引物、探针和试剂盒
文献发布时间:2023-06-19 10:05:17
技术领域
本发明涉及生物技术领域,公开了一种可用于EB病毒DNA定量检测的PCR引物、探针和试剂盒。
背景技术
EB病毒属于γ-疱疹病毒亚科淋巴浅隐病毒属,又被称为人类疱疹病毒4型 (HHV-4),于1964年首次自Burkitt淋巴瘤细胞中分离建株并在电镜下观察到病毒颗粒。EB病毒是线性双链DNA病毒,病毒基因组大小约为172KB,具有编码基因100 个,病毒基因组中含多个重复片段区域且不同毒株间基因组存在上千个SNV突变、indel 突变与其他类型突变。EB病毒感染在人群中普遍存在,在中国约90%的个体曾感染EB 病毒且很多个体在婴幼儿期就已感染。EB病毒的感染分为潜伏性感染或增殖性感染,大部分人感染EB病毒后,EB病毒处于潜伏感染状态,当机体免疫力下降时,EB病毒可由潜伏感染状态转化为增殖感染状态。EB病毒的感染与多种人类疾病相关,如鼻咽癌、传染性单核细胞增多症、Burkitt淋巴瘤等。
鼻咽癌是一种发生于人鼻咽部的上皮性恶性肿瘤,其发病率在世界范围内有明显的地域聚集性和种族易感性,我国和东南亚地区鼻咽癌年发病率高于20/100000,均属于鼻咽癌的高发地区且黄种人的鼻咽癌发病率明显高于白种人。研究表明鼻咽癌的发生与EB病毒的感染密切相关,超过90%的鼻咽癌患者肿瘤组织中可检出EB病毒。有研究表明在治疗前的鼻咽癌患者中,血浆EB病毒DNA载量与患者预后有关,载量越高预后越差。同时,鼻咽癌患者接受治疗后血浆EB病毒DNA是否转阴或转阴的速度也与患者的疗效和预后有关,治疗后病毒DNA转阴或载量快速下降提示患者治疗有效且预后较好。而治疗后再次检出血浆EB病毒DNA则提示鼻咽癌患者可能复发或出现远端转移,病毒DNA载量越高复发和远端转移的可能性越大。因此,血浆EB病毒DNA 定量检测对于鼻咽癌的辅助诊断、预后与疗效评估和复发转移评估具有重要意义并已在临床得到广泛应用。
目前EB病毒DNA定量检测方法主要为荧光定量PCR法。一项研究分析了之前的15项EB病毒DNA定量研究结果,分析结果显示15项研究中EB病毒DNA荧光定量PCR检测方法的灵敏度在53~96%之间,差异巨大。在另一项国际多中心Ⅲ期临床试验前的EB病毒DNA定量检测预研究中,40例鼻咽癌患者血浆样品被平均分为4套并分发至美国、中国香港特别行政区和台湾地区的4家EB病毒DNA定量检测实验室中,大家均采用荧光定量PCR法进行EB病毒DNA定量检测,结果显示各家实验室检测结果间的类内相关性 (Intraclass correlations,ICC)仅在0.59~0.70之间。以上研究结果表明当前EB病毒 DNA荧光定量PCR检测结果及其定量值差异明显,不能完全满足临床检测需求。另有研究显示鼻咽癌患者血浆标本中的EB病毒DNA主要以碎片化的游离DNA形式存在且浓度分布范围很广,这也对检测方法的合理设计和灵敏度提出了更高的要求。同时多项研究表明,鼻咽癌患者血浆标本中EB病毒DNA PCR检测方法的阳性率还与PCR扩增片段的大小呈负相关,扩增片段越大阳性率越低。2015年鉴于EB病毒DNA检测对于鼻咽癌诊疗的重要作用和目前EB病毒DNA检测存在的诸多问题,美国国家癌症研究所组织多方相关专家就鼻咽癌EB病毒DNA检测专题召开了研讨会。会议指出导致不同实验室间EB病毒DNA荧光定量PCR检测结果存在显著差异的原因除了以上指出的游离EB DNA片段化和PCR扩增片段大小差异外,至少还包括了合理检测靶区的选择、定量标准品的溯源、临床诊断阈值的确定和样品提取方法的差异等。而就符合标准化要求的EB病毒DNA荧光定量PCR检测方法的建立,会议给出了以下设计建议:首先应针对病毒的保守区合理设计引物与探针,在保证PCR扩增片段的保守性前提下尽量使扩增片段小于100bp。其次,检测方法的定量结果应可溯源至国际单位IU或使用内参基因标准化定量结果。再次,应对已建立的方法进行充分的性能评估,包括检测下限、检测线性范围、特异性和重复性等。最后,针对拟检测的实验样本类型和不同预期用途 (如疾病进展或预后评估等)设置不同临床诊断阈值。
综上所述,目前EB病毒DNA荧光定量PCR检测方法的建立还存在以下困难:1、 EB病毒的序列变异性:EB病毒感染在人群中普遍存在且在大多数感染人群中EB病毒处于潜伏感染状态,而在潜伏感染状态时EB病毒可以整合入人的细胞基因组,当机体免疫力下降时潜伏感染可以转化为增殖性感染。而病毒在传播时极易出现各种变异以适应新的环境和宿主,所以在不同感染人群中EB病毒基因组可能存在明显的序列变异性。当序列变异出现于检测靶区引物和探针结合位点时,EB病毒DNA荧光定量PCR检测就可能出现假阴性结果。目前关于EB病毒全基因组序列变异性的研究尚不多,但有研究结果显示在3个不同正常人来源的EB病毒基因组间就存在上千个SNV突变、indel 突变与其他类型突变。2、不同样本类型中EB病毒DNA状态的差异:目前可用于EB 病毒DNA荧光定量PCR检测的标本类型有组织样本、细胞样本和血浆样本等,在组织样品和细胞样品中存在完整的EB病毒的基因组DNA,此时对于扩增靶区、引物、探针和扩增子大小的要求较低,只要扩增引物和探针特异性良好就可检出EB病毒DNA。但当以血浆作为检测样品时,由于血浆中EB病毒DNA为碎片化的游离病毒DNA,故具有片段小、含量低和不同基因片段浓度在血浆中可能分布不均等特点。所以在定量 PCR检测方法建立是对扩增靶区、引物、探针和扩增子大小的要求较高,而针对不同扩增靶区设计的检测方法在检测低浓度样品时可能存在检测结果不一致的情况。3、EB 病毒基因拷贝数变异:在EB病毒基因组中存在多个多拷贝基因区域,如BamHI-W区等,在这些区域DNA序列存在5-11此次的重复,因此当PCR扩增目标区域位于此类区域时,理论上可更敏感的检出EB病毒DNA,但自然存在的拷贝数差异会影响定量结果的准确性而不利于统一临床诊断阈值的建立。4、检测灵敏度的问题:鉴于血浆中检出EB病毒DNA对于鼻咽癌筛查、预后和疗效评估等具有较高的应用价值,故临床期望能尽量灵敏的检出血浆EB病毒DNA。然而当PCR扩增目标区域选择在单拷贝基因区域时,理论上虽可准确定量但检测灵敏度却不及将目标检测区域设计在多拷贝区。然而虽然基因拷贝数的差异理论上可影响PCR检测的灵敏度,但由于DNA片段碱基构成和二级结构等因素的影响,实际上即使针对单拷贝基因区域PCR扩增的灵敏度还可以通过二次优选扩增靶区实现。
针对临床上对于EB病毒DNA荧光定量PCR检测的需求和现在EB病毒DNA荧光定量PCR检测还存在的问题,本发明设计开发了用于EB病毒DNA定量检测的荧光定量PCR引物、探针和试剂盒。
发明内容
本发明描述的一种用于EB病毒DNA定量检测的荧光定量PCR引物、探针和试剂盒。其包含以下几个方面特征:1、包含两套针对EB病毒EBNA1基因保守区的EB 病毒特异性PCR引物和探针。2、经过优化的PCR反应体系和反应条件。
首先,本发明优选设计了两套针对EB病毒的荧光定量PCR引物和探针。在选择特异性EB病毒DNA扩增靶区时,本发明优选了在EB病毒潜伏感染期和增值性感染期均可表达且在病毒基因组中为单拷贝存在的EBNA1基因作为目标扩增区域。通过序列组成筛选与Blast比对优选出两个EBNA1基因保守区域用于引物和探针的设计。通过相同引物筛选条件设置,利用Oligo 7.0软件设计两套荧光定量PCR引物和探针体系。具体引物与探针序列见表1。
通过如上引物与探针设计,可使该EB病毒DNA荧光定量PCR检测体系获得以下优势:1.可准确定量:该设计针对的目标扩增区域为单拷贝区域,因此该目标区域的 EB DNA片段拷贝数与EB病毒载量呈良好的一一对应关系且在人群中的生理变异小,故可用于EB病毒DNA的准确定量,利于临床统一诊断阈值的形成。2、灵敏度高:首先,该检测体系设计了两套针对EBNA1基因不同保守区域的PCR引物和探针体系并采用了双重PCR体系设计,因此当血浆样本中EB病毒DNA片段浓度很低位于临界点时,双检测位点设计可以提高弱阳性标本的检出率。同时两套引物所设计的扩增片段均约100bp左右,其不仅适用于组织、细胞样本的检测,也适合与血浆中碎片化EB病毒 DNA小片段的检测,可有效保证检测的阳性率。
其次,当两个检测位点中的一个出现可导致该位点检测失败的DNA序列变异时,另一检测位点也可成功检测EB病毒DNA。
再次,该双重PCR扩增体系使用了2条相同报告荧光基团标记的探针,因此其针对等量EB病毒DNA模板的信号强度增加了1倍,因此也可更高灵敏度的检出弱阳性样品。
表1 EB病毒DNA荧光定量PCR检测体系引物与探针序列表
本发明采用了经过优化的PCR反应体系和反应条件,具体见下文实施例。
附图说明
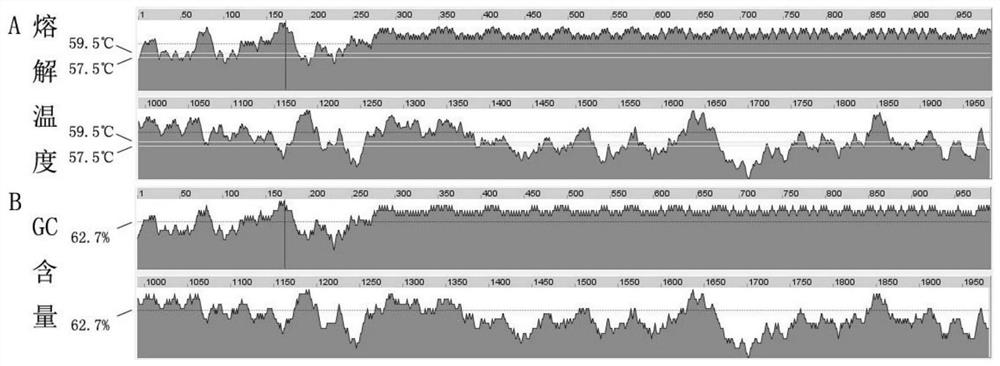
图1 :EB病毒EBNA1基因21号外显子序列特征分析图
图2: EB病毒DNA荧光定量PCR引物示意图
图3: EB病毒DNA荧光定量PCR检测体系的初步建立
图4:体系1、体系2和双重检测体系间血浆样本检测结果的比对
图5 :EB DNA双重荧光定量PCR体系检测梯度标准物质之荧光曲线与标准曲线
具体实施方式
下面通过具体实施例和附图对本发明技术方案进行详细说明,但是本发明的保护范围不局限于所述实施例。
实施例1:EB病毒EBNA1基因保守区筛选与序列特征分析
在NCBI网站(https://www.ncbi.nlm.nih.gov)搜索获得EB病毒EBNA1基因参考序列(NC_007605.1),分析其基因结构可见,EBNA1基因大小为86KB,约占EB病毒基因组的一半,位于基因组的11305~97654位置,与EBNA2、EBNA3a和EBNA3b/3c 等多个基因有重叠。在该基因上存在多个重复序列区域和序列变异区域,如重复序列区域21217-24288、82124-82201等,其大多位于基因中前部,又如高变序列区域 33127-40536、33356-40162、86839-89829等。鉴于上述EB病毒EBNA1基因的结构特征,该发明选择了位于96654-97654的EBNA1基因21外显子作为目标扩增区域,在该区域中95929-96636为高重复序列区,提示95654-95929和96636-97654区域可用于引物设计。利用NCBI网站对EBNA1基因21号外显子序列进行Blast分析显示与该段序列匹配的序列均为EB病毒序列,表明区域特异性良好,是EB病毒特有的保守序列区域。再利用Oligo 7.0软件对EBNA1基因21号外显子进行序列特征分析可见在 95929-96636高重复区,序列GC含量和熔解温度均较高故不适合进行引物设计,而95654-95929和96636-97654区域的部分区域其序列GC含量适中且溶解温度接近,故适合在这两个区域进行配对引物设计(见图1A,B)。最终,该发明选择将EBNA1基因21号外显子95654-95929和96636-97654区域作为选定的引物设计区域。
实施例2:EB病毒EBNA1荧光定量PCR引物与探针设计
利用Oligo 7.0软件在EBNA1基因21号外显子95654-95929和96636-97654区域分别设计一套引物和探针,详见表1(图2)。为了保证两套引物和探针体系在后期能成功构建为双重PCR检测体系,故在进行引物和探针设计与筛选时需保证以下几个原则: 1、引物与探针特异性好;2、引物与探针的Tm值等关键参数接近,以便于在相同的 PCR反应条件下可获得最接近的扩增性能;3、各条引物与探针间的假性错配和二级结构尽量少以保证最大可能的减少双重PCR体系间的相互影响,实现两体系合并后其性能1+1≥2;4、两条探针采用相同的报告基团标记保证其检测信号的有效叠加。通过相应设计优选,获得了针对两个不同目标区域的PCR扩增引物两套,各条引物间的Tm 值接近且未预测到引物间存在明显的假性错配和二级结构。然而两套PCR扩增引物所扩增的目标区域其序列平均Tm存在较大差异,这导致了两条探针和对应引物间的Tm 值梯度存在明显差异,由于在荧光定量PCR体系中对于荧光标记探针和配对引物间的 Tm值梯度有明确的要求,如Taqman荧光定量PCR法要求探针Tm值要比引物高 7~10℃,所以该发明设计的两套PCR引物和探针体系如采用相同荧光标记策略则无法保证其在相同反应条件下两套体系均能达到接近的检测效能。而不同荧光标记策略对于引物和探针间的Tm值梯度要求有所不同,如MGB荧光定量PCR则对探针和配对引物间的Tm值梯度差没有这么高的要求。因此该发明的体系1探针由于探针和配对引物间的Tm值梯度超过7℃采用了Taqman荧光标记方案而体系2探针由于Tm值梯度小则采用了MGB荧光标记方案,其中体系1探针还采取了简并引物设计方案。同时两套标记方案的报告基团均采用了FAM荧光基团标记,故两检测体系在设计上应可利用相同的反应条件实现高效PCR扩增,同时其检测信号可有效叠加。两检测体系扩增片段大小分别为109bp和80bp。最后利用NCBI网站对引物和探针进行Blast分析显示特异性良好。
实施例3:EB病毒DNA荧光定量PCR检测体系的初步建立
使用上海之江DNA提取试剂依说明书提取1例EB病毒DNA阳性血浆样本DNA 备用。使用表1之体系1和体系2的PCR扩增引物配置反应体系在ABI梯度PCR仪上对提取好的EB病毒DNA阳性血浆样本DNA进行PCR扩增,1.5%琼脂糖凝胶电泳鉴定。反应体系:Green Mix 10μl,上下游引物各1μl(10μM),样本DNA2μl,纯水6μl。反应条件:94℃3min;94℃5s,(58℃、60℃、62℃、64℃)30s,共40个循环。电泳结果(图3A)显示在58-64℃退火与延伸温度范围内体系1与体系2均能有效扩增单一片段,而各单一片段大小与设计扩增片段大小相符,提示体系1与体系2可有效并特异性扩增EB病毒DNA相应目的片段。在后续实验中选择60℃作为体系1与体系2统一的退火与延伸温度。
切胶纯化体系1与体系2的单一扩增片段,依大连宝生物TA克隆试剂盒说明书步骤将两个纯化后的扩增片段分别连接入质粒载体,将连接后质粒分别转化感受态细菌,铺板涂菌后挑取单克隆菌落置于液体培养基中摇菌增菌,利用上述扩增体系和扩增条件做菌液PCR,结果为阳性的菌液送华大基因克隆测序。测序结果显示体系1与体系2 (图3B)扩增产物序列与设计用模板序列相符,证明体系1与体系2有效且特异的扩增了EB病毒DNA相应目的片段。
使用上海之江DNA提取试剂依说明书再提取1例EB病毒DNA阴性血浆样本DNA 备用。使用表1之体系1和体系2的PCR扩增引物和相应探针配置反应体系在伯乐 CT1000荧光定量PCR仪上对提取好的EB病毒DNA阴、阳性血浆样本DNA进行定量 PCR检测。反应体系:2×Taq Fast Q-PCR mix 10μl,上下游引物各0.5μl(10μM),探针 0.5μl(10μM),样本DNA2μl,纯水6.5μl。反应条件:94℃3min;94℃5s,60℃30s (Fam通路荧光收集),共40个循环。结果显示体系1和体系2对阳性样本的扩增曲线均为明确的阳性曲线且CT值接近均为32左右,而两体系对阴性样本的扩增曲线也均为阴性曲线。表明体系1和体系2的引物和探针可有效并特异匹配,且两体系扩增效率接近(图3C)。至此EB病毒DNA荧光定量PCR检测体系1和体系2已初步建立。
其后利用体系1和体系2的PCR扩增引物和相应探针配置两重反应体系在伯乐CT1000荧光定量PCR仪上对提取好的EB病毒DNA阴、阳性血浆样本DNA进行定量 PCR检测。反应体系:2×Taq Fast Q-PCR mix 10μl,体系1与体系2上下游引物各0.5μl (10μM),体系1与体系2探针0.5μl(10μM),样本DNA2μl,纯水5μl。反应条件: 94℃3min;94℃5s,60℃30s(Fam通路荧光收集),共40个循环。结果显示双重PCR 反应体系对阳性样本的扩增曲线为明确的阳性曲线,其CT值也为32与体系1与体系2 单独扩增时一致,但其荧光信号强度较体系1与体系2高提示双重PCR反应体系提高了荧光信号效能。同时双重PCR反应体系对阴性样本的扩增曲线也均为阴性曲线(图 3C)。以上结果表明EB病毒DNA荧光定量双重PCR检测体系已初步建立。
实施例4:EB病毒DNA荧光定量PCR检测体系1、体系2和双重检测体系间的比对
选取临床鼻咽癌患者血浆样本10例,其中EB DNA阴性样本5例,不同浓度EB DNA阳性样本5例,使用上海之江DNA提取试剂依说明书提取样本DNA备用。利用已建立的EB病毒DNA荧光定量PCR检测体系1、体系2和双重复合检测体系检测上述10例样品,分析结果阴阳性是否一致,同时分析各检测体系间样本CT值的相关性,做三体系间样本CT值数据的单因素方差分析。结果显示体系1、体系2和双重复合检测体系检测10例血浆样本的结果阴阳性完全一致,双重复合检测体系对阳性样本的扩增曲线其相对荧光强度均高于体系1和体系2。单因素方差分析结果显示三体系间样本 CT值数据无显著性差异(P=0.968),另将三组CT值数据两两间做相关性分析,r值均大于0.99(P均<0.01,图4)。以上结果表明已建立的EB病毒DNA荧光定量PCR检测体系1、体系2的检测性能接近,如扩增效率等,两种检测体系对于不同EB DNA浓度范围临床样本定量结果一致。而将两检测体系组合为双重复合检测体系后,复合检测体系明显提高了EB DNA阳性样本扩增曲线的荧光强度,其在针对临界阳性样本进行检测时有望提高临界阳性样本检出率,另分析双重检测体系对阳性样本的CT值结果(也即定量检测结果)时可见与体系1与体系2相比,双重检测体系对于不同EB DNA浓度范围临床样本的定量结果与体系1和体系2一致。提示双重检测体系在提高了荧光定量PCR反应的荧光效率并有望提高临界阳性标本检出率的前提下也可对不同浓度范围的EB DNA阳性样本进行准确的定量检测。
实施例5:定量标准品的制备与标定
以实施例3中已连入体系1和体系2PCR扩增片段的质粒为待标定阳性标准品,以国家标准品EB病毒脱氧核糖核酸(EB DNA)标准物质GBW(E)090679(5.0×10
依据体系1和体系2质粒的量值,将高浓度的一个稀释至两者浓度相同,然后将相同量值的两个质粒等比例混合作为双重检测体系的待标定阳性标准品,重复以上赋值步骤和标准对该阳性标准品赋予量值,并将其作为双重检测体系的工作定量标准品。
实施例6:EB病毒DNA双重荧光定量PCR检测体系的性能评估
线性检测范围、检测下限:将EB病毒脱氧核糖核酸(EB DNA)标准物质 GBW(E)090681(5.4×10
重复性:对线性范围内5.4×10
实施例7:与商品试剂盒的比对实验
收集鼻咽癌患者血浆样本53例,以上海商品化EB病毒DNA荧光定量PCR检测试剂盒为对照试剂,利用该发明所建立的EB病毒DNA双重荧光定量PCR检测体系与商品化对照试剂对53例样本进行平行检测,分析两种试剂的检测结果的阳性率差异。由于对照试剂定量标准曲线的定量单位为Copies/ml,与本发明的IU/ml不同,故选用 CT值作为比较参数。结果显示双重检测体系与商品化对照试剂的检测阳性率分别为 26.4%(14/53)和22.6%(12/53),两试剂检测结果的符合率为92.5%(49/53),虽然双重检测体系检测阳性率略高于商品化对照试剂,但卡方检验显示两种试剂间检测阳性率无显著性差异(P=0.652),提示本发明自建的EB病毒DNA双重检测体系检测灵敏度已达到甚至略超过对照商品化试剂盒。以对照试剂定量结果为标准,在53例样本中EB DNA拷贝数大于1E3浓度的样品两检测体系检测结果均为阳性,而4例结果阴阳性有差异的样本不管任一体系检出的阳性结果均为无法进行准确定量的弱阳性结果。在所有两体系检测结果均为阳性的样本中,可见双重检测体系的样本平均CT值较对照试剂大 3左右,但不同样本间CT值差异并不均一,从2.51至5.12,这表明对照试剂检测的靶标区域应设计在多拷贝区域,因此其CT值均小于双重检测体系,同时由于不同样本间靶区域拷贝数的差异导致了CT值差异的不均一。而这也表明该双重检测体系虽然针对单拷贝靶区域进行设计,但其检测灵敏度达到了以多拷贝区域为靶区设计的检测试剂盒水平,同时在此基础上该双重检测体系还排除了多拷贝区域拷贝数变异对准确定量EB 病毒载量的影响,实现了对EB病毒载量的准确定量检测。
序列表
<110> 广州医科大学附属肿瘤医院
<120> 引物、探针和试剂盒
<160> 7
<170> SIPOSequenceListing 1.0
<210> 4
<211> 20
<212> DNA/RNA
<213> 体系1上游引物(Artificial Sequence)
<400> 4
acaggacctg gaaatggcct 20
<210> 3
<211> 20
<212> DNA
<213> 体系1下游引物(Artificial Sequence)
<400> 3
cccgtcctcg tccatggtta 20
<210> 3
<211> 25
<212> DNA
<213> 体系1简并探针1(Artificial Sequence)
<400> 3
tccggcggca gtggacctca aagaa 25
<210> 4
<211> 25
<212> DNA
<213> 体系1简并探针2(Artificial Sequence)
<400> 4
tccagcggca gtggacctca aagaa 25
<210> 5
<211> 21
<212> DNA
<213> 体系2上游引物(Artificial Sequence)
<400> 5
ccaacccgaa atttgagaac a 21
<210> 6
<211> 23
<212> DNA
<213> 体系2下游引物(Artificial Sequence)
<400> 6
ttcgtcggta gtcctttcta cgt 23
<210> 7
<211> 20
<212> DNA
<213> 体系2探针(Artificial Sequence)
<400> 7
aaggtttaag agctctcctg 20
- 用于检测SARS冠状病毒的引物和探针,包括该引物和/或探针的试剂盒及其检测方法
- 非洲猪瘟病毒荧光热对流PCR扩增引物对、探针引物、及制备的试剂盒