一种探针引物组合及其检测试剂盒
文献发布时间:2023-06-19 09:40:06
技术领域
本发明涉及核酸检测技术领域,具体涉及一种探针引物组合及其检测试剂盒。
背景技术
实时荧光PCR技术已广泛应用于检测和定量靶核酸序列。目前最常用的是TaqMan探针技术。在荧光PCR方法应用的诸多领域中,DNA基因分型检测是目前发展较快的一个重要领域。基因分型检测包含了基因型多态性和稀有突变检测,如病理样本的基因突变检测、遗传相关疾病的基因突变检测以及病毒基因分型检测等等。目前应用于基因分型检测的荧光PCR技术主要有以下几种:1、荧光探针分型检测。该方法针对不同的基因型设计不同的荧光标记探针,如TaqMan探针、分子信标等。2、熔解曲线分析法。此类方法通过探针与扩增产物的杂交温度差异来区分不同的基因型。例如邻近杂交探针,以及基于双标记TaqMan探针或分子信标的熔解曲线分析方法。
然而,这些方法均有一定的局限性。TaqMan探针或分子信标的熔解曲线分析方法的应用较为广泛,而且,TaqMan探针在杂交状态和游离状态下的信号差异较小,尤其是当探针长度较长时,这种信号差异更小,很难与背景信号区分。特异性和灵敏度均存在较大问题。邻近杂交探针需要两条探针与扩增产物杂交,对探针的设计要求较高,限制了此方法的应用范围。近年来,出现了一种单标记探针(U.S.Pat.No.6635427),该方法利用互补链上的鸟苷酸碱基淬灭探针上的荧光基团,杂交信号弱,也难与背景信号区分,对探针序列设计也有一定要求,应用有限。有的现有技术中,将引物为茎环结构,结构复杂,设计难度较大。
现有的实时荧光PCR方法使用双分子杂交技术,包括类似于蝎形探针的分子内杂交以及探针与扩增产物的分子间杂交,杂交体结构复杂,设计难度较大。比如,当两处杂交同时存在错配时,会干扰检测结果的判断,甚至导致假阴性结果。另外,现有方法涉及到分子内和分子间两次杂交,扩增产物的分子内杂交(环状结构)的Tm值必须远大于探针与扩增产物杂交的Tm值,否则在溶解曲线分析程序中,探针上标记的荧光基团不能被有效淬灭。这也给该方法的应用带来了一定的局限性。
发明内容
本发明提供一种探针引物组合及其检测试剂盒。
根据第一方面,一种实施例中提供一种探针引物组合,包括可与靶核苷酸序列杂交的探针寡核苷酸、用于扩增包含靶核苷酸序列的核酸区域的第一引物寡核苷酸、第二引物寡核苷酸,所述探针寡核苷酸连接有第一标记分子,所述第一引物寡核苷酸或所述第二引物寡核苷酸连接有第二标记分子,所述探针寡核苷酸互补于所述靶核苷酸序列的一条单链,连接有所述第二标记分子的引物寡核苷酸互补于所述靶核苷酸序列的另一条单链,所述第一标记分子、第二标记分子提供不同的可检测信号,所述第一标记分子、第二标记分子物理靠近时发生荧光能量共振转移。
在一些实施例中,所述第一标记分子连接至所述探针寡核苷酸的3’端。
在一些实施例中,所述第二标记分子所连接的碱基靠近该碱基所在的引物寡核苷酸的3’端。
在一些实施例中,所述第二标记分子所连接的碱基距离其所在引物寡核苷酸的3’端8个碱基以内,第二标记分子所连接的碱基尽量靠近该碱基所在的引物寡核苷酸的3’端。
在一些实施例中,所述第二标记分子所连接的碱基为A、T、C、G碱基中的任一种。
在一些实施例中,所述探针寡核苷酸的序列长度为40nt以内,还可以为15-30nt。
在一些实施例中,所述探针寡核苷酸的Tm值为55℃-90℃,还可以为60℃-80℃。
在一些实施例中,所述第一引物寡核苷酸、第二引物寡核苷酸的序列长度均为30nt以内,还可以为20-25nt。
在一些实施例中,所述靶核苷酸序列为单链核苷酸、双链核苷酸中的至少一种。
在一些实施例中,所述靶核苷酸序列为DNA序列、RNA序列中的至少一种。
在一些实施例中,所述靶核苷酸序列为单链核苷酸时,连接有所述第二标记分子的引物寡核苷酸互补于所述靶核苷酸序列的互补序列。
在一些实施例中,所述第一标记分子为荧光报告基团,所述第二标记分子为荧光淬灭基团,或,所述第一标记分子为荧光淬灭基团,所述第二标记分子为荧光报告基团。
在一些实施例中,所述探针寡核苷酸的杂交位置的3’端靠近所述第二标记分子。
在一些实施例中,所述荧光报告基团选自FAM、HEX、VIC、ROX、Cy5中的至少一种;
在一些实施例中,所述荧光淬灭基团选自BHQ1、BHQ2中的至少一种。
根据第二方面,一种实施例中提供一种检测试剂盒,含有如第一方面所述探针引物组合。
在一些实施例中,所述检测试剂盒还含有不对称PCR反应体系。
在一些实施例中,所述不对称PCR反应体系选自PCR体系、逆转录PCR体系中的任一种。PCR体系主要用于扩增DNA靶序列,逆转录PCR体系主要用逆转录并扩增RNA靶序列。
在一些实施例中,所述PCR体系含有PCR Master Mix、热稳定的聚合酶、dNTPs中的至少一种。
在一些实施例中,所述逆转录PCR体系含有RT-PCR Master、热稳定的聚合酶、逆转录酶、dNTPs中的至少一种。
在一些实施例中,所述热稳定的聚合酶选自Taq DNA聚合酶、Tth DNA聚合酶中的一种。
在一些实施例中,所述不对称PCR反应体系中探针寡核苷酸的摩尔终浓度为200nM-800nM。
在一些实施例中,连接有所述第二标记分子的引物寡核苷酸的摩尔终浓度>未连接所述第二标记分子的引物寡核苷酸的摩尔终浓度。
在一些实施例中,所述不对称PCR反应体系中,连接有所述第二标记分子的引物寡核苷酸的摩尔终浓度为未连接所述第二标记分子的引物寡核苷酸摩尔终浓度的5-20倍。
在一些实施例中,所述不对称PCR反应体系的体积为10-100μL,具体可以为25μL。
根据第三方面,一种实施例中提供如第一方面所述探针引物组合或如第二方面所述检测试剂盒在基因分型检测中的应用。
在一些实施例中,所述基因分型检测包括但不限于单核苷酸多态性(SNPs)位点检测、病毒基因分型等等。
根据第四方面,一种实施例中提供如第一方面所述探针引物组合或如第二方面所述检测试剂盒在不对称熔解曲线PCR检测中的应用。
在一些实施例中,所述不对称熔解曲线PCR检测方法的反应程序如下:
95℃,1-10min;45×(95℃,5-10s;55℃,30-60s);95℃,1min;55℃,1min;55℃-95℃连续收集荧光信号。
在一实施例中,通过单荧光标记探针(或荧光淬灭基团标记探针),与荧光淬灭基团(或荧光报告基团)标记的寡核苷酸链杂交产生荧光信号。本发明设计的单标记探针不会因构象变化产生非特异的荧光信号,背景更低。
附图说明
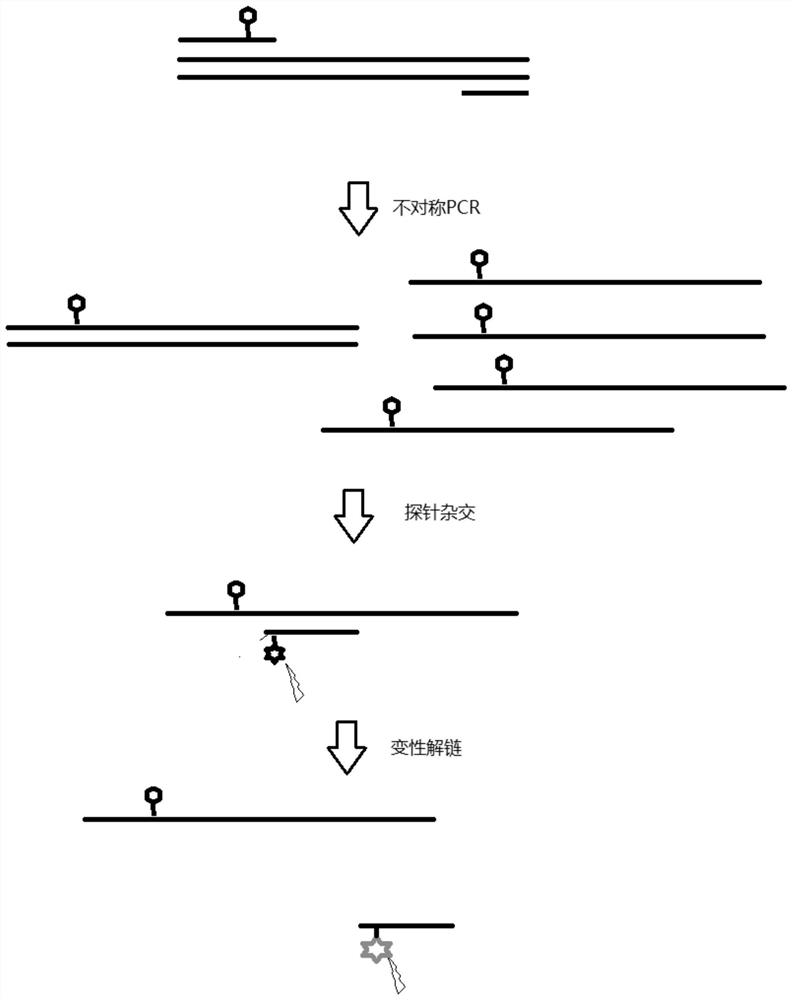
图1显示为本发明实施例的反应原理图;
图2显示为本发明实施例1的不对称PCR的反应程序设置界面图;
图3显示为本发明实施例1的单标记探针熔解曲线图。
图4显示为本发明实施例1的双标记探针熔解曲线图。
图5显示为本发明实施例2的MTHFR基因677C纯合子检测结果图,探针杂交Tm值约为64℃。
图6显示为本发明实施例2的MTHFR基因677T纯合子检测结果图,探针杂交Tm值约为55℃。
图7显示为本发明实施例2的MTHFR基因677C/T杂合子检测结果图,杂交探针的Tm值约为54.5℃和64.5℃。
图8显示为rs13182883A/A纯合子检测结果,R1:对比专利CN 102321765 A的方法;R2:本发明实施例3的方法。
图9显示为rs13182883G/G纯合子检测结果,R1:对比专利CN 102321765 A的方法;R2:本发明实施例3的方法。
图10显示为rs13182883A/G杂合子检测结果,R1:对比专利CN 102321765 A的方法;R2:本发明实施例3的方法。
具体实施方式
下面通过具体实施方式结合附图对本发明作进一步详细说明。其中不同实施方式中类似元件采用了相关联的类似的元件标号。在以下的实施方式中,很多细节描述是为了使得本申请能被更好的理解。然而,本领域技术人员可以毫不费力的认识到,其中部分特征在不同情况下是可以省略的,或者可以由其他元件、材料、方法所替代。在某些情况下,本申请相关的一些操作并没有在说明书中显示或者描述,这是为了避免本申请的核心部分被过多的描述所淹没,而对于本领域技术人员而言,详细描述这些相关操作并不是必要的,他们根据说明书中的描述以及本领域的一般技术知识即可完整了解相关操作。
另外,说明书中所描述的特点、操作或者特征可以以任意适当的方式结合形成各种实施方式。同时,方法描述中的各步骤或者动作也可以按照本领域技术人员所能显而易见的方式进行顺序调换或调整。因此,说明书和附图中的各种顺序只是为了清楚描述某一个实施例,并不意味着是必须的顺序,除非另有说明其中某个顺序是必须遵循的。
定义
此处所使用的术语和短语具有其本领域公认的含义,可以通过参考本领域技术人员已知的标准教科书和期刊中找到。提供下列定义以阐明它们在本发明的上下文中的特定用途。
本文中为技术特征所编序号本身,例如“第一”、“第二”等,仅用于区分所描述的对象,不具有任何顺序或技术含义。
术语“互补”是指在寡核苷酸序列中足够数量的相匹配的碱基对与被扩增或检测到的靶核酸序列发生特异性(杂交)的相互作用。在本领域中,杂交的特异性和灵敏度需要非常高度的互补,虽然它不一定是100%。
术语“变性”是指互补的DNA链的伸展和分离,并可以通过热或变性剂处理来完成。
术语“DNA”是指以单链或双链的状态聚合的脱氧核糖核苷酸(腺嘌呤、鸟嘌呤、胸腺嘧啶或胞嘧啶),并且包括线性或环状DNA分子。在讨论DNA分子时,序列仅可通过给出5′到3′方向的序列的约定进行描述。
术语“DNA扩增”和“扩增”是指任何方法,通过酶促扩增增加一个特定的DNA序列的复制。通常使用的过程是聚合酶链反应(PCR)。PCR包括使用一种热稳定的DNA聚合酶、引物和加热循环,这能够使DNA链分开,并指数扩增感兴趣的基因区域。也可使用任何类型的PCR,如定量PCR、RT-PCR、热启动PCR、LAPCR、多重PCR、降落式PCR等。有利的是,可使用实时PCR。链式反应的延伸产物将是含有对应于所使用的引物的末端的终端的不连续的核酸双链。
术语“酶促扩增”或“扩增”是指DNA扩增。目前最常用的方法是聚合酶链反应(PCR)。其它扩增方法包括LCR(连接酶链式反应)、链置换扩增(SDA);Qβ复制酶扩增(QβRA);自我持续复制(3SR);以及NASBA(基于核酸序列的扩增),其可以在RNA和DNA中进行。
术语“荧光报告基团”是指其存在可以通过其发光特性检测的任何报告基团。
术语“荧光淬灭剂”或“淬灭剂”是指干扰或吸收由附近的荧光团发出的荧光的分子。典型的淬灭剂包括,但不限于,无荧光芳族分子的DABSYL或Black holeQuencher.RTM。淬灭剂也可以是TAMRA(羧基四甲基罗丹明),其在不同的波长发射。
术语“杂交”是指两个核酸链结合的过程,以通过相对的链之间的稳定氢键形成反平行双链。术语“杂交”和“结合”可互换使用,并且是指两个多核苷酸片段的核苷酸序列之间的互补的A-T和C-G碱基对的形成。杂交链被称为“双链”。
术语“解链温度”(Tm)是指杂交双链解杂交并返回到它们的单链状态的温度。同样地,杂交不会在两链之间的温度高于所得到的双链的解链温度下发生。
术语“核苷酸”是指核酸的一个子单元(无论DNA或RNA或其类似物),其可以包括,但不限于,磷酸基、糖基、含氮基以及这些子单元的类似物。术语“核苷酸”和“核苷”包括不仅含有天然存在的嘌呤和嘧啶碱基,例如腺嘌呤(A)、胸腺嘧啶(T)、胞嘧啶(C)、鸟嘌呤(G)或尿嘧啶(U)等基团,而且要修饰或模拟本领域技术人员已知的碱基。
术语“寡核苷酸”是指包含PCR反应中使用的足够数量的核苷酸碱基的一系列连接的核苷酸残基。短的寡核苷酸序列可以是基于或由基因组或cDNA序列设计并用于扩增、确定,或揭示特定的细胞或组织中相同、相似或互补的DNA或RNA的存在。寡核苷酸可以被化学合成,并且可以被用作引物或探针。此处所使用的术语“寡核苷酸”和“多核苷酸”也可以是指修饰或未修饰的RNA或DNA。
此处所使用的术语“聚合酶链反应”或“PCR”是指一种热循环的、聚合酶介导的DNA扩增反应,其使用与模板分子、热稳定的DNA聚合酶,以及脱氧核糖核苷酸互补的模板分子、寡核苷酸引物。而且它涉及三个重复过程(变性、杂交和引物延伸),它们在不同的温度和步骤下进行。在许多实施例中,杂交和延伸过程可以同时进行。扩增的其它方法包括但不限于NASBR、SDA、3SR、TSA和滚环式复制。
术语“聚合酶”是指将连续加入的单体单元催化为一种聚合物链的酶。在本发明的有利实施例中,“聚合酶”将通过加入其特性是由一个特定序列的互补模板确定的单体单元起作用。DNA聚合酶如DNA聚合酶1和Taq聚合酶以模板依赖的方式添加脱氧核糖核苷酸到多核苷酸链中的3′端,从而合成一个互补的核酸。聚合酶可能会延长引物一次,或使用两个引物可以重复扩增两条互补链。
术语“引物”是指与被扩增或复制的DNA片段互补的寡核苷酸。通常引物用于PCR。一种引物与模板DNA杂交(或“退火”),并通过聚合酶作为复制/扩增过程的起点被使用。通过“互补”是指该引物序列可与模板形成稳定的氢键复合物。
引物被选择为“基本上”互补于靶DNA序列的不同链,但它们不必反映模板的确切序列。例如,非互补的核苷酸片段可连接到引物的5′端,引物的其余部分与链是互补的。或者,非互补碱基可以穿插入引物,只要与靶杂交有足够的互补性并引发延伸产物。
术语“探针”是指可变长度的核酸序列的寡核苷酸,适用于相同、相似或互补的核酸序列的杂交检测。用作检测探针的寡核苷酸序列可以标记有可检测的基团。各种标记基团是本领域已知的,例如放射性的、荧光性的、化学发光的或电化学发光的化合物。
术语“淬火”或“淬火的”或“猝灭”或“猝灭的”是指减少由一个分子产生的信号。它包括,但不限于,降低产生的信号至零或低于检测限。因此,一个给定的分子可以被另一个分子“猝灭”,尽管该信号被大大降低,仍产生检测信号。
术语“定量PCR”是指实时聚合酶链反应,也称为被用于扩增和同时进行有针对性检测靶DNA分子的量的定量实时聚合酶链反应(Q-PCR/qPCR/qrt-PCR)。量可被表示为大量的复制或规范化为输入DNA的相对量。检测不像标准的PCR随着反应实时进行,其中反应产物是在其结束点进行检测。实时PCR中用于检测产品的两种常用方法有:(1)插入任何双链DNA的非特异的荧光染料,和(2)标记有荧光受体的序列特异的寡核苷酸和与互补的DNA靶杂交后的许可检测。
术语“靶”和“靶核苷酸序列”是指期望检测的寡核苷酸。用于公开方法的靶分析物可能是一个分离的寡核苷酸,固定在载体上或在自由溶液中的寡核苷酸。对于应用到本发明的方法,“靶”可指来自植物、动物或人类个体、细菌、病毒或单细胞真核生物,或者是来自整个生物体,及其组织,或者是来自培养细胞或细胞的任何核酸。
术语“模板”是指与靶多核苷酸链,例如,但不限于,未修饰的天然存在的DNA链,其中聚合酶用作识别哪些核苷酸应该下一个合并到一个不断增长的链以聚合天然存在的链的互补链。模板可以是单链或双链。在本发明要求聚合的重复循环的应用中,例如,聚合酶链反应(PCR),模板链本身可以经修饰的核苷酸的并入而修饰,但仍作为聚合酶的模板来合成另外的多核苷酸。
术语“热循环反应”是指多步反应,其中至少两个步骤是通过改变反应温度来实现。
术语“热稳定的聚合酶”是指可以承受极高温度,例如接近100℃的DNA或RNA聚合酶。耐热聚合酶的例子包括Taq、Tth、Pfu、Vent和deep vent。
本文中,术语“第一引物寡核苷酸”也可称为上游引物,术语“第二引物寡核苷酸”也可称为下游引物。
本文中,单标记探针是指只连接有一个标记分子的探针。
除非另有定义,本文所使用的所有技术和科学术语具有分子生物学的领域中的普通技术人员通常所理解的相同含义。方法类似或等同于本文描述的材料可以用于本发明的实践中使用,合适的方法和材料如本文所述。
缩略语
SNPs(Single Nucleotide Polymorphisms),单核苷酸多态性。
现有的探针熔解曲线方法利用双标记探针在游离状态和杂交状态下的构象变化产生荧光信号上的差异(即检测信号),这种荧光信号差异往往比较弱。探针构象变化和核苷酸序列有很大的关系,因此,为了得到比较好的检测效果,需要对探针序列进行改进,这在一定程度上增加了探针的设计难度。而且探针自身容易产生非特异的荧光信号,导致假阳性结果。信噪比也比较低。
在一些实施例中,本发明主要解决的技术问题是如何提升用于核酸检测的探针的信号差异,使得阳性检测信号和背景信号容易区分,并显著降低探针和引物的设计难度。
第一方面,一种实施例中提供一种探针引物组合,包括可与靶核苷酸序列杂交的探针寡核苷酸、用于扩增包含靶核苷酸序列的核酸区域的第一引物寡核苷酸、第二引物寡核苷酸,所述探针寡核苷酸连接有第一标记分子,所述第一引物寡核苷酸或所述第二引物寡核苷酸连接有第二标记分子,所述探针寡核苷酸互补于所述靶核苷酸序列的一条单链互补,连接有所述第二标记分子的引物寡核苷酸互补于所述靶核苷酸序列的另一条单链,所述第一标记分子、第二标记分子提供不同的可检测信号,所述第一标记分子、第二标记分子物理靠近时发生荧光能量共振转移。显著提升用于核酸检测的探针的信号差异,使得阳性检测信号和背景信号容易区分,并显著降低探针和引物的设计难度。
在一些实施例中,所述第一标记分子可以连接至所述探针寡核苷酸的3’端或5’端。
在一优选的实施例中,所述第一标记分子连接至所述探针寡核苷酸的3’端。
在一些实施例中,所述第二标记分子所连接的碱基靠近该碱基所在的引物寡核苷酸的3’端或5’端。
在一优选的实施例中,所述第二标记分子所连接的碱基靠近该碱基所在的引物寡核苷酸的3’端。
在一些实施例中,所述第二标记分子所连接的碱基距离其所在引物寡核苷酸的3’端8个碱基以内(包含本数),具体可以是8个碱基以内、7个碱基以内、6个碱基以内、5个碱基以内、4个碱基以内、3个碱基以内、2个碱基以内、1个碱基以内等等。
在一些实施例中,所述第一引物寡核苷酸与所述靶核苷酸序列的一条单链互补,所述第二引物寡核苷酸与所述靶核苷酸序列的另一条单链互补。
在一些实施例中,所述第二标记分子所连接的碱基为A、T、C、G碱基中的任一种。对于本发明的方法而言,各修饰碱基没有显著差别,只是dT修饰淬灭基团在合成上用的较多,可能跟原料的合成难度有关。即使引物3’端序列没有T,也可用替换的方式引入,对结果影响不大。
在一些实施例中,所述第二标记分子所连接的碱基为T碱基。
当目标序列存在时,标记有第一标记分子的所述探针寡核苷酸杂交于经靶核苷酸不对称PCR扩增得到的标记有第二标记分子的DNA单链,所述第一标记分子、第二标记分子物理靠近,相互作用,发生荧光能量共振转移,具体可以是发生荧光淬灭。当目标序列不存在时,所述第一标记分子、第二标记分子不会发生相互作用,进而不会发生荧光淬灭。
在一些实施例中,本发明通过单标记探针与荧光淬灭基团或荧光报告基团标记的DNA单链杂交产生荧光信号。本发明设计的单标记探针不会因构象变化产生非特异的荧光信号,背景更低。探针与DNA单链模板杂交产生的荧光信号变化也较传统方法更高,更稳定,这些特性使得该方法的信噪比更高,特异性更好。同时,探针也更加容易设计。
在一些实施例中,本发明的探针引物组合可用于各种基因分型检测,包括但不限于单核苷酸多态性(SNPs)检测、病毒基因分型等等。同时通过标记不同的荧光基团和淬灭基团,可以实现在不同荧光通道中的多重熔解曲线分析。探针寡核苷酸序列、第一引物寡核苷酸序列、第二引物寡核苷酸序列可根据具体的检测样本进行设计,探针、引物的结构简单,不存在茎-环结构,设计难度小。
在一些实施例中,所述探针寡核苷酸的序列长度为40nt以内,还可以为15-30nt。
在一些实施例中,所述探针寡核苷酸的序列长度包括但不限于5nt、6nt、7nt、8nt、9nt、10nt、11nt、12nt、13nt、14nt、15nt、16nt、17nt、18nt、19nt、20nt、21nt、22nt、23nt、24nt、25nt、26nt、27nt、28nt、29nt、30nt、31nt、32nt、33nt、34nt、35nt、36nt、37nt、38nt、39nt、40nt等等。
在一些实施例中,所述探针寡核苷酸的Tm值为55℃-90℃,还可以为60℃-80℃。
在一些实施例中,所述探针寡核苷酸的Tm值包括但不限于55℃、56℃、57℃、58℃、59℃、60℃、61℃、62℃、63℃、64℃、65℃、66℃、67℃、68℃、69℃、70℃、71℃、72℃、73℃、74℃、75℃、76℃、77℃、78℃、79℃、80℃、81℃、82℃、83℃、84℃、85℃、86℃、87℃、88℃、89℃、90℃等等。
上述Tm值又称熔解温度(melting temperature,Tm),是指核苷酸链的吸光值增加到最大值的一半时的温度。
在一些实施例中,所述第一引物寡核苷酸、第二引物寡核苷酸的序列长度均为30nt以内,还可以为20-25nt。
在一些实施例中,所述第一引物寡核苷酸、第二引物寡核苷酸的序列长度包括但不限于10nt、11nt、12nt、13nt、14nt、15nt、16nt、17nt、18nt、19nt、20nt、21nt、22nt、23nt、24nt、25nt、26nt、27nt、28nt、29nt、30nt等等。
在一些实施例中,所述靶核苷酸序列为单链核苷酸、双链核苷酸中的至少一种。
在一些实施例中,所述靶核苷酸序列为DNA序列、RNA序列中的至少一种。
在一些实施例中,所述靶核苷酸序列为单链核苷酸时,连接有所述第二标记分子的引物寡核苷酸互补于所述靶核苷酸序列的互补序列。
在一些实施例中,本发明所检测的对象,即靶核苷酸序列包括但不限于双链DNA、单链DNA序列、双链RNA序列、单链RNA序列等等,上述对象可以是从自然界提取,也可以是人工合成,通过一定的生物学手段,转化成可供检测的靶核苷酸序列。
在一些实施例中,不论靶核苷酸序列为单链还是双链,均可以在反应体系实现双链的合成,用于后续的不对称PCR,不用额外合成双链。设计引物探针也只需要单链序列即可。如果靶核苷酸序列为单链,可根据该单链及其互补链设计对应的上下游引物。将设计的引物加入反应体系后,在相关酶的作用下,会合成可用于不对称PCR的双链。
在一些实施例中,所述靶核苷酸序列可以由自然界中已有的物种分离出来或者通过人工的方法合成得到。
在一些实施例中,所述靶核苷酸序列包括但不限于冠状病毒。
冠状病毒为不分节段的单股正链RNA病毒,属于巢病毒目(Nidovirales)冠状病毒科(Coronaviridae)正冠状病毒亚科(Orthocoronavirinae),根据血清型和基因组特点冠状病毒亚科被分为α、β、γ和δ四个属,可以感染许多动物物种,包括人、蝙蝠、狗、猪、老鼠、鸟、牛、鲸、马、山羊、猴子等。已知感染人的冠状病毒有6种,包括α属的229E和NL63,β属的OC43和HKU1、中东呼吸综合征相关冠状病毒(MERSr-CoV)和严重急性呼吸综合征相关冠状病毒(SARSr-CoV)。冠状病毒检测中存在的假阳性将会导致严重误判。
在一些实施例中,所述靶核苷酸序列为冠状病毒229E型核酸。
本领域技术人员可以理解,上述靶核苷酸序列仅仅是示例性列举,本发明适用于各种基因分型检测,如单核苷酸多态性(SNPs)位点检测、病毒基因分型等等。
在一些实施例中,所述探针寡核苷酸含有如下核苷酸序列:
5’-CCATTGGCCACAACACCTGCACTTCC-3’(SEQ ID NO:1)。
在一些实施例中,所述第一引物寡核苷酸含有如下核苷酸序列:
5’-CCCCAGAGACCT(Int BHQ1 dT)GACCACAA-3’(SEQ ID NO:2)。
在一些实施例中,所述第二标记分子连接在SEQ ID NO:2所示序列中靠近3’的T碱基上。
在一些实施例中,所述第二标记分子为BHQ1。
在一些实施例中,所述第二引物寡核苷酸含有如下核苷酸序列:
5’-CACAAGCTCAGCAAATTGTGGATA-3’(SEQ ID NO:3)。
在一些实施例中,所述第一标记分子为荧光报告基团,所述第二标记分子为荧光淬灭基团,或,所述第一标记分子为荧光淬灭基团,所述第二标记分子为荧光报告基团。
在一些实施例中,所述第一标记分子为荧光报告基团,所述第二标记分子为荧光淬灭基团。
在一些实施例中,本发明涉及的荧光能量共振转移(FRET)现象为如下情况中的任意一种:1、当荧光基团与淬灭基团靠近时,其直观表现为荧光基团的荧光信号被淬灭,较其单独存在时信号大大降低;2、当两种不同的荧光基团离的较近,且其中一种基团(供体)的发射谱与另一种基团(受体)的激发谱有相当程度的重叠时,当供体被激发时,受体会因供体激发能的转移而被激发,其直观表现就是供体产生的荧光强度较其单独存在时要低的多,而受体发射的荧光却大大增强。
在另一些实施例中,所述第一标记分子为荧光淬灭基团,所述第二标记分子为荧光报告基团。
在一些实施例中,当连接有所述第一标记分子的所述探针寡核苷酸杂交于连接有所述第二标记分子的扩增产物时,所述第一标记分子靠近所述第二标记分子。
在一些实施例中,当连接有所述第一标记分子的所述探针寡核苷酸杂交于连接有所述第二标记分子的扩增产物,且所述探针寡核苷酸的3’段连接有第一标记分子时,所述探针寡核苷酸的杂交位置的3’端靠近所述第二标记分子,两个基团距离越近,淬灭的效果越好,信号越强。在一些实施例中,在20nt甚至更远都是有效的。当然在设计上,一般都能满足较短的距离。
在一些实施例中,所述扩增产物是在第一引物寡核苷酸、第二引物寡核苷酸存在的条件下,经所述靶核苷酸序列不对称PCR扩增得到的、连接有第二标记分子的单链DNA。
在一些实施例中,所述第一引物寡核苷酸为上游引物寡核苷酸,所述第二引物寡核苷酸为下游引物寡核苷酸。
在一些实施例中,所述探针寡核苷酸、第一引物寡核苷酸、第二引物寡核苷酸均根据靶核苷酸序列进行设计。
在一些实施例中,所述荧光报告基团包括但不限于FAM(6-carboxy-fluorescein,6-羧基荧光素,绿色)、HEX、VIC、ROX、Cy5等等,荧光报告基团可以是前述所列举基团中的至少一种,或者是其中的任一种。
在一些实施例中,所述荧光淬灭基团包括但不限于BHQ1(黑洞猝灭基团)、BHQ2等等,荧光淬灭基团可以是前述所列举基团中的至少一种,或者是其中的任一种。
本领域技术人员可以理解,上述探针寡核苷酸、第一引物寡核苷酸、第二引物寡核苷酸序列仅仅是示例性列举,在靶核苷酸序列不同时,探针寡核苷酸、第一引物寡核苷酸、第二引物寡核苷酸序列均根据靶核苷酸序列具体设计。
第二方面,一种实施例中提供一种检测试剂盒,其包括第一方面所述探针引物组合。
在一些实施例中,所述检测试剂盒包括不对称PCR反应体系。
在一些实施例中,所述不对称PCR反应体系选自PCR体系、逆转录PCR体系中的任一种。
在一些实施例中,所述逆转录PCR体系可以为一步法逆转录PCR体系,无需分多步进行,一步完成,操作简单。
在一些实施例中,所述PCR体系含有PCR Master Mix、热稳定的聚合酶、dNTPs等组分。
在一些实施例中,所述逆转录PCR体系含有RT-PCR Master Mix、热稳定的聚合酶、逆转录酶、dNTPs等组分。
在一些实施例中,本文提及的RT-PCR Master Mix、热稳定的聚合酶、逆转录酶、dNTPs均可从市场上购买得到。例如,可以购自美国Life公司、宝生物工程(大连)有限公司、珠海宝锐生物科技有限公司、深圳市菲鹏生物股份有限公司等等。
在一些实施例中,所述热稳定的聚合酶包括但不限于Taq DNA聚合酶、Tth DNA聚合酶中的至少一种。
在一些实施例中,所述不对称PCR反应体系中探针寡核苷酸的摩尔终浓度为200-800nM,包括但不限于200nM、250nM、300nM、350nM、400nM、450nM、500nM、550nM、600nM、650nM、700nM、750nM、800nM等等。通常情况下,探针浓度对结果的影响不大,在合理范围内即可,探针浓度过高,背景高,成本也高,一般会在不影响检测结果的前提下尽量降低探针浓度。
在一些实施例中,连接有第二标记分子的引物寡核苷酸的摩尔终浓度>未连接第二标记分子的引物寡核苷酸的浓度。
在一些实施例中,连接有所述第二标记分子的引物寡核苷酸的摩尔终浓度为未连接所述第二标记分子的引物寡核苷酸摩尔终浓度的5-20倍,包括但不限于5倍、6倍、7倍、8倍、9倍、10倍、11倍、12倍、13倍、14倍、15倍、16倍、17倍、18倍、19倍、20倍等等,优选为10倍。
在一些实施例中,连接有第二标记分子的引物寡核苷酸的浓度为200nM-800nM,包括但不限于200nM、250nM、300nM、350nM、400nM、450nM、500nM、550nM、600nM、650nM、700nM、750nM、800nM等等。
在一些实施例中,未连接第二标记分子的引物寡核苷酸的浓度为10nM-100nM,包括但不限于10nM、15nM、20nM、25nM、30nM、35nM、40nM、45nM、50nM、55nM、60nM、65nM、70nM、75nM、80nM、85nM、90nM、95nM、100nM。
在一些实施例中,所述试剂盒还包括模板、水。
在一些实施例中,所述模板可以为RNA模板、DNA模板。示例但非限制性地,模板具体可以是单链DNA病毒、单链RNA病毒,还可以是原核生物、原生生物、动物、植物、真菌的基因组DNA等等。
在一些实施例中,所述模板可以为人类或动物基因组DNA。
在一些实施例中,所述Taq DNA聚合酶在试剂盒中的终浓度为0.01-0.5U/μL,包括但不限于0.01U/μL、0.05U/μL、0.1U/μL、0.15U/μL、1U/μL、1.5U/μL、2U/μL、2.5U/μL、3U/μL、3.5U/μL、4U/μL、4.5U/μL、5U/μL等等。
在一些实施例中,所述逆转录酶在试剂盒中的终浓度为0.01-0.5U/μL,包括但不限于0.01U/μL、0.05U/μL、0.1U/μL、0.15U/μL、1U/μL、1.5U/μL、2U/μL、2.5U/μL、3U/μL、3.5U/μL、4U/μL、4.5U/μL、5U/μL等等。
在一些实施例中,所述检测试剂盒的总体积为10-100μL,包括但不限于10μL、15μL、20μL、25μL、30μL、35μL、40μL、45μL、50μL、55μL、60μL、65μL、70μL、75μL、80μL、85μL、90μL、95μL、100μL等等,优选为25μL。
第三方面,一种实施例中提供如第一方面所述探针引物组合或如第二方面所述检测试剂盒在基因分型检测中的应用。需要说明的是,此处的基因分型检测结果仅仅为中间参考结果,临床诊断中,还需要根据受试者的的主观感受症状、既往病史、家族遗传史等信息,才能得出最后的诊断结果或健康状况,因此,上述应用不属于疾病的诊断方法,更不属于疾病的治疗方法。上述应用还可以应用于其他多种非诊断治疗目的。
在一些实施例中,所述基因分型检测包括但不限于单核苷酸多态性(SNPs)位点检测、病毒基因分型。
SNPs,即单核苷酸多态性(Single Nucleotide Polymorphisms,简称SNPs),主要是指在基因组水平上由单个核苷酸的变异所引起的DNA序列多态性。
第四方面,一种实施例中提供如第一方面所述探针引物组合或如第二方面所述检测试剂盒在不对称熔解曲线PCR检测中的应用。
在一些实施例中,所述不对称熔解曲线PCR检测的反应程序如下:95℃,1-10min;45×(95℃,5-10s;55℃,30-60s);95℃,1min;55℃,1min;95℃-50℃连续收集荧光信号。
在一些实施例中,本发明的原理图如图1所示,本发明的单标记探针熔解曲线核酸检测方法包括:
1、通过在上游引物或下游引物靠近3’端的位置标记荧光淬灭基团,使得后续的PCR扩增产物的一条链上引入荧光淬灭基团。
2、探针序列和标记有荧光淬灭基团的上游引物序列分别互补于DNA的两条链。也可在下游引物靠近3’端的位置标记荧光淬灭基团,探针序列和标记有荧光淬灭基团的下游引物序列分别互补于DNA的两条链。
3、在探针的3’端标记荧光报告基团,探针的杂交位置需使3’端尽量靠近荧光淬灭基团,具体设计方法可参照图1所示原理图。
4、检测过程分为两步,第一步是不对称PCR过程,第二步是熔解曲线分析过程。
5、不对称PCR:当PCR反应体系中存在目标序列时(阳性样本),不对称PCR反应产生出大量带有荧光淬灭基团的DNA单链扩增产物;而当PCR反应体系中无目标序列时(阴性样本),PCR反应不会发生,无DNA单链扩增产物。
6、熔解曲线分析:如存在带有荧光淬灭基团的DNA单链扩增产物,在较低温度下,单标记探针与带有荧光淬灭基团的DNA单链杂交,荧光基团发出的荧光被互补DNA链上的淬灭基团吸收。当温度逐渐上升时,探针与DNA单链分子解链,探针上的荧光基团不再被互补链上的淬灭基团淬灭,荧光信号在熔解温度附近迅速增强。通过二阶导数法对熔解曲线分析,可以得到探针的熔解温度。而当PCR反应体系中无目标序列时,PCR反应不会发生,探针也不能特异的与单链DNA杂交,熔解曲线的二阶导数图没有特异的峰产生。
本发明的探针、引物设计遵循一般的探针、引物设计原则,比如,探针、引物设计原则可以参考《实时荧光PCR技术》(第二版,科学出版社,李金明著),具体设计可使用ABIPrimer Express等软件。
以下实施例的探针、引物均由申请人通过人工合成的方式获得。
以下实施例的模板来自病毒样本或人基因组样本。
实施例1
单标记探针熔解曲线法与双标记探针熔解曲线法结果比较
以冠状病毒229E型核酸检测为例,设计以下探针引物。分别采用单标记探针熔解曲线法和双标记探针熔解曲线法检测相同的病原核酸。两种方法的探针引物序列相同,但标记不同。
两种方法的探针、引物序列如表1所示。
表1
不对称PCR的反应体系如表2所示:
表2
反应程序如图2所示,具体如下:
50℃,30min;95℃,5min;45×(95℃,5s;55℃,40s);95℃,1min;55℃,1min;55℃-95℃连续收集荧光信号。
模板序列如下:
上述模板序列中,下划单直线标记的序列为与上游引物相同的序列,这部分序列的互补链互补于上游引物;下划单波浪线标记的序列为与探针序列互补的序列;下划双直线标记的序列为与下游引物互补的序列。
图3显示为单标记探针熔解曲线图,图4显示为双标记探针熔解曲线图。深黑色线为阳性样本检测结果,灰色线为阴性样本检测结果。
结果显示,单标记熔解曲线法,阳性样本和阴性样本的结果信号差异更大,特异性更好。而双标记探针,由于探针序列等问题,阴性样本产生了非特异性的熔解曲线峰,难以与阳性结果相区分。
实施例2
本实施例提供人类MTHFR基因rs1801133SNP位点检测。
设计以下探针引物序列,用于检测人类MTHFR基因上的rs1801133SNP位点(677C/T),该SNP位点与叶酸代谢能力有关。探针引物序列如表3所示。
表3
不对称PCR的反应体系如表4所示:
表4
模板序列(MTHFR_677C/T)如下:
上述模板序列中,“C(T)”为SNP位点,两个等位基因上,该位点的碱基分别为C、T。
上述模板序列中,下划单直线标记的序列为与上游引物相同的序列,这部分序列的互补链互补于上游引物;下划单波浪线标记的序列为与探针序列互补的序列;下划双直线标记的序列为与下游引物互补的序列。
反应程序为:95℃,5min;45×(95℃,5s;55℃,30s);95℃,1min;40℃,1min;40℃-80℃连续收集荧光信号。
图5-7为荧光探针溶解曲线法检测结果。
图5显示为MTHFR基因677C纯合子检测结果,探针杂交Tm值约为64.5℃。
图6显示为MTHFR基因677T纯合子检测结果,探针杂交Tm值约为55℃。
图7显示为MTHFR基因677C/T杂合子检测结果,杂交探针的Tm值约为54℃和64.5℃。
结果显示,本方法可以很好的分辨MTHFR基因677位的三种SNP类型。
实施例3及对比例1
本对比例参照公开号为CN 102321765 A(专利名称为《一种实时荧光PCR方法及用途》)的专利(以下简称对比专利)进行,设计与对比专利文件实施例2相同的探针、引物,检测SNP位点rs13182883。按照对比专利描述的方法合成探针引物。同时应用本发明的技术原理设计一条下游引物(rs13182883R2)作比较。
具体如表5所示。
表5
为了严格对比两种方法,两种方法共用探针Pb1和下游引物R1,只是上游引物不同。
不对称PCR的反应体系同表4。
反应程序参照对比专利方法设置:95℃,5min;40×(95℃,15s;55℃,30s;72℃,20s);95℃,1min;40℃,1min;45℃-90℃连续收集荧光信号。
检测人类基因组样本,结果如图8~图10所示。
图8显示为rs13182883A/A纯合子检测结果,R1:对比专利CN 102321765 A方法;R2:本发明实施例3的方法。
图9显示为rs13182883G/G纯合子检测结果,R1:对比专利CN 102321765 A方法;R2:本发明实施例3的方法。
图10显示为rs13182883A/G杂合子检测结果,R1:对比专利CN 102321765 A方法;R2:本发明实施例3的方法。
参照对比专利设计的颈环结构序列如下:
上述序列中,下划单直线标示的序列为与rs13182883R1(下游引物)相同的序列,该序列的互补链互补于rs13182883R1(下游引物);下划波浪线标示的序列为与rs13182883Pb1(探针)相同的序列,该序列的互补链互补于rs13182883Pb1(探针),其中,加粗的G碱基所对应的靶序列位点为变异位点;下划双直线所标示的序列为与rs13182883F2(上游引物2)互补的序列。
采用对比专利、本实施例3的方法对同一DNA模板进行检测,DNA模板序列信息如下:
rs13182883[Homo sapiens]
Variant type:
SNV。
Alleles:
G>A[Hide Flanks]
ATGTTTTAAGGAGACTATGAGGTGTGTCTCTCTTTTGTGAGGGGAGGGGT;
CCCTTCTGGCCTAGTAGAGGGCCTGGCCTGCAGTGAGCATTCAAATCCTC。
[G/A]
AGGAACAGGGTGGGGAGGTGGGACAAAGGCAGGAAGAAAGTAACGGAGAG;
CCTGGGGAGACATGGTAGGGCACAAACATGAGCAGACCAAGGATTGTCAG。
Chromosome:
5:137297649(GRCh38);
5:136633338(GRCh37)。
Gene:
SPOCK1(Varview)。
Functional Consequence:
intron_variant。
Validated:
by frequency,by cluster。
MAF:
A=0.288333/173(NorthernSweden);
A=0.29308/1313(Estonian);
A=0.357605/1326(TWINSUK);
A=0.36274/1398(ALSPAC);
A=0.371427/11616(GnomAD);
A=0.398031/49980(TOPMED);
A=0.415735/2082(1000Genomes);
A=0.425119/33439(PAGE_STUDY)。
HGVS:
NC_000005.10:g.137297649G>A,NC_000005.9:g.136633338G>A,NG_034127.1:g.206681C>T。
搜索引擎:PubMedLitVar。
结合图8~图10的检测结果可以看出,本发明实施例3的信号更强。并且,对比专利的方法在探针引物设计上较为复杂,要求产物自身形成二级结构,且二级结构的Tm值需大于探针Tm值,SNP位点临近序列Tm值较低,可能对探针引物设计带来一定的难度和局限性,探针Tm值很难自由选择。
在一实施例中,本发明通过单荧光标记探针(或荧光淬灭基团标记探针),与荧光淬灭基团(或荧光报告基团)标记的DNA单链杂交产生荧光信号。本发明中涉及的单标记探针,不会因构象变化产生非特异的荧光信号,背景更低。探针与DNA单链模板杂交产生的荧光信号变化也较传统方法更高,更稳定,这些特性使得该方法的信噪比更高,特异性更好。同时,探针也更加容易设计。
在一些实施例中,本发明在探针和引物上分别引入荧光基团和淬灭基团,可通过不对称PCR技术扩增出大量单链DNA分子,探针与标记有淬灭基团的单链DNA分子杂交后可产生荧光信号的变化,通过熔解曲线法检测和分析目的序列。
以上应用了具体个例对本发明进行阐述,只是用于帮助理解本发明,并不用以限制本发明。对于本发明所属技术领域的技术人员,依据本发明的思想,还可以做出若干简单推演、变形或替换。
Organization Applicant
----------------------
Street :
City :
State :
Country :
PostalCode :
PhoneNumber :
FaxNumber :
EmailAddress :
<110> OrganizationName : 深圳澳东检验检测科技有限公司
Application Project
-------------------
<120> Title : 一种探针引物组合及其检测试剂盒
<130> AppFileReference : 20I25365
<140> CurrentAppNumber :
<141> CurrentFilingDate : ____-__-__
Sequence
--------
<213> OrganismName : Artificial Sequence
<400> PreSequenceString :
ccattggcca caacacctgc acttcc 26
<212> Type : DNA
<211> Length : 26
SequenceName : 1
SequenceDescription :
Sequence
--------
<213> OrganismName : Artificial Sequence
<400> PreSequenceString :
ccccagagac cttgaccaca a 21
<212> Type : DNA
<211> Length : 21
SequenceName : 2
SequenceDescription :
Sequence
--------
<213> OrganismName : Artificial Sequence
<400> PreSequenceString :
cacaagctca gcaaattgtg gata 24
<212> Type : DNA
<211> Length : 24
SequenceName : 3
SequenceDescription :
Sequence
--------
<213> OrganismName : Artificial Sequence
<400> PreSequenceString :
cagcctaatg atgatgtgac atctaatgtc acacaatgtt ttggccccag agaccttgac 60
cacaactttg gaagtgcagg tgttgtggcc aatggtgtta aagctaaagg ctatccacaa 120
tttgctgagc ttgtgccgtc aacagctgct atgctgtttg atagtcacat tgtttccaa 179
<212> Type : DNA
<211> Length : 179
SequenceName : 4
SequenceDescription :
Sequence
--------
<213> OrganismName : Artificial Sequence
<400> PreSequenceString :
tgtctgcggg agccgatttc a 21
<212> Type : DNA
<211> Length : 21
SequenceName : 5
SequenceDescription :
Sequence
--------
<213> OrganismName : Artificial Sequence
<400> PreSequenceString :
gagctttgag gctgacctga a 21
<212> Type : DNA
<211> Length : 21
SequenceName : 6
SequenceDescription :
Sequence
--------
<213> OrganismName : Artificial Sequence
<400> PreSequenceString :
tcaaagaaaa gctgcgtgat ga 22
<212> Type : DNA
<211> Length : 22
SequenceName : 7
SequenceDescription :
Sequence
--------
<213> OrganismName : Artificial Sequence
<400> PreSequenceString :
caggtggagg ccagcctctc ctgactgtca tccctattgg caggttaccc caaaggccac 60
cccgaagcag ggagctttga ggctgacctg aagcacttga aggagaaggt gtctgcggga 120
gctcgatttc atcatcacgc agcttttctt tgaggctgac acattcttcc gctttgtgaa 180
ggcatgcacc gacatgggca tcacttgccc catcgtcccc gggatggcat agtggggtgg 240
tgaata 246
<212> Type : DNA
<211> Length : 246
SequenceName : 8
SequenceDescription :
Sequence
--------
<213> OrganismName : Artificial Sequence
<400> PreSequenceString :
accctgttcc tcgaggattt ga 22
<212> Type : DNA
<211> Length : 22
SequenceName : 9
SequenceDescription :
Sequence
--------
<213> OrganismName : Artificial Sequence
<400> PreSequenceString :
caggctctcc gttactttct tc 22
<212> Type : DNA
<211> Length : 22
SequenceName : 10
SequenceDescription :
Sequence
--------
<213> OrganismName : Artificial Sequence
<400> PreSequenceString :
ggatgctcac tgcctagtag agggcctggc ct 32
<212> Type : DNA
<211> Length : 32
SequenceName : 11
SequenceDescription :
Sequence
--------
<213> OrganismName : Artificial Sequence
<400> PreSequenceString :
ctggcctgca gtgagcatt 19
<212> Type : DNA
<211> Length : 19
SequenceName : 12
SequenceDescription :
Sequence
--------
<213> OrganismName : Artificial Sequence
<400> PreSequenceString :
cacctcgatt gaagacattc actgacctcc tagatcttag tgagataact tctgacaatc 60
cttggtctgc tcatgtttgt gccctaccat gtctccccag gctctccgtt actttcttcc 120
tgcctttgtc ccacctcccc accctgttcc tcgaggattt gaatgctcac tgcaggccag 180
gccctctact aggccagaag ggacccctcc cctcacaaaa gagagacaca cctcatagtc 240
tccttaaaac atagtctgat acttatcgtt gttaaaacaa t 281
<212> Type : DNA
<211> Length : 281
SequenceName : 13
SequenceDescription :