基于全基因组选择的大黄鱼抗刺激隐核虫病的选育方法
文献发布时间:2023-06-19 09:43:16
技术领域
本发明属于动物抗病育种领域,尤其是涉及利用全基因组选择技术进行大黄鱼抗刺激隐核虫培育的一种基于全基因组选择的大黄鱼抗刺激隐核虫病的选育方法。
背景技术
大黄鱼(Pseudosciaena crocea),隶属于鲈形目(Perciformes),石首鱼科(Sciaenidae),黄鱼属(Larimichthys),主要分布于太平洋西南海域,即中国东南沿海区域。大黄鱼的野生资源由于过度捕捞趋于枯竭,但在科技工作者突破大黄鱼的人工繁殖后,其养殖规模逐年增加,现以成为中国东南沿海各省,尤其是福建省的主要养殖的海水品种,也是中国海水养殖单产量最高的养殖品种。近些年来,由于大黄鱼养殖产业膨胀式的发展,导致沿海内湾网箱养殖区域的水质极度恶化,导致病害频发。在诸多病害中,刺激隐核虫造成的危害最大,俗称白点病,每年的春夏转交之际,由于水温突升,导致其爆发,给大黄鱼养殖产业造成了巨大的经济损失。至今,没有有效的药物可以治疗白点病,而通过遗传选育提升大黄鱼刺激隐核虫抗性便具有极大的实践意义,也避免了药物治疗对水域的进一步污染。
遗传育种的发展经过了漫长了历史,育种技术在不断的革新,使其能够满足不同需求的选育目的。现代育种技术的代表作之一,而且还在蓬勃发展的全基因组选择技术(GS),自从2002年提出以来,经过快20年的发展,已经在动植物遗传选育研究中得到了广泛的应用,使一些遗传力较低,较难测量的性状的选育成为了可能,例如抗病抗逆等性状。在水产动物的抗病遗传选育领域中,很少存在有将GS技术成功应用的案例,而仅仅局限于理论研究。在此技术背景下,本发明聚焦大黄鱼抗刺激隐核虫遗传改良,并将GS技术应用其中,形成一套独特的培育流程,实现了理论成果向实践应用转化。本发明需要大量的测序任务,虽然成本较高,但是随着测序技术的更新换代,测序成本在急速的下降,导致个体分型成本也在下降,这也使本发明在实践过程中,成本可进一步降低。相比于本发明的资金投入,其减少的刺激隐核虫的对养殖产业造成的经济损失是不可估量的。而且,本发明是一个完整的体系,操作清晰,实用性强,可以在水产动物抗病育种工作中进行推广。
发明内容
本发明的目的在于提供有效解决复杂性状选育中亲本无法进行抗性测评等问题的一种基于全基因组选择的大黄鱼抗刺激隐核虫病优良品系的选育方法。
本发明包括以下步骤:
1)建立参考群体;
2)对参考群体进行抗性性状测量;
3)建立选育群体;
4)对参考群体和选育群体进行基因分型;
5)建立最佳全基因组选择模型;
6)对选育群体的基因组育种值(GEBV)进行估算;
7)以一定的选择强度选取选育群体GEBV值排在前面的个体作为亲鱼,并繁殖产生抗刺激隐核虫子一代;
8)对抗性子一代进行刺激隐核虫攻毒验证。
在步骤1)中,所述建立参考群体的具体步骤可为:
①选取来源广泛的大黄鱼2龄健康性成熟亲鱼至少500尾,保证其基因多样性;
②亲鱼不经过刺激隐核虫攻毒挑选,强化培育后,催产并繁殖后代;
③后代即为参考群体,按照商品鱼培育体系进行参考群体的养殖。
在步骤2)中,所述对参考群体进行抗性性状测量的具体步骤可为:
①选取参考群体6月龄幼鱼至少2000尾,将这批鱼进行岸上养殖室内暂养,按照正常的室内养殖规章进行管理,暂养时间为两周;
②培育刺激隐核虫幼虫,并在孵化高峰期计数;
③从暂养的上述大黄鱼个体中,选取至少1000尾进行攻毒实验,分为四组,每组放入3t规格的实验桶,攻毒浓度为每5升水每尾鱼5000个刺激隐核虫幼虫;
④自攻毒起,开始不间断的跟踪每组鱼的攻毒状况;将失去平衡,且10s内不能恢复的个体视为死亡样本,记录死亡时间,并将其捞出实验桶外,剪其背部肌肉保存于纯酒精中;观察时间持续120h,实验结束后将所有存活的个体转移至新的实验桶,进行攻毒后恢复;恢复两周后,仍然存活的个体视为存活样本;如此,大黄鱼抗刺激隐核虫的表型性状采集模式为二分类性状,存活-死亡。
在步骤3)中,所述建立选育群体的具体步骤可为:
①选取即将性成熟且健康的2龄大黄鱼至少500尾作为选育群体;
②对上述选育群体打入电子标签,并剪取鳍条保存于酒精;
③将其转移入新的网箱进行产前强化培养。
在步骤4)中,所述对参考群体和选育群体进行基因分型的具体步骤可为:
①选取保存在酒精中的参考群体死亡样本和存活样本中的鳍条至少各200尾,选育群体样本至少350尾进行DNA提取,方法为酚氯仿法;
②对DNA样品进行基因分型,可以采取任何形式的测序策略,包括简化基因组测序,全基因组重测序,基因芯片等;
③将原始分型数据经过进一步清洗和质控,最终获得可以进行全基因组选择育种分析的可用分型数据集。
在步骤5)中,所述建立最佳全基因组选择模型的具体步骤可为:
①基于参考群体的二分类抗性表型和基因型进行全基因组关联分析,所使用软件为R语言环境下的gaston包,得到所有SNP位点的显著性P值,并将其由小到大排序;
②使用R语言环境下BGLR包中的Bayesian LASSO分析模型建立全基因组选择模型,以10个标记为增量,按照已经排好序的SNP标记,逐渐增加排在前面的标记使用数量,并利用5折交叉验证计算每个标记量下的模型预测准确性;
③判断模型预测准确性最高时,对应的标记数量,相应的此标记密度下的建立的模型即是最佳的全基因组选择模型。
在步骤6)中,所述对选育群体的基因组育种值(GEBV)进行估算的具体步骤可为:
①在最佳全基因组选择模型下,对选育群体的GEBV进行估计,并对其进行排名;
②利用人类中应用十分广泛的多基因风险评分来佐证上述得到的GEBV值的准确性,所使用的软件为PRSice软件。
在步骤7)中,所述以一定的选择强度选取选育群体GEBV值排在前面的个体作为亲鱼,并繁殖产生抗刺激隐核虫子一代的具体步骤可为:
①按照一定的选择强度,选择选育群体GEBV排在前面的个体作为亲鱼繁殖后代,如此,这些亲鱼的后代即为抗性子一代;
②按照正常的商品鱼养殖体系培育抗性子一代。
在步骤8)中,所述对抗性子一代进行刺激隐核虫攻毒验证的具体步骤可为:
①在刺激隐核虫爆发的季节,对抗虫子一代进行攻毒实验,实验流程同参考群体的抗性性状测评的攻毒流程一致;
②与对照组相比,评价其抗虫性。
本发明提供了一种基于全基因组选择的大黄鱼抗刺激隐核虫病优良品系的选育方法,利用本发明提供的培育流程,可直接将其应用到大黄鱼抗刺激隐核虫的选育工作中,也为后续培育稳定的抗虫品系提供最初的原始群体,即抗性子一代。与现有技术相比,本发明至少有以下优点及技术效果:
(1)本发明首次将GS技术应用到大黄鱼抗刺激隐核虫遗传育种中,有效的解决了复杂性状选育中亲本无法进行抗性测评的问题。
(2)本发明在两年之内就可获得抗虫子一代,大大缩短了育种年限。
(3)本发明得到的抗虫子一代的抗虫性能显著高于对照组。
(4)本发明可大大降低刺激隐核虫对大黄鱼养殖产业造成的经济损失。
附图说明
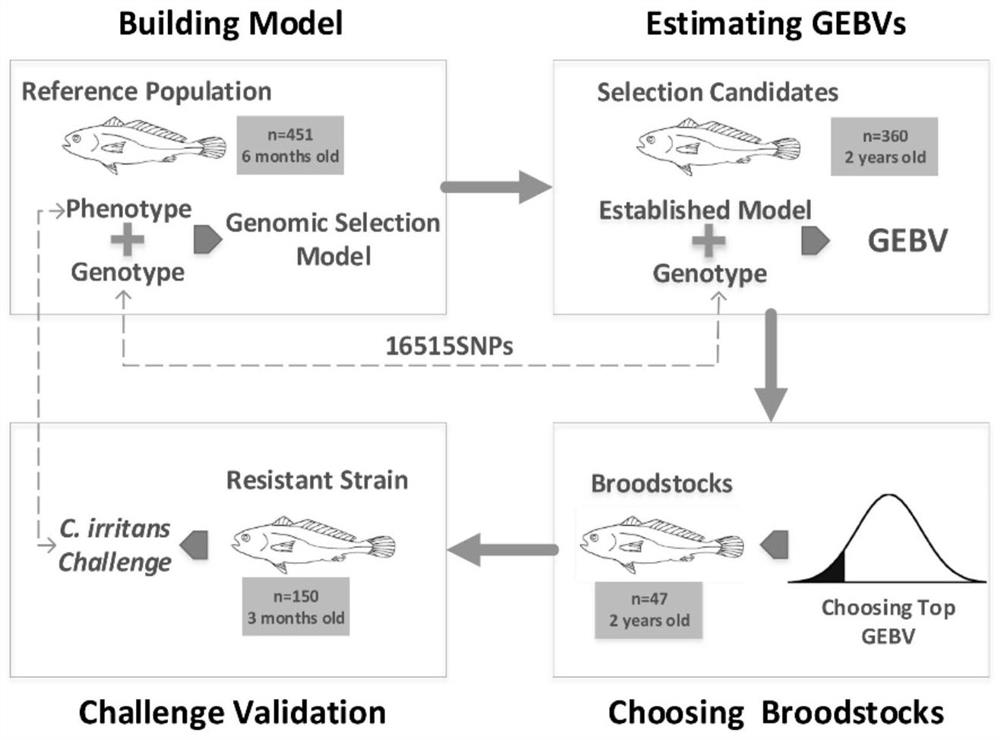
图1为本发明在具体实施过程中的示意图。
图2为本发明在具体实施过程中第⑵个步骤中参考群体抗性性状测量中死亡个体的死亡时间分布图。
图3为本发明在具体实施过程中第⑸个步骤中判断全基因组模型最佳标记的结果图。横坐标表示标记数量,图中上方的图中纵坐标代表模型拟合度(Goodness of Fit),下方的图中纵坐标表示模型预测准确性(predictability)。
图4为本发明在具体实施过程中第⑹个步骤中使用PRSice软件对R语言BGLR包得到的GEBV结果的佐证结果。横坐标表示使用BGLR包得到的选育群体的死亡概率,纵坐标表示使用PRSice软件得到的选育群体多基因死亡风险评分。
图5为本发明在具体实施过程中第⑻个步骤中抗虫子一代的刺激隐核虫攻毒验证结果。横坐标代表死亡时间,纵坐标代表生存概率,CS代表对照组,RS代表抗性子一代。
具体实施方式
下面将结合附图对本发明作进一步的说明和详细描述。
参见图1~5,本发明实施例包括以下步骤:
⑴建立参考群体;
①2017年12月份在福建某海域网箱养殖区,从各鱼排上选取来源广泛的大黄鱼2龄健康性成熟亲鱼500尾,亲鱼来源广泛保证其基因多样性。
②亲鱼不经过刺激隐核虫攻毒挑选,强化培育后,催产并繁殖后代。
③这批亲鱼的后代即为参考群体,按照商品鱼培育体系进行参考群体的养殖。
⑵对参考群体进行抗性性状测量;
①2018年6月中旬选取参考群体6月龄幼鱼4000尾作为攻毒实验材料,鱼的体重规格为6.80±2.52g,体长规格为7.8±1.1cm。将这批鱼进行岸上养殖室内暂养,按照正常的室内养殖规章进行管理,暂养时间为两周。
②培育刺激隐核虫幼虫,具体流程为:收集网箱养殖患病的大黄鱼,将其转移至岸上的养殖桶内;待其包囊脱落并粘附于桶底后,用刷子将包囊与桶底分离,并将其收集并清洗之后转移到1000mL大烧杯中,杯中注满过滤海水,并将其放置于避光的恒温水浴锅中孵化,温度调节为26℃;期间每隔2h观察包囊孵化情况,大约2天就可达到孵化高峰期,此时,利用显微镜计数孵化的幼虫密度。
③从暂养的实验鱼中,选取2000尾进行攻毒实验,分为四组,攻毒实验前一天将其放入3t规格的实验桶,并停止投喂。攻毒时,将计算好的携带有幼虫的培养液倒入实验桶内,并搅拌均匀,攻毒浓度为每5升水每尾鱼5000个刺激隐核虫幼虫。
④自攻毒起,开始不间断的跟踪每组鱼的攻毒状况。将失去平衡,且10s内不能恢复的个体视为死亡样本,记录死亡时间,并将其捞出实验桶外,剪其背部肌肉保存于纯酒精中。观察时间持续120h,实验结束后将所有存活的个体转移至新的实验桶,进行攻毒后恢复。恢复两周后,仍然存活的个体视为存活样本。如此,大黄鱼抗刺激隐核虫的表型性状采集模式为二分类性状,存活-死亡。
⑶建立选育群体;
①2018年12月份中旬,在福建某海域网箱养殖区选取即将性成熟且体表无伤口健康的2龄大黄鱼500尾作为选育群体。
②对上述选育群体打入电子标签,并剪取鳍条保存于酒精。
③进行完上述工作后,将其转移入新的网箱进行产前强化培养,而这些亲鱼即是抗虫子一代的候选亲鱼群体。
⑷对参考群体和选育群体进行基因分型;
①选取保存在酒精中的参考群体死亡样本和存活样本中的鳍条各240尾,还有384尾选育群体样本进行DNA提取,方法为酚氯仿法。
②对DNA样品进行建库测序,建库方式为ddRAD,测序平台为HiSeq2000。
③将原始测序数据经过清洗,获得纯净测序数据,利用stacks2软件将所有混在一起的个体基于不同的barcode分开;之后利用bwa程序将每个个体的测序clean reads与大黄鱼基因组(PRJNA505758)比对,之后利用samtools软件统计测序深度、基因组覆盖率和基因组比对率;之后使用stacks2软件对所有个体进行分型工作,生成vcf文件;之后利用plink软件对分型后的SNP位点进行质控,质控条件为SNP位点检出率>0.9(geno=0.1),个体SNP位点检出率>0.9(mind=0.1),SNP最小等位基因频率>0.05(maf=0.05);之后利用beagle4软件进行缺失基因型填补;最后使用haploview软件搜索tag SNP(高度连锁区域的代表SNP),最终获得可以进行全基因组选择育种分析的可用分型数据集。可用分型数据集包括16515个SNP,以及811个个体,包括451个参考群体个体以及360个选育群体个体。这部分工作在Linux系统下运行,以下是这部分工作的shell执行脚本。
##使用stacks2软件进行分个体工作
pre="path"
n=0
cmd="#!/bin/bash\n"
for i in`ls$pre|grep FKD`
do
f=$(echo$i|awk'{x=substr($1,1,4);y=substr($1,26,1);print x""y}')
cmd=$cmd"process_radtags-P-p$pre/$i/-b$pre/clean.data/adaptor.list/${f}_new_new1-o$pre/clean.data/140bp/--inline_null-c-r-q--renz_1ecoRI-t 130--barcode_dist_2 1-i gzfastq\n"
n=$(($n+1))
if[[$n-ge 16]]
then
echo-e$cmd|qsub-N${f}.tag-o$pre/clean.data/log/rad.tag.log/${f}.log-joe-l nodes=2:ppn=4-q high-
cmd="#!/bin/bash\n"
n=0
fi
done
##利用bwa程序与大黄鱼基因组比对并形成bam文件,利用samtools软件生成.sort和.depth文件
pre="path"
n=0
cmd="#!/bin/bash\n"
for i in`ls$pre/130bp|grep rem-v|grep fq|sed's/\..\.fq\.gz//g'|grepfq-v|sort-k 1,1-u`
do
cmd=$cmd\
bwa aln LG_Chr_genome.fasta$pre/130bp/${i}.1.fq.gz-k 2-t30>$pre/130bp/${i}_read1.sai\nbwa aln LG_Chr_genome.fasta$pre/130bp/${i}.2.fq.gz-k 2-t 30>$pre/130bp/${i}_read2.sai\n bwa sampeLG_Chr_genome.fasta$pre/130bp/${i}_read1.sai$pre/130bp/${i}_read2.sai$pre/130bp/${i}.1.fq.gz$pre/130bp/${i}.2.fq.gz|samtools view-bS->$pre/bam/${i}.bam\n cmd=$cmd"samtools sort-o$pre/sort/${i}.sort$pre/bam/${i}.bam\n/public/tools/sam_utils/samtools-1.8/samtoolsdepth$pre/sort/${i}.sort>$pre/depth/${i}.depth\n"
n=$(($n+1))
if[[$n-ge 49]]
then
echo-e$cmd|qsub-N${i}.process-o$pre/log/process.log/${i}.log-j oe-lnodes=2:ppn=4-q high-
cmd="#!/bin/bash\n"
n=0
fi
done
##使用samtools软件计算基因组比对率、测序深度以及基因组覆盖率
pre="path"
f=""
for i in`ls$pre/bam|sed's/\.bam//g'`
do
f=$f"$i\n"
echo$f
samtools-1.8/samtools flagstat$pre/bam/${i}.bam>>$pre/stat.summary.total/mapping.raw
done
grep"mapped("$pre/stat.summary.total/mapping.raw|awk'{print$5}'|sed's/(//g'>$pre/stat.summary.total/txt
echo-e$f|paste-$pre/stat.summary.total/txt>$pre/stat.summary.total/mappingratio
size=`grep-v">"LG_Chr_genome.fasta|tr"\n""@"|sed's/@//g'|wc|awk'{print$3}'`
for i in`ls$pre/depth|sed's/\.depth//g'`
do
awk-v a=$size-v id=$i'BEGIN{sum=0}{sum+=$3}END{print id"\t"NR/a"\t"sum/NR}'$pre/depth/${i}.depth>>$pre/stat.summary.total/depth
done
##使用stacks2软件进行基因分型工作,并生成vcf文件
pre="path"
stacks2/bin/ref_map.pl-T 6-o$pre/stacks--popmap$pre/popmap--samples$pre/
stacks2/bin/populations-O$pre/population--vcf-M$pre/popmap-P$pre/stacks##利用plink软件基因分型后的数据质控
pre="path"
vcftools--vcf$pre/populations.snps.vcf--plink--out$pre/lc.ci.final
plink--file$pre/lc.ci.final--allow-extra-chr-geno 0.10--recode--out$pre/lc.ci.final
plink--file$pre/lc.ci.final--allow-extra-chr-mind 0.10--recode--out$pre/lc.ci.final
plink--file$pre/lc.ci.final--allow-extra-chr-maf 0.05--recode--out$pre/lc.ci.final
##利用beagle4软件进行缺失基因型填补
pre="path"
plink--file$pre/lc.ci.final--allow-extra-chr--make-bed--out$pre/lc.ci.final plink--bfile$pre/lc.ci.final--allow-extra-chr--recode vcf-iid--out$pre/lc.ci.final
java–jarbeagle/beagle4.jar gt=$pre/lc.ci.final.vcf out=$pre/lc.ci.input.final nthreads=30
gunzip$pre/lc.ci.input.final.vcf.gz
vcftools--vcf$pre/lc.ci.input.final.vcf--plink--out$pre/lc.ci.input.final
##利用Haploview软件找Tag SNP
pre="path"
mkdir$pre/LG
for i in$(seq 1 25)
do
plink--file$pre/lc.ci.input.final--allow-extra-chr--chr LG${i}--recode--out$pre/LG/LG${i}
awk'{print$2"\t"$4}'$pre/LG/LG${i}.map>$pre/LG/LG${i}.map1
mv$pre/LG/LG${i}.map1$pre/LG/LG${i}.map
sed-i's/-9/0/g'$pre/LG/LG${i}.ped
java-jar haploview/Haploview.jar-nogui-pedfile$pre/LG/LG${i}.ped-info$pre/LG/LG${i}.map-pairwiseTagging-log$pre/LG/LG${i}.log-out$pre/LG/LG${i}
cat$pre/LG/LG${i}.TESTS>>$pre/tag.SNP
done
plink--file$pre/lc.ci.input.final--allow-extra-chr--extract$pre/tag.SNP--recode--out$pre/lc.ci.input.tag.final
plink--file$pre/lc.ci.input.tag.final--allow-extra-chr--maf 0.05--recode--out$pre/lc.ci.input.tag.final
⑸建立最佳全基因组选择模型;
①基于参考群体的二分类抗性表型和基因型进行全基因组关联分析,所使用软件为R语言环境下的gaston包,得到所有SNP位点的显著性P值,并将其由小到大排序。
②使用R语言环境下BGLR包中的Bayesian LASSO分析模型建立全基因组选择模型,以10个标记为增量,按照已经排好序的SNP标记,逐渐增加排在前面的标记使用数量,并利用5折交叉验证计算每个标记量下的模型预测准确性。5折交叉验证将参考群体随机分为5份,其中4份为训练集,1份为测试集,即利用训练集建立的模型来预测测试集,预测准确性为测试集的GEBV与真实的观察值之间的相关系数的平方。最终发现,在600个标记下,预测准确性就达到最高。
③判断模型预测准确性最高时,对应的标记数量,相应的此标记密度下的建立的模型即是最佳的全基因组选择模型。
这部分分析工作在linux系统下运行,并配合R语言脚本。以下是具体的执行脚本。
##shell脚本,生成R语言脚本所必须的输入文件
pre="path";file="lc.ci.input.tag.final"
awk'{if(/^[34]/){$6=2;print}else if(/^S/){$6=1;print}else{$6="NA";print}}'$pre/${file}.ped>$pre/test.ped
mv$pre/test.ped$pre/${file}.ped
awk'{if(/HX/){}else{print$1"\t"$2}}'$pre/${file}.ped>$pre/reference.id
plink--file$pre/$file--allow-extra-chr--keep$pre/reference.id--recode--out$pre/${file}.ref#生成只有参考群体的plink格式
plink--file$pre/${file}.ref--allow-extra-chr--maf 0.05--recode--out$pre/${file}.ref
cut-f 2$pre/${file}.ref.map>$pre/test.id
plink--file$pre/$file--allow-extra-chr--extract$pre/test.id--recode--out$pre/$file
rm$pre/test.id
plink--file$pre/$file--allow-extra-chr--make-bed--recode--out$pre/$file
plink--file$pre/$file--allow-extra-chr--freq--out$pre/$file
awk'{printf$1"\t";for(i=7;i<=NF;i=i+2){printf"%s%s\t",$i,$(i+1)};print""}'$pre/${file}.ped|sed's//\//g'|awk'{if(NR==FNR){a[FNR-1]=$3}else{printf$1;for(i=2;i<=NF;i++){split($i,b,"/");printf""(a[i-1]==b[1])+(a[i-1]==b[2])};print""}}'$pre/${file}.frq->$pre/${file}.geno
printf"\nstart generating pheno-data\n"#产生表型文件
awk'{print$1"\t"$6}'$pre/${file}.ped>$pre/${file}.pheno
Rscript$pre/script/binary.gs.best.marker.R$pre/$file
##R语言脚本,binary.gs.best.marker.R。主要分析任务是判断最佳标记数量。
args<-commandArgs(T)
lc.geno<-read.table(paste(args[1],".geno",sep=""),sep="")
lc.map<-read.table(paste(args[1],".map",sep=""),sep="\t")
lc.pheno<-read.table(paste(args[1],".pheno",sep=""),sep="\t")
colnames(lc.pheno)<-c("Taxa","staus")
lc.map<-lc.map[c(2,1,4)]
colnames(lc.map)<-c("SNP","Chromosome","Position")
colnames(lc.geno)<-c("taxa",as.character(lc.map$SNP))
rownames(lc.geno)<-lc.geno$taxa
lc.geno<-lc.geno[-1]
rownames(lc.pheno)<-lc.pheno$Taxa
all.id<-rownames(lc.pheno)
ref.id<-all.id[-grep("HX",all.id)]
sur.id<-ref.id[grep("S",ref.id)];death.id<-ref.id[-grep("S",ref.id)]
lc.pheno[sur.id,2]<-1;lc.pheno[death.id,2]<-0
head(lc.pheno)
lc.geno[1:5,1:5]
library(BGLR);library(sommer);library(gaston)
lc.bim<-read.table(paste(args[1],".bim",sep=""),sep="\t")
lc.fam<-read.table(paste(args[1],".fam",sep=""),sep="")
names(lc.bim)<-c("chr","id","dist","pos","A1","A2")
names(lc.fam)<-c("famid","id","father","mother","sex","pheno")
rownames(lc.fam)<-lc.fam$id
lc.fam[sur.id,6]<-1;lc.fam[death.id,6]<-0
lc.bim$chr<-gsub("LG","",lc.bim$chr)
options(gaston.auto.set.stats=
TRUE,gaston.autosomes=c(1:25),gaston.chr.x=FALSE,gaston.chr.y=FALSE)
lc.geno<-as.matrix(lc.geno)
lc.geno1<-lc.geno[ref.id,]
lc.fam1<-lc.fam[ref.id,]
lc.bed.matrix<-as.bed.matrix(lc.geno1,lc.fam1,lc.bim)
gaston.result<-association.test(lc.bed.matrix,K=GRM(lc.bed.matrix),method="lmm",response="binary")
head(gaston.result)
plink.result<-read.table(paste(args[1],".ref.assoc",sep=""),sep="",header=TRUE)cor(gaston.result$p,plink.result$P)
write.table(gaston.result,paste(args[1],".gaston.result",sep=""),quote=F,row.names=F)
gaston.result<-gaston.result[order(gaston.result$p),]
cat("\n--------OK-------\n")
library(foreach)
library(doParallel)
cl<-makeCluster(40)
registerDoParallel(cores=40)
lc.pheno1<-lc.pheno[ref.id,]
y<-10
x<-floor(nrow(lc.map)/y)
z<-floor(nrow(lc.pheno1)/5)
m<-40
cor<-list()
fit<-list()
for(j in 1:x){
geno<-lc.geno1[,as.character(gaston.result$id[1:(y*j)])]
foreach(i=1:m)%dopar%sample(1:nrow(lc.geno1),z)->tst
foreach(i=1:m)%dopar%lc.pheno1->trait
for(n in 1:m){trait[[n]][tst[[n]],2]<-NA}
foreach(i=1:m)%dopar%BGLR(y=trait[[i]][,2],response_type='ordinal',ETA=list(list(X=geno,model='BL')),nIter=1200,burnIn=200,verbose=F)->ans
foreach(i=1:m,.combine="c")%dopar%cor(ans[[i]]$probs[-tst[[i]],2],lc.pheno1[-tst[[i]],2])->fittness
foreach(i=1:m,.combine="c")%dopar%cor(ans[[i]]$probs[tst[[i]],2],lc.pheno1[tst[[i]],2])->predict
fit[[j]]<-fittness;cor[[j]]<-predict
cat(paste("\nthis is",j,"time\n\n"))
}
stopCluster(cl)
predict<-data.frame(marker=sapply(cor,"[",1))
for(i in 1:m){predict[i]<-sapply(cor,"[",i)}
names(predict)<-paste("rep",1:m,sep="")
predict<-data.frame(number=(1:x)*y,predict)
fitness<-data.frame(marker=sapply(fit,"[",1))
for(i in 1:m){fitness[i]<-sapply(fit,"[",i)}
names(fitness)<-paste("rep",1:m,sep="")
fitness<-data.frame(number=(1:x)*y,fitness)
write.table(predict,paste(args[1],".predict",sep=""),sep="\t",quote=F,row.names=F)
write.table(fitness,paste(args[1],".fitness",sep=""),sep="\t",quote=F,row.names=F)
⑹对选育群体的基因组育种值(GEBV)进行估算;
①在最佳全基因组选择模型下,对选育群体的GEBV进行估计,并对其进行排名。
②利用人类中应用十分广泛的多基因风险评分来佐证上述得到的GEBV值的准确性,所使用的软件为PRSice软件。
这部分工作由以下执行脚本完成。
##R语言脚本,binary.gs.GEBV.R。最佳标记数量下,建立全基因组模型,并计算选育群体GEBV。
args<-commandArgs(T)
lc.geno<-read.table(paste(args[1],".geno",sep=""),sep="")
lc.map<-read.table(paste(args[1],".map",sep=""),sep="\t")
lc.pheno<-read.table(paste(args[1],".pheno",sep=""),sep="\t")
colnames(lc.pheno)<-c("Taxa","staus")
lc.map<-lc.map[c(2,1,4)]
colnames(lc.map)<-c("SNP","Chromosome","Position")
colnames(lc.geno)<-c("taxa",as.character(lc.map$SNP))
rownames(lc.geno)<-lc.geno$taxa
lc.geno<-lc.geno[-1]
rownames(lc.pheno)<-lc.pheno$Taxa
all.id<-rownames(lc.pheno)
slect.id<-all.id[grep("HX",all.id)]
ref.id<-all.id[-grep("HX",all.id)]
sur.id<-ref.id[grep("S",ref.id)];death.id<-ref.id[-grep("S",ref.id)]
lc.pheno[sur.id,2]<-1;lc.pheno[death.id,2]<-0;lc.pheno[slect.id,2]<-NA
print(lc.pheno)
cat("\n--------OK-------\n")
predict<-read.table(paste(args[1],".predict",sep=""),row.names="number",header=T,sep="\t")
predict.mean<-apply(predict,1,mean)
predict.sd<-apply(predict,1,sd)
best.number<-which.max(predict.mean)*10
print(best.number)
cat("\n--------OK-------\n")
gaston.result<-read.table(paste(args[1],".gaston.result",sep=""),header=T)
head(gaston.result)
gaston.result<-gaston.result[order(gaston.result$p),]
head(gaston.result)
lc.geno1<-lc.geno[ref.id,]
lc.pheno1<-lc.pheno[ref.id,]
z<-floor(nrow(lc.pheno1)/5)
library(BGLR)
lc.pheno1->trait
tst<-sample(1:nrow(lc.geno1),z);trait[tst,2]<-NA
geno<-lc.geno1[,as.character(gaston.result$id[1:best.number])]
BGLR(y=trait[,2],response_type='ordinal',
ETA=list(list(X=geno,model='BL')),nIter=1200,burnIn=200,verbose=F)->ans
class(ans$probs);dim(ans$probs)
cv_5<-data.frame(lc.pheno1,ans$probs,stat="train",stringsAsFactors=F)
print(cv_5)
cat("\n--------OK-------\n")
cv_5[tst,"stat"]<-"tst";names(cv_5)[c(3,4)]<-c("death_prob","survival_prob")write.table(cv_5,paste(args[1],".cv_5",sep=""),sep="\t",quote=F,row.names=F)cor(ans$probs[-tst,2],lc.pheno1[-tst,2]);cor(ans$probs[tst,2],lc.pheno1[tst,2])cat("\n--------OK-------\n")
geno<-lc.geno[,as.character(gaston.result$id[1:best.number])]
BGLR(y=lc.pheno[,2],response_type='ordinal',
ETA=list(list(X=geno,model='BL')),nIter=1200,burnIn=200,verbose=F)->ans
GEBV<-data.frame(lc.pheno,ans$probs,stat="train",stringsAsFactors=F)
head(rownames(GEBV));GEBV[slect.id,"stat"]<-"slection"
names(GEBV)[c(3,4)]<-c("death_prob","survival_prob")
write.table(GEBV,paste(args[1],".GEBV",sep=""),sep="\t",quote=F,row.names=F)cat("\n--------OK-------\n")
##shell脚本执行多基因风险评分分析任务
sort-k 8,8g$pre/${file}.gaston.result|cut-f 3-d""|sed 1d|head-670>$pre/effect.snp.id
plink--file$pre/$file--allow-extra-chr--extract$pre/effect.snp.id--make-bed--recode--out$pre/${file}.target
grep-f$pre/effect.snp.id-F-w$pre/${file}.ref.assoc|cut-f 1,2,3,4,7,9,10-d"">$pre/${file}.train.BASE
cut-f 1,2,3,4,7,9,10-d""$pre/${file}.ref.assoc|head-1|cat-$pre/${file}.train.BASE>$pre/txt
mv$pre/txt$pre/${file}.train.BASE
awk'{if(NR>1){$4=1;$5=2;print}else{print}}'$pre/${file}.train.BASE>$pre/txt
mv$pre/txt$pre/${file}.train.BASE
sed-i's/^LG//g'$pre/${file}.train.BASE
awk-v OFS="\t"'{$5=1;$6=2;print}'$pre/${file}.target.bim>$pre/txt
mv$pre/txt$pre/${file}.target.bim
sed-i's/^LG//g'$pre/${file}.target.bim
awk'{$5=1;print}'$pre/${file}.target.fam>$pre/txt
mv$pre/txt$pre/${file}.target.fam
sh$pre/script/binary.gs.PRS.sh$pre/$file
cut-f 4-d""$pre/${file}.prs.best|paste$pre/${file}.GEBV-|cut-f1,2,4,5,6>$pre/${file}.gs.result
awk'{if(NR>1&&$4=="slection"){print}else if(NR==1){print}}'
$pre/${file}.gs.result>$pre/${file}.gs.selection.result
pre=/public/home/fgl/public/softwares/PRSice/
Rscript$pre/PRSice.R--dir$pre\
--prsice$pre/PRSice_linux\
--base${1}.train.BASE\
--target${1}.target\
--thread 1\
--no-clump\
--upper 1\
--scatter-r2\
--stat OR\
--out${1}.prs\
--binary-target T
⑺以一定的选择强度选取选育群体GEBV值排在前面的个体作为亲鱼,并繁殖产生抗刺激隐核虫子一代;
①2019年4月中旬,从360尾有基因型的选育群体中根据电子标签挑选GEBV排名前47的个体作为抗性子一代的亲鱼,如此,选择强度为47/360=13%。挑选完剩余的亲鱼繁殖的后代作为对照组。
②按照正常的商品鱼养殖体系培育抗性子一代和对照组。在苗种培育流程中,抗性子一代已经表现出耐操作刺激等优良表现。
⑻对抗性子一代进行刺激隐核虫攻毒验证。
①在刺激隐核虫爆发的季节,2019年6月中旬,选取抗虫子一代和对照组2月龄幼鱼各150尾进行刺激隐核虫攻毒实验,实验流程同参考群体的抗性性状测评的攻毒流程一致。
②对攻毒结果进行生存分析发现,抗性子一代与对照组存在显著的抗性差异,表现为在96h时,抗虫子一代存活率为59.2%,而对照组为9.9%;在120h,抗虫子一代存活率为40.8%,而对照组已全部死亡;在160h,抗性子一代仍存活34.4%,且已从患病状态中恢复。
以上实施情况,表明本发明提供的基于全基因组选择的大黄鱼抗刺激隐核虫病优良品系的选育方法的可实用性和高效性。
图1为本发明在具体实施过程中的示意图。图2为本发明在具体实施过程中第⑵个步骤中参考群体抗性性状测量中死亡个体的死亡时间分布图。图3为本发明在具体实施过程中第⑸个步骤中判断全基因组模型最佳标记的结果图。结果表明,前600个标记(标记经过P值显著性排名)下,模型预测准确性达到最高。图4为本发明在具体实施过程中第⑹个步骤中使用PRSice软件对R语言BGLR包得到的GEBV结果的佐证结果,结果表明二者具有高度相关性,也佐证了对选育群体GEBV计算的准确性。图5为本发明在具体实施过程中第⑻个步骤中抗虫子一代的刺激隐核虫攻毒验证结果,结果表明,本发明培育出的抗性子一代抗虫性有显著的提升。
本发明将基因组育种技术首次应用到大黄鱼抗刺激隐核虫病的良种选育中。包括:建立参考群体;对参考群体群体进行抗性性状测量;建立选育群体;对参考群体和选育群体进行基因分型;建立最佳全基因组选择模型;对选育群体的基因组育种值GEBV进行估算;以一定的选择强度选取选育群体GEBV值排在前面的个体作为亲鱼,并繁殖产生抗刺激隐核虫子一代;对抗性子一代进行刺激隐核虫攻毒验证。利用该方法培育的抗刺激隐核虫子一代在攻毒验证中,96h存活率较对照组高出49.3%,抗虫性有显著的提升。提高对选育群体基因组育种值的估计准确性,大大缩短了育种年限,在一代之内就可获得高抗性的后代,减少刺激隐核虫对大黄鱼养殖产业造成的经济损失,同时也为其他鱼类的抗病育种提供借鉴和基础,具有广阔的应用前景。
- 基于全基因组选择的大黄鱼抗刺激隐核虫病的选育方法
- 基于全基因组选择的大黄鱼抗刺激隐核虫病的选育方法