基于ATAC测序数据筛选子宫内膜癌关键基因的方法及应用
文献发布时间:2023-06-19 10:24:22
技术领域
本发明涉及基因筛选技术领域,具体而言,涉及基于ATAC测序数据筛选子宫内膜癌关键基因的方法及应用。
背景技术
目前,肿瘤关键基因筛选方法主要包括以下三种:(1)利用突变小鼠,进行正向基因筛查并确定与人类癌症高度相关的候选驱动基因;(2)基于蛋白质序列、功能注释和相互作用网络等方面通过研究基因在肿瘤中的特征以分选出候选肿瘤基因;(3)基于公共数据库提取差异表达基因,结合蛋白互作网络(PPI)进行关键基因的筛选。
但上述肿瘤关键基因筛选方法都具有明显的缺陷和不足。首先,由于物种间的差异显著,导致在利用突变小鼠进行关键基因筛查时得到的候选驱动基因在人体中的作用较小鼠具有显著差异,不够准确,同时利用突变小鼠进行关键基因筛查的方法主要针对的是反复突变的“显著突变基因”,那些罕见突变基因无法被获取到。
其次,由于蛋白质序列只能反映编码区的基因组成,因此,基于蛋白质序列、功能注释和相互作用网络等方面通过研究基因在肿瘤中的特征以分选出候选肿瘤基因的方法对基因的功能注释主要集中在基因的编码区,而占比约97%的基因非编码区同样对肿瘤发生和发展具有重要作用,而这一方法无法对非编码区进行功能注释,同时,该方法对具有已知功能注释的基因进行研究筛选时存在偏差。
随着基因数据库的逐渐丰富,利用公共数据库提取差异表达基因,结合蛋白互作网络进行关键基因筛选的方法逐渐成为肿瘤关键基因筛选的重要手段,但由于数据来源受实验条件、技术以及对照组选择标准的制约,一组或者几组筛选数据并不能代表广泛的病例。
鉴于此,特提出本发明。
发明内容
本发明的目的在于提供一种子宫内膜癌关键基因的筛选方法,该方法基于ATAC测序数据,通过选取多个判断指标对目的肿瘤的关键基因进行筛选,可以避免数据处理过程中人为因素的干扰,具有更高的可靠性。
本发明还提供了上述子宫内膜癌关键基因筛选方法在制备筛选子宫内膜癌关键基因产品中的应用。
本发明是这样实现的:
本发明提供了一种基于ATAC测序数据筛选子宫内膜癌关键基因的方法,该方法包括,首先选取两个以上的指标将ATAC测序所得的峰位进行排序;然后将不同指标的峰位排序结果按照峰位数量均分成组,选择至少一组作为疑似包含肿瘤关键基因的初筛峰位分组;再筛选出不同指标得到的初筛峰位分组中共同含有的峰位构成次筛峰位分组;最后根据子宫内膜癌的基因表达值,对得到的次筛峰位分组中各峰位对应的基因进行最终筛选,得到肿瘤关键基因。
ATAC测序所得峰位数量巨大,在对峰位数据进行分析之前,首先进行分类处理是十分必要的,本发明首先选择两个以上的指标将ATAC测序所得峰位排列成不同的顺序。
在可选的实施方式中,可用于对ATAC峰位数据进行排序的指标包括转录因子数目、染色质开放信号强度、peaks的大小或peak在基因组上的位置(如启动子、增强子或CPG岛)中的两种以上组合。
优选地,所述指标为转录因子数目和染色质开放信号强度。
真核生物转录起始过程十分复杂,往往需要多种蛋白因子的协助,转录因子与RNA聚合酶形成转录起始复合体,共同参与转录起始过程。转录因子是一群能够与基因5’端上游特定序列专一性结合,从而保证目的基因以特定的强度在特定的时间与空间表达的蛋白质分子。许多转录因子充当着主调节因子和选择基因的角色,控制着细胞类型的决定、发育模式和特定途径控制(如免疫反应)的过程。在实验室中,转录因子可以促进细胞分化、去分化和转分化。含有转录因子数目越多的基因序列的开放程度越高或者说越活跃,而转录因子和转录因子结合位点突变是人类致病(或致癌)的主要因素。突变基因的转录产物可能增强或丧失了某些功能,这种转录失调可能在肿瘤的形成及恶性进展方面具有重要作用,因此,含有转录因子数目多的序列区域能够作为肿瘤关键基因的筛选区域。
在可选的实施方式中,所述转录因子数目的获取方法包括转录因子结合位点的识别方法。
优选地,所述转录因子结合位点的识别方法包括通过处理TFBS数据库的转录因子数据得到对应的转录因子基序,再通过iForm识别ATAC-seq开放染色质区域的转录因子结合位点。
优选地,所述TFBS数据库包括TRANSFAC、JASPAR或UniPROBE数据库中的一种或两种以上组合,该三种数据库均能够同时兼顾基因编码区以及基因非编码区,适合用于识别转录因子结合位点。
进一步优选地,所述TFBS数据库为TRANSFAC数据库,TRANSFAC数据库是一个全面的真核生物转录调控的数据库,包含了转录因子及其在基因组上的结合位点和相应的靶基因的信息,其专业版中共包括了12795个转录因子,26589个转录因子结合位点和51325个调节基因,其中包括miRNA及其靶序列、ChIP-chip实验序列片段,以及所有收录数据的相关参考文献、启动子序列等信息,所含转录因子信息全面,尤其适合用于转录因子结合位点的识别。
进一步优选地,所述TFBS数据库为TRANSFAC、JASPAR和UniPROBE数据库,当选用三个数据库时,涵盖基因组范围更广泛,可以找到更多有价值的靶点。
上述染色质开放信号强度是ATAC测序检测过程中能够获取的用来表示特定区域染色质开放程度的数值,染色质开放信号越强与基因启动子区域的距离越近,且该区域富集更多的高表达基因。这些转录活跃peaks中基因启动子区域的高突变比例将直接影响该基因转录调控功能,同样可能是潜在的致癌因素。
由于肿瘤的形成与正常基因的错误突变有关,因此,本发明将基因的突变比例高低作为该基因是否可以作为目的肿瘤关键基因的判断标准,具有更多突变比例的基因与具有较低突变比例的基因相比,其引发肿瘤发生的概率更高。在此基础上,可以通过判断上述指标对于基因突变比例的影响,从而判断该指标是否能够用于肿瘤关键基因的筛选。例如,随着转录因子数目的增加,位于基因启动子区ATAC-peaks的碱基突变比例呈下降趋势,而随着染色质开放信号强度的增加,ATAC-seq数据中总体突变比例逐渐增加,因此,上述的转录子数目和染色质开放信号强度均可作为肿瘤关键基因筛选的指标,而且转录子数目和染色质开放信号强度分别与基因突变比例呈负比例关系和正比例关系相关,将转录子数目和染色质开放信号强度同时作为筛选指标时能够起到互相校正的作用,从而提高筛选准确率。
ATAC峰位经过不同的指标排序后,在不同排序中的先后位置代表了该峰位在该选定指标下包含肿瘤关键基因可能性的高低程度,通过设定合适阈值,即能够从每个指标所得的ATAC峰位排序中筛选出疑似包含肿瘤关键基因的初筛峰位分组,通过比较,筛选出得到的不同初筛峰位分组中共同含有的峰位即可大大地避免采用单一数据进行关键基因筛选时带来的不准确性。
而由于所处理的ATAC数据量巨大,对于每个指标直接设定出既能保证筛选准确率,又能减低工作量的阈值难度很大,因此,在本发明可选的实施方式中,均分成组的组别数量至少为十组。
由于ATAC测序所得峰位数量巨大,将不同指标得到的峰位分成十组之后,通过选取合适的峰位分组和进行后续检测的分组数量,可以在保证基因筛选准确性的基础上,调整得到的初筛峰位分组中ATAC的峰位数量,从而保证工作量适度。例如,当筛选目的是为了得到子宫内膜癌最为关键的少数(例如三个或者五个)几个基因时,则可以仅选择包含关键基因可能性最大的一个分组,使得获得的ATAC的峰位数量在保证准确性的前提下,尽量少;而当筛选目的是为了更加全面的了解与子宫内膜癌相关的关键基因时,则可以选择多个分组,扩大获得的ATAC的峰位数量使得筛选过程更加全面,从而避免罕见突变形成的肿瘤关键基因被遗漏的情况发生,因此,通过选取不同阈值能够得到不同数量的关键基因,且能够清楚地判断不同阈值得到的关键基因对于子宫内膜癌的重要程度的高低。
在可选实施方式中,所述子宫内膜癌的基因表达值包括子宫内膜癌的基因表达量和/或目的肿瘤的基因差异表达量。
目前获取某一基因在子宫内膜癌中的表达量的检测方法包括有RNA测序、RT-PCR或Western blot,在本发明可选实施方式中,所述子宫内膜癌的基因表达量数据来自于TCGA数据库,通过检索TCGA数据库(肿瘤基因组图谱)获取某一基因在子宫内膜癌细胞中表达量,所述的TCGA数据库是由美国NCI(National Cancer Institute)和NHGRI(NationalHuman Genome Research Institute)于2006年联合启动的项目,目前共计研究36种癌症类型,收录了各种人类癌症(包括亚型在内的肿瘤)的临床数据、基因组变异、mRNA表达、miRNA表达和甲基化等数据,是癌症研究者很重要的数据来源。是目前获取肿瘤基因表达量最快捷且全面的途径,同时能够有效降低筛选中的假阳性率。
优选地,通过TCGA数据库检索时,选取FPKM>10的基因。
所述的FPKM为每1百万个map上的fragments中map到外显子的每1K个碱基上的fragments个数。一般认为FPKM大于1认为有意义,其FPKM大于10代表该基因在肿瘤中的表达水平较高,适合用于本发明可避免由于检测误差等引起的假阳性率。
子宫内膜癌的基因差异表达是指在正常组织与肿瘤组织中目的基因表达量水平的变化情况,通常限定log
优选地,所述的子宫内膜癌的基因差异表达量来自于检索GEPIA数据库,所述检索GEPIA数据库过程中,设定log
进一步优选地,检索GEPIA数据库过程中,设定log
log
在可选实施方式中,所述筛选方法还包括得到肿瘤关键基因的验证步骤。
可选实施方式中,所述验证步骤包括生存分析和/或文献对比。
所述的生存分析是研究影响因素与生存时间和结局关系的方法,通过对筛选出的肿瘤关键基因的生存分析,即可验证筛选得到的关键基因的表达水平的高低在子宫内膜癌的发生、发展过程中是否起着关键作用,该验证方法准确性高。
所述文献对比是指,针对筛选出的子宫内膜癌关键基因,进行广泛的文献检索,查阅现有报道中是否具有该基因为子宫内膜癌关键基因的工作,如果有则可以节省大量的、重复性的验证工作。
本发明还提供了上述的筛选方法在制备筛选肿瘤关键基因产品中的应用。
本发明具有以下有益效果:
本发明提供了基于ATAC测序数据的肿瘤关键基因的筛选方法,采用多种指标对ATAC测序得到的峰位进行全面的评价分析,得到子宫内膜癌的关键基因,由于目前ATAC测序技术已经十分成熟,因此,本方法的准确度高,且可以避免传统筛选关键基因时,数据处理过程中人为因素的干扰。
本发明提供的肿瘤关键基因的筛选方法能够应用于制备筛选肿瘤关键基因的产品中,为提高基因筛选工作的效率及准确率提供了新途径。
附图说明
为了更清楚地说明本发明实施例的技术方案,下面将对实施例中所需要使用的附图作简单地介绍,应当理解,以下附图仅示出了本发明的某些实施例,因此不应被看作是对范围的限定,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他相关的附图。
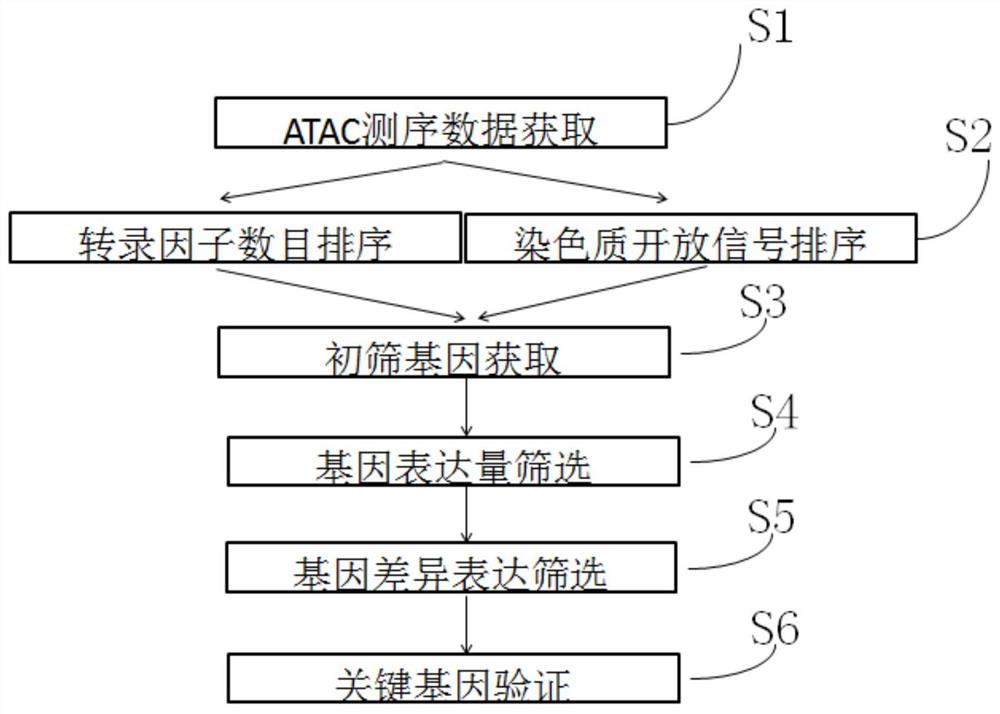
图1为实施例1的子宫内膜癌关键基因的筛选步骤;
图2为实施例1得到的210个初筛基因;
图3为实施例1得到的207个初筛基因;
图4为实施例1基因表达量筛选得到的148个基因;
图5为实施例1差异表达基因筛选得到的16个基因;
图6为实施例1中SCGB2A1基因与SCGB1D2基因生存分析结果;
图7为实施例2中得到的268个初筛基因;
图8为实施例2中基因表达量筛选得到的80个基因;
图9为实施例2中差异表达基因筛选得到的5个基因;
图10为实施例3中670个初筛基因的A部分;
图11为实施例3中670个初筛基因的B部分;
图12为实施例3中670个初筛基因的C部分;
图13为实施例3中基因表达量筛选获得的220个基因的A部分;
图14为实施例3中基因表达量筛选获得的220个基因的B部分;
图15为实施例3中基因差异表达筛选获得的20个关键基因。
具体实施方式
为使本发明实施例的目的、技术方案和优点更加清楚,下面将对本发明实施例中的技术方案进行清楚、完整地描述。实施例中未注明具体条件者,按照常规条件或制造商建议的条件进行。所用试剂或仪器未注明生产厂商者,均为可以通过市售购买获得的常规产品。
以下结合实施例对本发明的特征和性能作进一步的详细描述。
实施例1
本实施例的子宫内膜癌关键基因的筛选步骤如图1所示,包括:
S1:ATAC测序数据获取
本实施例从TCGA数据库中获取子宫内膜癌ATAC测序数据,去除比对到Y染色体上的错误测序结果,共得到包含104723个peaks用于本实施例的进一步分析。
上述peaks的基因组坐标为hg38,为了下一步与TFBS数据进行对比,将上述基因组由hg38转换为hg19,转换后共得到104400个peaks。
S2:转录因子数目和染色质开放信号强度排序
通过扫描S1中得到的ATAC-seq peaks,并与TRANSFAC、JASPAR和UniPROBE三个数据库得到的共计488个转录因子的结合位点信息进行比对,得到各峰位的转录因子数目,并将各峰位的转录因子数目定义为转录因子复杂度TC值。
再将各峰位的染色质开放信号强度Score值定义为SC值;将TC、SC均按照数值大小平均分为10个组别,分别为TC0~TC9和SC0~SC9,得到两个代表该区间不同维度信息的指标。
S3:初筛基因获取
结合上述S2得到的ATAC-seq数据中两个不同维度指标的特征,筛选得到同时属于TC0,SC9的ATAC-seq peaks共588个,并得到位于这些peaks中的初筛基因417个,如图2和图3所示,图2中示出了其中210个初筛基因,图3示出了另外207个初筛基因。
S4:基因表达量筛选
利用TCGA数据库中肿瘤基因表达量数据,依据FPKM>10对上述初筛基因进行筛选,得到148个基因,如4所示。
S5:基因差异表达筛选
对上述步骤S4所得到的148个基因,利用GEPIA数据库中差异表达基因数据,选取log
S6:关键基因验证
通过已发表文献查阅得到基因SCGB2A1(10.1111/j.1525-1438.2007.01137.x)、TPX2(10.3892/or.2020.7648)、UBE2C(10.1158/1541-7786.MCR-19-0561)、DUSP1(10.4103/0366-6999.181954)、IGSF9(10.1155/2018/2439527)、PAX8(10.1089/dna.2019.5148)、S100A14(10.1016/j.intimp.2020.106735)、TXNIP(10.21873/anticanres.13664)与子宫内膜癌的增殖与侵袭、肌层浸润及分化程度、FIGO分期进展、雌激素受体(ER)表达以及预后不良等显著相关,证明了本实施例提供的筛选方法能够准确获得子宫内膜癌的关键基因。
而基因SCNN1A(10.1089/cbr.2019.2824)的高表达与卵巢癌患者总体生存不良和无进展生存有关;基因CDCA8(10.7717/peerj.9078)高表达在膀胱癌中被报道与患者预后差相关,可以促进肿瘤的发展;基因HSPB6(10.1371/journal.pone.0151907)具有诱导的肝癌细胞迁移和侵袭的能力;基因KRT7(10.1016/j.gene.2020.144947)在卵巢癌中高表达,并可能与生存率降低相关;PRSS22(10.1186/1471-2407-6-250)可能作为结肠癌早期检测的标志。SCGB1D2(10.1186/1471-2407-6-88)在妇科恶性肿瘤中大量表达;基因SYNE4有文献报道与听力损伤相关(10.4274/balkanmedj.2017.0946),对于上述7个基因,虽然没有明确证据证明其与子宫内膜癌直接相关,但是均为致病基因,同时大部分均与肿瘤相关,尤其是与妇科肿瘤相关,因此,存在作为子宫内膜癌关键基因的可能性,为后续的研究提供了新的开发目标。对于PRSS8(10.1038/s41388-018-0453-3;10.1159/000453136)基因,文献中报道仅表明该基因可能是新型肿瘤抑制基因,并在多种肿瘤中报道了PRSS8基因降低与恶性进展以及EMT相关,但并没有报道指出该基因与子宫内膜癌有关,可见本发明提供的肿瘤关键基因筛选方法为发现肿瘤关键基因提供了新途径,并且取得了切实的效果。
为了进一步验证本实施例提供的筛选方法的有效性,本实施例同时对上述8个有明确报道为子宫内膜癌关键基因中的SCGB2A1基因与上述8个可能为子宫内膜癌基因中的SCGB1D2共同进行了生存分析,通过GEPIA数据库采用生存分析方法验证得到SCGB2A1基因的P=0.044(如图6中A所示)与SCGB1D2基因的P=0.011(如图6中B所示),均小于0.05,证明了SCGB2A1基因和SCGB1D2基因都与子宫内膜癌患者预后显著相关。
实施例2
本实施例提供了一种与实施例1相同的筛选方法,区别在于,本实施例S3步骤中选取TC0和SC0进行关键基因的筛选,具体步骤如下:
S3:初筛基因获取
参考实施例1中的峰位分组结果,筛选得到同时属于TC0,SC0的ATAC-seq peaks1240个,得到位于这些peaks中的初筛基因268个,如图7所示。
S4:基因表达量筛选
利用TCGA数据库中肿瘤基因表达量数据,依据FPKM>10对上述初筛基因进行筛选,得到80个基因,如图8所示。
S5:基因差异表达筛选
对上述步骤S4步骤所得到的80个基因,利用GEPIA数据库中差异表达基因数据,选取log
S6:关键基因验证
通过已发表文献查阅得到仅有一篇文献报道了基因NID1与子宫内膜癌具有明确的相关性(10.1007/s10585-015-9720-7);基因ACTA2(10.1002/cbin.11451;10.1186/s12935-020-01471-w;10.3390/ijms21124509)与基因SAPCD2(10.26355/eurrev_202004_20844;10.1186/s12935-020-1121-6;10.1002/cam4.2227)被报道与多种肿瘤相关,如肺癌,宫颈癌,乳腺癌、胃癌等;基因PRR15L(10.1007/s00428-019-02604-x)仅报道与乙状结肠癌具有密切联系;基因CHMP2C未见相关报道。
通过GEPIA数据库进行是生存分析验证,各基因与子宫内膜癌患者生存期无显著相关性。
实施例3
本实施例提供了一种与实施例1相同的筛选方法,区别在于,本实施例S3步骤中选取TC0、SC8和SC9进行关键基因的筛选,具体步骤如下:
S3:初筛基因获取
参考实施例1中的峰位分组结果,筛选得到同时属于TC0,SC8和SC9的ATAC-seqpeaks 1447个,得到位于这些peaks中的初筛基因670个,如图10~12所示,分别为所得670个基因中的一部分(A、B和C)。
S4:基因表达量筛选
利用TCGA数据库中肿瘤基因表达量数据,依据FPKM>10对上述初筛基因进行筛选,得到220个基因,如图13和图14所示,分别为所得220个基因的A部分和B部分。
S5:基因差异表达筛选
对上述步骤S4步骤所得到的220个基因,利用GEPIA数据库中差异表达基因数据,选取log
S6:关键基因验证
对于MUC1基因,有文献报道(10.1046/j.1365-2559.2002.01316.x)子宫内膜癌中MUC1表达的丧失与预后良好相关;有文献报道(10.1309/AJCPTK87EMMIKPFS)S100A1基因是子宫内膜样癌亚型预后不良的标志物,而对于CCNB2基因和THBS基因,并未见相关报道与子宫内膜癌相关,但有研究预测CCNB2基因可能是内膜癌关键基因,因此,可以进一步开展对CCNB2基因和THBS基因的研究。
上述实施例1~3提供的子宫内膜癌关键基因的筛选结果如表1所示。
表1实施例1~3所得筛选结果对比表(单位:个)
可见,选取不同组别可以获得不同数量的子宫内膜癌关键基因,所以本发明提供的子宫内膜癌关键基因的筛选方法能够适应在相关领域研究中的多种需求。
以上所述仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 基于ATAC测序数据筛选子宫内膜癌关键基因的方法及应用
- 一种基于高通量测序检测子宫内膜癌相关基因突变的文库构建方法