法
12)填写与发送严重不良事件报告
一旦研究者获知患者发生了SAE,需要按11)中列出的要求,在24小时内向研究的发起单位报告。SAE报告应始终尽可能填写完整,记录所有已获知的SAE细节,并在指定的时间窗内传送给研究的发起单位或指定人员。
如果研究者未获知SAE的所有详尽信息,不能等到收到补充信息之后才将该SAE通知研究的发起单位或指定人员并填写表格。在收到额外信息后,该表会被更新。
研究者必须按照12)中的描述,为每个SAE提供因果关系评估。
研究的发起单位将提供一个接收SAE的联系信息。
13)严重不良事件报告的监管要求
根据11)和12)中详述的程序,研究者需要快速向研究的发起单位报告所有SAE。
研究者或符合当地要求的负责人员,将遵循与SAE报告有关的适用当地监管要求,向适用的监管机构和/或机构评审委员会/独立伦理委员会(IRB/IEC)报告SAE。
研究中心从发起单位收到初始或随访安全报告或其他安全信息(如经修订的IB)时,研究者或指定负责人须遵循适用的当地监管要求,及时通知适用的机构单位。研究者应当将研究发起单位提供的安全性报告副本保存在试验者研究中心文件中。
(12)统计分析
分析集
·意向治疗(ITT)分析集-包括所有随机患者。患者将根据随机治疗组(即R-CHOP-X组或R-CHOP组)进行分析。这将是所有有效性分析的主要分析集。
·符合方案(PP)分析集-包括所有接受了所分配的研究药物(R-CHOP-X或R-CHOP)≥1次剂量并且没有重大方案偏离的随机患者。重大方案偏离将在主要分析的数据库锁定之前确定和记录。(当有超过10%的ITT患者存在重大方案偏离时,这将是所有有效性分析的次要分析集。)
·安全性分析集-包括接受了所分配的研究药物的≥1次剂量的所有患者(R-CHOP-X或R-CHOP)。这将是所有安全性分析的分析集。
1)一般方法
对于治疗前数据进行统计描述。对于两组间基线的可比性比较,主要采用χ
2)主要及次要研究终点分析
主要研究终点分析
主要终点PFS将采用Kaplan-Meier(KM)法,估计每个治疗组的PFS中位时间、2年PFS率及其95%置信区间,并绘制生存曲线,采用Log-rank法对组间差异进行统计检验,并通过Cox比例风险回归模型估计组间风险比及其95%置信区间。PFS删失规则将遵循美国食品与药品管理局(FDA)抗癌药物和生物制剂审批临床试验终点行业指南(FDA2018)。
次要研究终点分析
完全缓解率
基于ITT分析集的两个治疗组之间的CR差异的统计学显著性将采用alpha水平为0.05(双侧)、以分层因子实际值作为分层的Cochran-Mantel-Haenszel(CMH)卡方检验进行。将计算比值比和CR差异的双侧95%CI,以及各组内CR的Clopper-Pearson95%CI。
总生存期
使用以分层因子的实际值作为分层的对数秩检验来比较ITT分析集中两组之间的总生存期。将采用Kaplan-Meier法估算2年OS率,将计算每个治疗组中OS率,并给出双侧95%CI。
客观缓解率
基于ITT分析集的两个治疗组之间的差异的统计学ORR显著性将采用alpha水平为0.05(双侧)、CMH卡方检验进行。将计算比值比和ORR差异的双侧95%CI,以及各组内ORR的ClopperPearson95%CI。
可能针对不同亚组进行疗效分析,包括分子亚型、IPI评分、年龄、ECOG评分、疾病分期、不良事件分级、药物暴露量等。
3)安全性分析
将基于安全性分析集,对药物暴露量进行汇总,包括暴露持续时间、剂量和剂量强度。不良事件的逐字描述基于国际医学用语词典
4)基线描述性分析
基于ITT分析集,采用描述性统计对各组间基线时的人口学特征与实验室指标进行比较
5)亚组分析
对主要研究终点按DLBCL分子分型分组,进行亚组分析;并进行多中心间研究终点的比较,以评价中心间存在的差异。
(13)支持性文件与注意事项
1)知情同意过程
受试者在入选试验前,研究者必须向他们解释试验目的,方法,可能的受益,潜在的风险和可能出现的不适。告知受试者参与试验是自愿的,可以在任何时候退出,是否参与试验对其疾病的治疗没有任何影响。受试者的隐私权将受到保护。
受试者或监护人员应有足够时间来阅读知情同意书和提出疑问。在入选前,受试者或其监护人必须签署知情同意书,受试者保留知情同意书的复印件。
2)隐私保护
本次试验中只收集局限于对研究方案的有效性,安全性等研究所需要的信息。收集和使用这些数据时将遵守相关的保护隐私权的法律和规定。
3)标本及资料的收集与使用
根据GCP要求进行标本及资料的收集与使用。
4)质量控制与质量保证
根据GCP要求进行质量控制与质量保证。
5)数据处理与记录保存
数据收集与管理
在分析实施前,需要根据研究方案和eCRF收集的数据特点,起草详细的数据管理计划书,包括数据质量审核等内容,并建立相应的电子数据库,完整、准确录入所有预期收集的数据。eCRF的数据录入到数据库中并核查双录入完全一致后,需要对数据库中的数据质量进行核查。将采用计算机程序结合人工审核的方法对数据的一致性,逻辑性等进行检查。产生的任何疑问将由研究者解答,返回数据管理中心,数据管理人员将据此对数据库进行修改和更新。所有检查程序需要重复多次,直到没有任何质疑。所有的修改及更新都需进行记录及归档。
数据收集由临床研究人员在负责人监督下进行,负责人将对报告数据的准确性、完整性、及时性负责。所有数据应清晰以确保准确的解释,并保证其可溯源性。临床数据将建立数据库保管,数据库应有密码保护,数据库建立时应设立逻辑校对程序。
研究数据保留
依照GCP原则,研究者应保存受试者所有的详细原始文件,并在eCRF中记录有关试验进程、用药情况、实验室检查数据、安全性数据和疗效评定等方面的内容,记录的数据应保证完整、及时、清晰。eCRF、原始文件、医学记录等应清楚、详细并易被参加此临床试验的人员辨识。主要研究者至少须在eCRF的入选确认页和完成页签名,以证实所有数据的准确性和完整性。eCRF及原始文件只能由研究者进行修改。对eCRF及原始文件的任何修改都不得将原始数据涂抹掉。正确的修改方法是在原数据上划单线,再将修改后的数据写在原数据的旁边,并签署日期及修改人员的姓名缩写。试验资料由研究者保留至研究结束后5年。
- 一种检测弥漫大B细胞淋巴瘤的试剂盒及其应用
- 一种用于检测原发性乳腺弥漫性大B细胞淋巴瘤的标志物及其试剂盒和应用